Time to Harmonize Dengue Nomenclature and Classification

 , , , ,
, , , , {kind=link}
Abstract
:1. Letter
- (a)
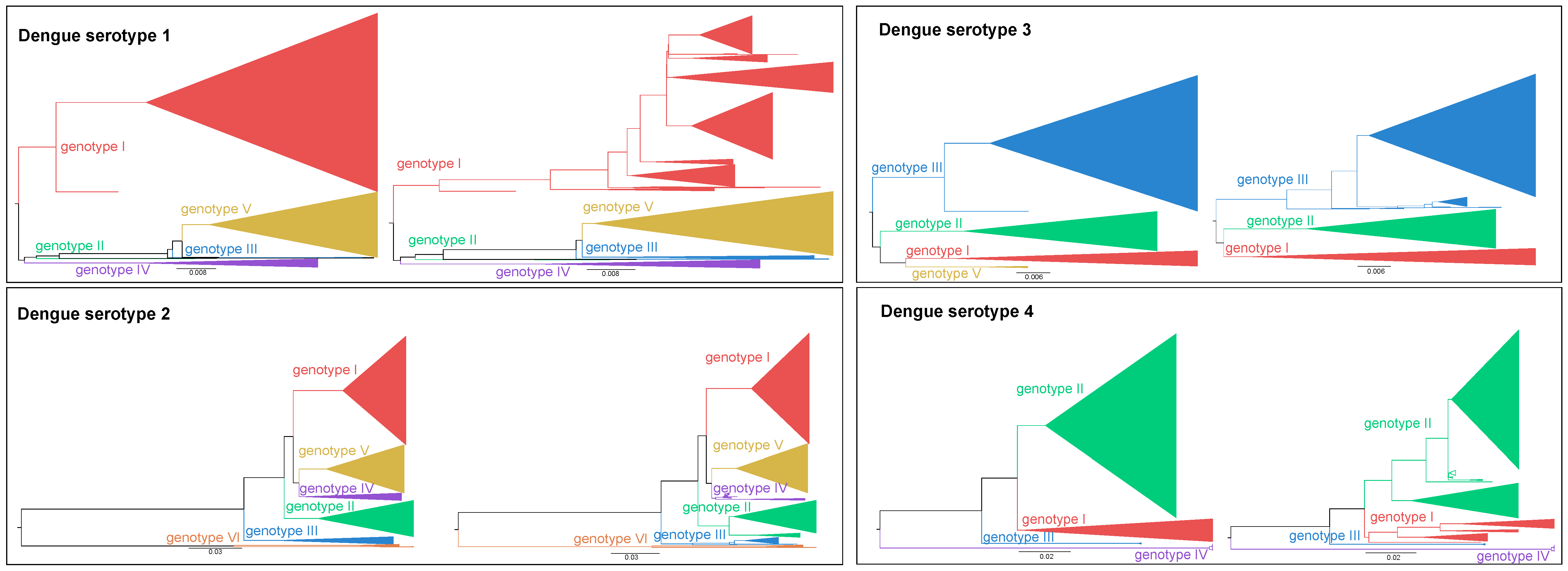
- The phylogenetic signal of the different DENV genes was assessed (see Supporting Information: Figure S1a). This signal is a measure of statistical dependence among species due to their phylogenetic relationships and is associated with the accuracy of phylogenetic studies [23]. The results of this analysis indicate that only parts of the DENV genome are suitable to classify sequences, at genotype and sub-genotype level, with high confidence. Although the envelope gene region is most often used for classification purposes, given its historical, diagnostic and functional importance, our evaluation shows that other genetic regions, such as NS1, NS3, and NS5 exhibit higher phylogenetic support. Whole-genome sequences provide superior classification precision and their availability is expected to increase in the near future when next-generation sequencing becomes routine practice, which will create an opportunity to harmonize DENV classification. Therefore, a detailed analysis of the classification potential of different genomic regions (as well as combinations of such genomic regions) is imperative to propose an adequate classification protocol.
- (b)
- We identified that particular clades are not clustering consistently over the entire genome and established these to be clades with a potential recombinant origin (see Supplementary Materials: Figure S1b). This highlights the need for any future classification protocol to assess the recombination signal of strains by identifying recombinant breakpoints prior to their classification. In addition, it illustrates the necessity to carefully select reference strains to perform consistent and sound classifications, in contrast to the widespread ad hoc classification in much of the current literature.
- (c)
- Certain whole-genome strains do not cluster with any known genotype. As we verified that these strains are not inter-genotypic recombinants (for details see Supplementary Materials: “Dataset and alignment” section), these strains appear to be outliers that the currently described genotypes fail to cover (see Supplementary Materials: Figure S1c and Table S1). To improve our understanding of their origin and whether these outliers indicate the source of novel genotypic or sub-genotypic clades, an in-depth analysis considering both a representative dataset and formal genotype definition is warranted.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gyawali, N.; Bradbury, R.S.; Taylor-Robinson, A.W. The epidemiology of dengue infection: Harnessing past experience and current knowledge to support implementation of future control strategies. J. Vector Brone Dis. 2016, 53, 293–304. [Google Scholar]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepard, D.S.; Undurraga, E.A.; Halasa, Y.A.; Stanaway, J.D. The global economic burden of dengue: A systematic analysis. Lancet Infect. Dis. 2016, 16, 935–941. [Google Scholar] [CrossRef]
- Messina, J.P.; Brady, O.J.; Scott, T.W.; Zou, C.; Pigott, D.M.; Duda, K.A.; Bhatt, S.; Katzelnick, L.; Howes, R.E.; Battle, K.E.; et al. Global spread of dengue virus types: Mapping the 70 year history. Trends Microbiol. 2014, 22, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Tatem, A.J.; Lemey, P. Virus evolution and transmission in an ever more connected world. Proc. Biol. Sci. 2015, 282, 20142878. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.E.; Quam, M.B.; Wilder-Smith, A. Epidemiology of dengue: Past, present and future prospects. Clin. Epidemiol. 2013, 5, 229–309. [Google Scholar] [CrossRef]
- Nunes, M.R.; Palacios, G.; Faria, N.R.; Sousa, E.C., Jr.; Pantoja, J.A.; Rodrigues, S.G.; Carvalho, V.L.; Medeiros, D.B.; Savji, N.; Baele, G.; et al. Air travel is associated with intracontinental spread of dengue virus serotypes 1-3 in Brazil. PLoS Negl. Trop. Dis. 2014, 8, e2769. [Google Scholar] [CrossRef] [PubMed]
- Medlock, J.M.; Leach, S.A. Effect of climate change on vector-borne disease risk in the UK. Lancet Infect. Dis. 2015, 15, 721–730. [Google Scholar] [CrossRef]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Rico-Hesse, R. Microevolution and virulence of dengue viruses. Adv. Virus Res. 2003, 59, 315–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rico-Hesse, R. Dengue virus markers of virulence and pathogenicity. Future Virol. 2009, 4, 581–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rico-Hesse, R. Dengue virus virulence and transmission determinants. Curr. Top. Microbiol. Immunol. 2010, 338, 45–55. [Google Scholar] [CrossRef] [PubMed]
- De Melo, F.L.; Romano, C.M.; de Andrade Zanotto, P.M. Introduction of dengue virus 4 (DENV-4) genotype I into Brazil from Asia? PLoS Negl. Trop. Dis. 2009, 3, e390. [Google Scholar] [CrossRef] [PubMed]
- Trout, A.; Baracco, G.; Rodriguez, M. Locally acquired dengue, Key West, Florida 2009–2010. Morb. Mortal. Wkly. Rep. 2010, 59, 577–581. [Google Scholar]
- Rico-Hesse, R. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology 1990, 174, 479–493. [Google Scholar] [CrossRef]
- Twiddy, S.S.; Farrar, J.J.; Vinh Chau, N.; Wills, B.; Gould, E.A.; Gritsun, T.; Lloyd, G.; Holmes, E.C. Phylogenetic relationships and differential selection pressures among genotypes of dengue-2 virus. Virology 2002, 298, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Libin, P.; Deforche, K.; Theys, K.; Abecasis, A. VIRULIGN: Fast codon-correct alignment and annotation of viral genomes. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Kraemer, M.U.G.; Hill, S.C.; de Jesus, J.G.; de Aguiar, R.S.; Iani, F.C.; Xavier, J.; Quick, J.; de Plessis, L.; Dellicour, S.; et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 2018, 361, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Ragonnet-Cronin, M.; Hodcroft, E.; Hué, S.; Fearnhill, E.; Delpech, V.; Brown, A.J.; Lycett, S. Automated analysis of phylogenetic clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef] [PubMed]
- Santagio, G.A.; McElroy-Horne, K.; Lennon, N.J.; Santiago, L.M.; Birren, B.W.; Henn, M.R.; Muñoz-Jordán, J.L. Reemergence and decline of dengue virus serotype 3 in Puerto Rico. J. Infect. Dis. 2012, 206, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Twiddy, S.S.; Holmes, E.C.; Rambaut, A. Inferring the rate and time-scale of dengue virus evolution. Mol. Biol. Evol. 2003, 20, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Brady, O.J.; Pigott, D.M.; Golding, N.; Kraemer, M.U.; Scott, T.W.; Wint, G.W.; Smith, D.L.; Hay, S.I. The many projected futures of dengue. Nat. Rev. Microbiol. 2015, 13, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Revell, L.J.; Harmon, L.J.; Collar, D.C. Phylogenetic signal, evolutionary process, and rate. Syst. Biol. 2008, 57, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Afreen, N.; Naqvi, I.H.; Broor, S.; Ahmed, A.; Parveen, S. Phylogenetic and molecular clock analysis of dengue serotype 1 and 3 from New Delhi, India. PLoS ONE 2015, 10, e0141628. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.R.; Cruz, A.C.; Vallinoto, M.; Melo, D.D.; Ramos, R.T.; Medeiros, D.B.; Silva, E.V.; Vasconcelos, P.F. Molecular characterization of dengue virus type 1 reveals lineage replacement during circulation in Brazilian territory. Mem. Inst. Oswaldo Cruz 2012, 107, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Katzelnick, L.C.; Fonville, J.M.; Gromowski, G.D.; Arriaga, J.B.; Green, A.; James, S.L.; Lau, L.; Montoya, M.; Wang, C.; VanBlargan, L.A.; et al. Dengue viruses cluster antigenically but not as discrete serotypes. Science 2015, 349, 1338–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuypers, L.; Libin, P.J.K.; Simmonds, P.; Nowé, A.; Muñoz-Jordán, J.; Alcantara, L.C.J.; Vandamme, A.-M.; Santiago, G.A.; Theys, K. Time to Harmonize Dengue Nomenclature and Classification. Viruses 2018, 10, 569. https://0-doi-org.brum.beds.ac.uk/10.3390/v10100569
Cuypers L, Libin PJK, Simmonds P, Nowé A, Muñoz-Jordán J, Alcantara LCJ, Vandamme A-M, Santiago GA, Theys K. Time to Harmonize Dengue Nomenclature and Classification. Viruses. 2018; 10(10):569. https://0-doi-org.brum.beds.ac.uk/10.3390/v10100569
Chicago/Turabian StyleCuypers, Lize, Pieter J. K. Libin, Peter Simmonds, Ann Nowé, Jorge Muñoz-Jordán, Luiz Carlos Junior Alcantara, Anne-Mieke Vandamme, Gilberto A. Santiago, and Kristof Theys. 2018. "Time to Harmonize Dengue Nomenclature and Classification" Viruses 10, no. 10: 569. https://0-doi-org.brum.beds.ac.uk/10.3390/v10100569