Gene Gangs of the Chloroviruses: Conserved Clusters of Collinear Monocistronic Genes

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Assembly of the Chlorovirus Dataset
2.2. Determination of Single-Copy Core Gene Pair Distances
2.2.1. Strictly Constrained Bin Analysis
2.2.2. Growing Bin Analysis
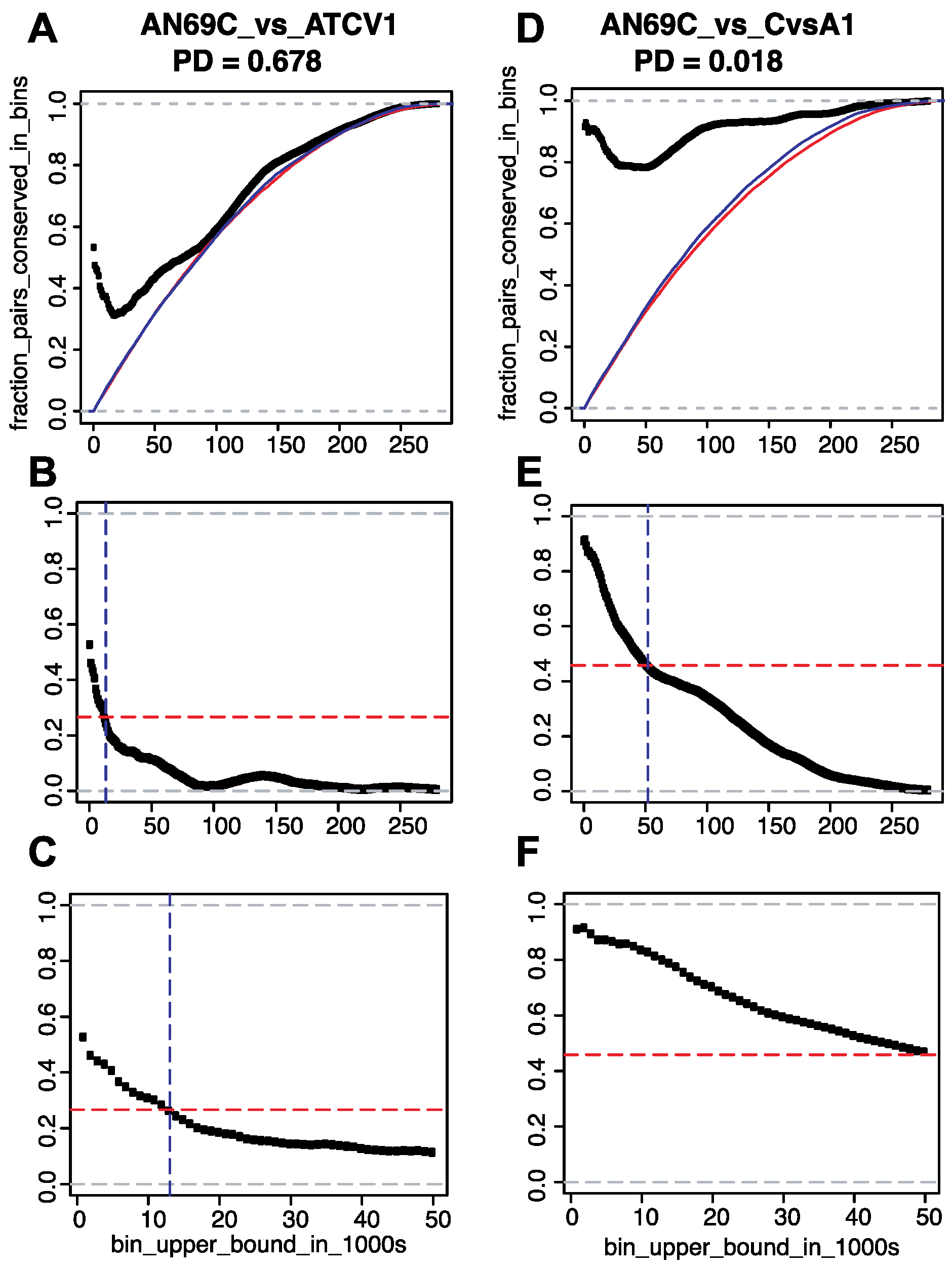
2.3. Determination of Phylogenetic Distances
2.4. Identification of Gene Gangs
2.5. Functional Annotation Methods
3. Results
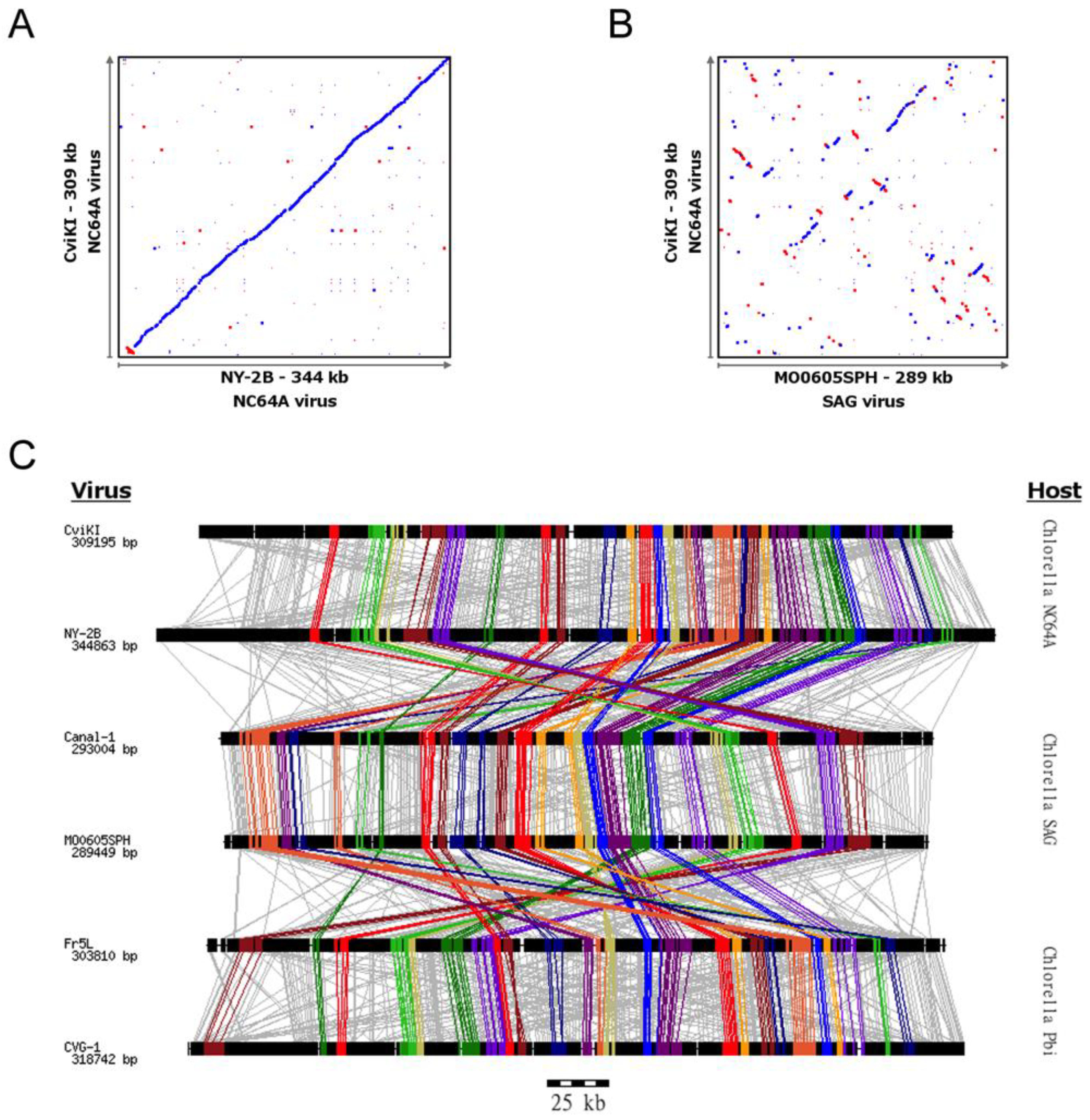
3.1. Chlorovirus Genomic Stability
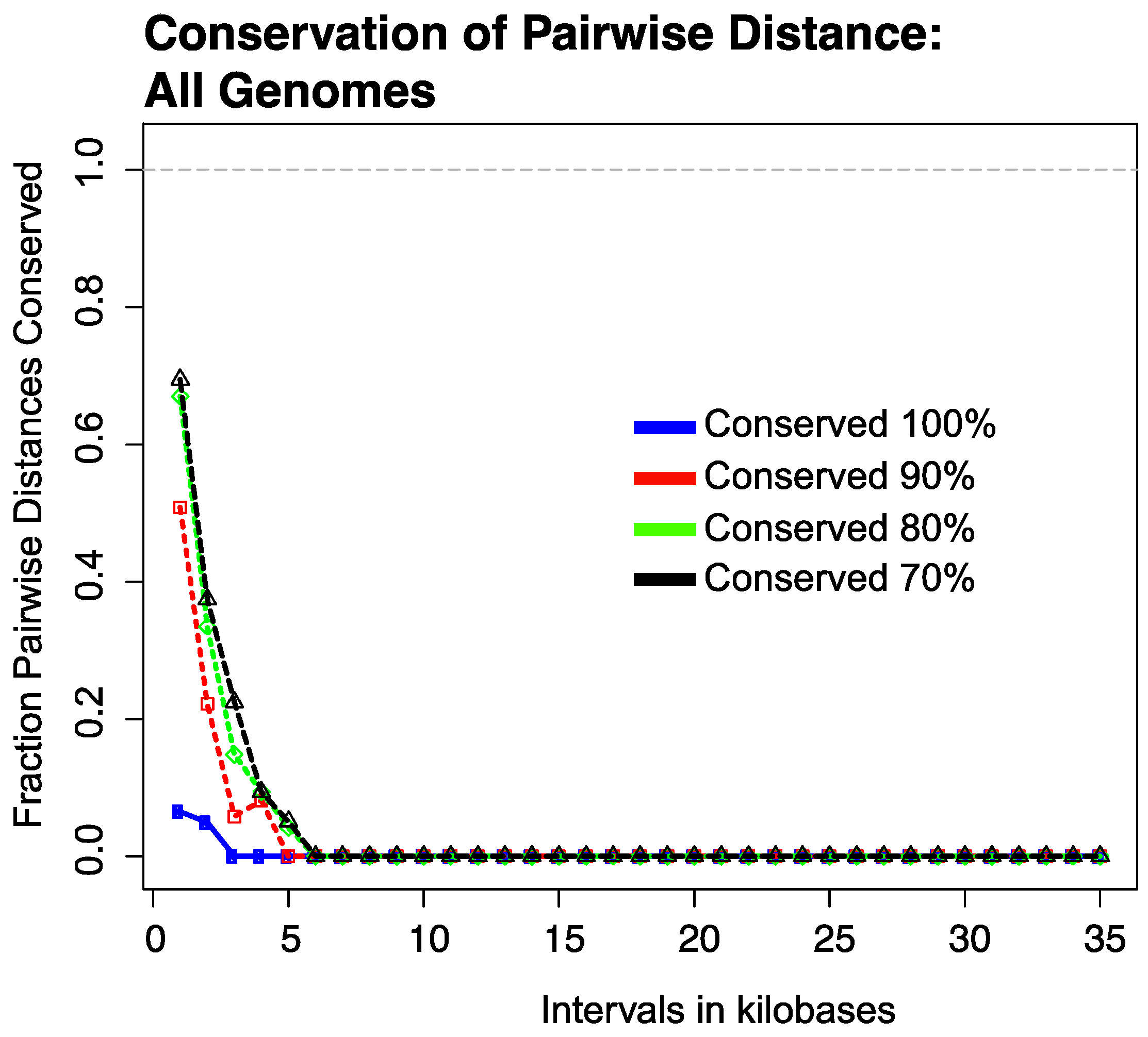
3.2. Conservation of Distance between Single Copy Core Genes
3.2.1. Results of Strictly Constrained Bin Analysis
3.2.2. Results of Growing Bin Analysis or Pairwise Distances
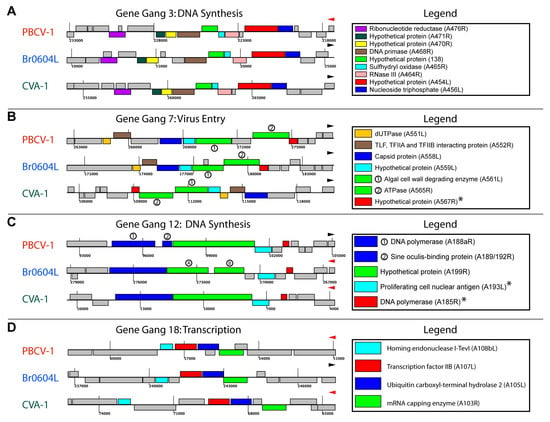
3.3. Identification of Gene Gangs
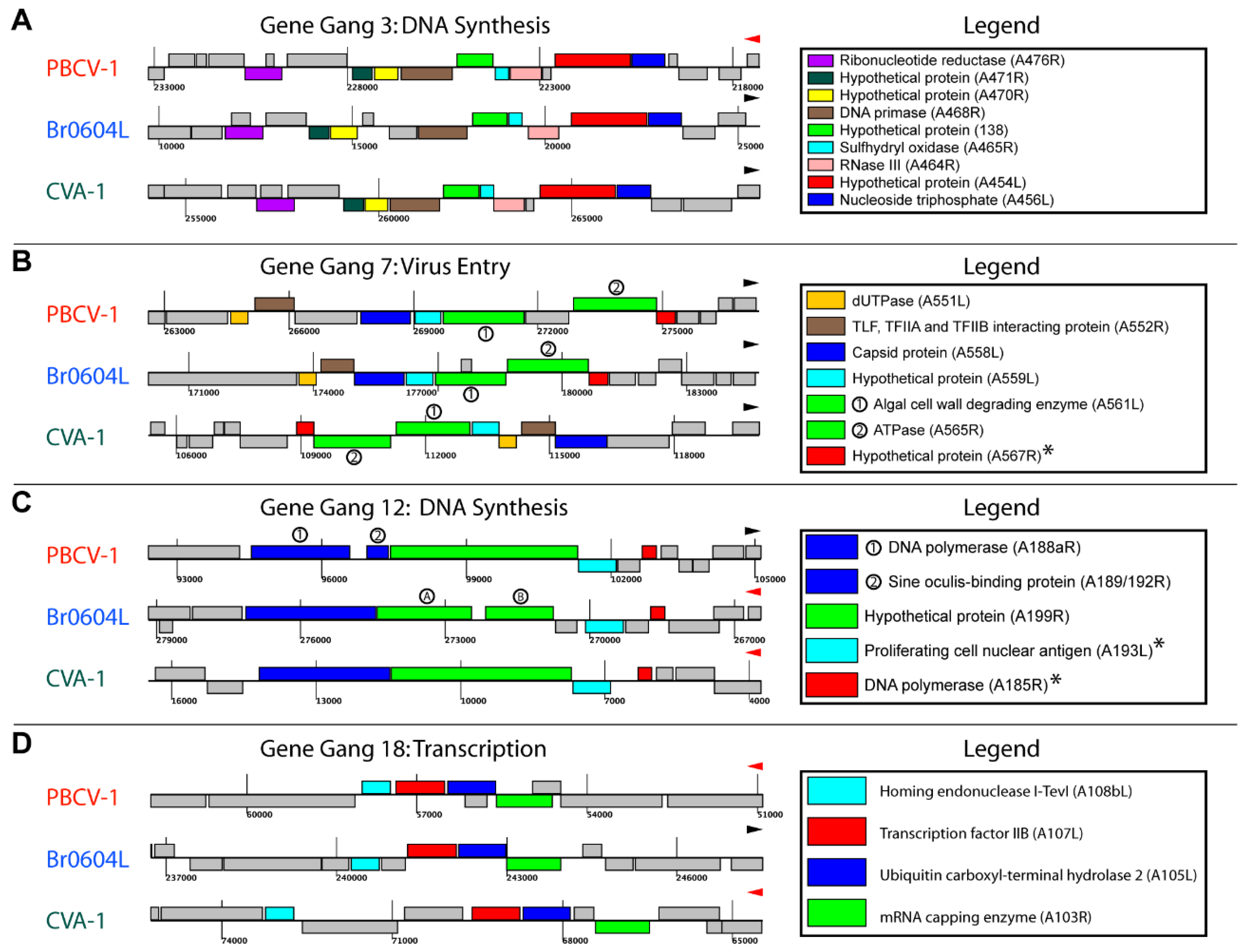
3.4. Topological Features of Selected Gene Gangs
3.5. Functional Evaluation of Gene Gangs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dunigan, D.D.; Fitzgerald, L.; Van Etten, J.L. Phycodnaviruses: A peek at genetic diversity. Virus Res. 2006, 117, 119–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Etten, J.L.; Dunigan, D.D. Chloroviruses: Not your everyday plant virus. Trends Plant Sci. 2012, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Dunigan, D.D. Giant Chloroviruses: Five Easy Questions. PLoS Pathog. 2016, 12, e1005751. [Google Scholar] [CrossRef] [PubMed]
- Jeanniard, A.; Dunigan, D.D.; Gurnon, J.R.; Agarkova, I.V.; Kang, M.; Vitek, J.; Duncan, G.; McClung, O.W.; Larsen, M.; Claverie, J.M.; et al. Towards defining the chloroviruses: A genomic journey through a genus of large DNA viruses. BMC Genom. 2013, 14, 158. [Google Scholar] [CrossRef] [PubMed]
- Seitzer, P.; Huynh, T.A.; Facciotti, M.T. JContextExplorer: A tree-based approach to facilitate cross-species genomic context comparison. BMC Bioinform. 2013, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A language and environment for statistical computing. The R Project for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 11 July 2016).
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Hackathon, R.; Bolker, B.; Butler, M.; Cowan, P.; de Vienne, D.; Eddelbuettel, D.; Holder, M.; Jombart, T.; Kembel, S.; Michonneau, F.; et al. phylobase: Base Package for Phylogenetic Structures and Comparative Data. R package version 0.8.4. Available online: https://CRAN.Rproject.org/package=phylobase (accessed on 11 July 2016).
- Dragulescu, A.A.; Arendt, C. xlsx: Read, Write, Format Excel 2007 and Excel97/2000/XP/2003 Files. R package version 0.6.1. Available online: https://CRAN.R-project.org/package=xlsx (accessed on 11 July 2016).
- Jombart, T.; Dray, S. Adephylo: Exploratory analyses for the phylogenetic comparative method. Bioinformatics 2010, 26, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Dunigan, D.D.; Cerny, R.L.; Bauman, A.T.; Roach, J.C.; Lane, L.C.; Agarkova, I.V.; Wulser, K.; Yanai-Balser, G.M.; Gurnon, J.R.; Vitek, J.C.; et al. Paramecium bursaria chlorella virus 1 proteome reveals novel architectural and regulatory features of a giant virus. J. Virol. 2012, 86, 8821–8834. [Google Scholar] [CrossRef] [PubMed]
- Yanai-Balser, G.M.; Duncan, G.A.; Eudy, J.D.; Wang, D.; Li, X.; Agarkova, I.V.; Dunigan, D.D.; Van Etten, J.L. Microarray analysis of Paramecium bursaria chlorella virus 1 transcription. J. Virol. 2010, 84, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Mozar, M.; Agarkova, I.V.; Gurnon, J.R.; Yanai-Balser, G.; Rowe, J.M.; Xia, Y.; Riethoven, J.J.; Dunigan, D.D.; Van Etten, J.L. Deep RNA sequencing reveals hidden features and dynamics of early gene transcription in Paramecium bursaria chlorella virus 1. PLoS ONE 2014, 9, e90989. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, L.A.; Boucher, P.T.; Yanai-Balser, G.M.; Suhre, K.; Graves, M.V.; Van Etten, J.L. Putative gene promoter sequences in the chlorella viruses. Virology 2008, 380, 388–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.J.P.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Fox, N.K.; Brenner, S.E.; Chandonia, J.M. SCOPe: Structural Classification of Proteins—Extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic. Acids. Res. 2014, 42, D304–D309. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S. Guilt-by-association goes global. Nature 2000, 403, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Vey, G. Metagenomic guilt by association: An operonic perspective. PLoS ONE 2013, 8, e71484. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, P.; Gillis, J. Progress and challenges in the computational prediction of gene function using networks: 2012–2013 update. F1000Res. 2013, 2, 230. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kumar, R.; Sakai, A.; Vetting, M.W.; Wood, B.M.; Brown, S.; Bonanno, J.B.; Hillerich, B.S.; Seidel, R.D.; Babbitt, P.C.; et al. Discovery of new enzymes and metabolic pathways by using structure and genome context. Nature 2013, 502, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, S.; Korczynska, M.; Wichelecki, D.J.; San Francisco, B.; Zhao, S.; Rodionov, D.A.; Vetting, M.W.; Al-Obaidi, N.F.; Lin, H.; O’Meara, M.J.; et al. Prediction of enzymatic pathways by integrative pathway mapping. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Wang, X.; Fang, Q.; Van Etten, J.L.; Rossmann, M.G.; Rao, Z.; Zhang, X. Pushing the resolution limit by correcting the Ewald sphere effect in single-particle Cryo-EM reconstructions. Nat. Commun. 2018, 9, 1552. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Tanji, Y.; Hori, K.; Unno, H. Evidence for a novel Chlorella virus-encoded alginate lyase. FEMS Microbiol. Lett. 1999, 180, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, I.; Hiramatsu, S.; Murakami, D.; Fuie, M.; Usami, S.; Yamada, T. Algal-lytic activities encoded by Chlorella virus CVK2. Virology 2000, 277, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Agarkova, I.V.; Dunigan, D.D.; Van Etten, J.L. Virion-associated restriction endonucleases of chloroviruses. J. Virol. 2006, 80, 8114–8123. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.K.; Gong, C.; Shuman, S. RNA triphosphatase component of the mRNA capping apparatus of Paramecium bursaria chlorella virus 1. J. Virol. 2001, 75, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Perrin, D.; Sanchez, C.; Monod, J. The operon: A group of genes whose expression is coordinated by an operator. Science 1960, 250, 1727–1729. [Google Scholar]
- Che, D.; Li, G.; Mao, F.; Wu, H.; Xu, Y. Detecting uber-operons in prokaryotic genomes. Nucleic. Acids. Res. 2006, 34, 2418–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, X.; He, X.; Xin, D. Detecting gene clusters under evolutionary constraint in a large number of genomes. Bioinformatics 2009, 25, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luc, N.; Risler, J.L.; Bergeron, A.; Raffinot, M. Gene teams: A new formalization of gene clusters for comparative genomics. Comput. Biol. Chem. 2003, 27, 59–67. [Google Scholar] [CrossRef]
- Lathe, W.C., III; Snel, B.; Bork, P. Gene context conservation of a higher order than operons. Trends Biochem. Sci. 2000, 25, 474–479. [Google Scholar] [CrossRef]
- Carter, D.; Chakalova, L.; Osborne, C.S.; Dai, Y.F.; Fraser, P. Long-range chromatin regulatory interactions in vivo. Nat. Genet. 2002, 32, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Francastel, C.; Schübeler, D.; Martin, D.I.K.; Groudine, M. Nuclear compartmentalization and gene activity. Nat. Rev. Mol. Cell Biol. 2000, 1, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Spilianakis, C.G.; Lalioti, M.D.; Town, T.; Lee, G.R.; Flavell, R.A. Interchromosomal associations between alternatively expressed loci. Nature 2005, 435, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Arkin, A.P.; Alm, E.J. The life-cycle of operons. PLoS Genet. 2006, 2, e96. [Google Scholar] [CrossRef] [PubMed]
- Vallenet, D.; Belda, E.; Calteau, A.; Cruveiller, S.; Engelen, S.; Lajus, A.; Le Fevre, F.; Longin, C.; Momico, D.; Roche, D.; et al. MicroScope—An integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic. Acids. Res. 2013, 41, D636–D647. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.G.; Roth, J.R. Selfish operons: Horizontal transfer may drive the evolution of gene clusters. Genetics 1996, 143, 1843–1860. [Google Scholar] [PubMed]
- Fang, G.; Rocha, E.P.C.; Danchin, A. Persistence drives gene clustering in bacterial genomes. BMC Genom. 2008, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Huang, K.H.; Arkin, A.P.; Alm, E.J. Operon formation is driven by co-regulation and not by horizontal gene transfer. Genome Res. 2005, 15, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, F.; Monod, J. On the Regulation of Gene Activity. Cold Spring Harbor Symp. Quant. Biol. 1961, 26, 193–211. [Google Scholar] [CrossRef]
- Ballouz, S.; Francis, A.R.; Lan, R.; Tanaka, M.M. Conditions for the evolution of gene clusters in bacterial genomes. PLoS Comput. Biol. 2010, 6, e1000672. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes of Interest | Ruliness | ||||||
|---|---|---|---|---|---|---|---|
| 0.826 | 0.854 | 0.902 | 0.927 | 0.951 | 0.976 | 1.00 | |
| Core Genes | 66.9% | 65.5% | 63.5% | 60.4% | 60.4% | 57.7% | 53.1% |
| All Genes | 35.2% | 33.7% | 31.5% | 29.3% | 29.0% | 27.7% | 24.8% |
| Gang | Maximum Number of Gang Members | Consensus Number of Gene Members of the Gang in Block Diagram $ | Number of Viruses That Are Ruly (100% Conserved in Gene Content), Total = 41 | Number of Viruses That Maintain Synteny, Total = 41 | Number of Viruses with Strand Conservation, Total = 41 | Number of Viruses with Infiltrated Non-Gang (Alien) Genes, Total = 41 |
|---|---|---|---|---|---|---|
| 1 | 11 | 10 | 38 | 41 | 39 | 41 |
| 2 | 9 | 8 | 40 | 40 | 40 | 41 |
| 3 | 9 | 9 | 41 | 41 | 27 | 38 |
| 4 | 8 | 8 | 38 | 27 | 27 & | 17 |
| 5 * | 7 | 6 | 37 | Virus type-specific | Virus type-specific | 23 |
| 6 | 6 | 6 | 34 | 40 | 40 | 41 |
| 7 | 7 | 7 | 40 | 28 | 28 | 21 |
| 8 | 6 | 6 | 35 | 27 | 27 | 25 |
| 9 | 6 | 5 | 39 | 39 | 28 | 37 |
| 10 | 5 | 5 | 33 | 33 | 33 | 3 |
| 11 | 5 | 5 | 34 | 34 | 34 | 41 |
| 12 | 5 | 4 | 40 | 40 | 40 | 35 |
| 13 | 4 | 4 | 40 | 40 | 27 | 13 |
| 14 | 4 | 4 | 35 | 35 | 35 | 38 |
| 15 | 4 | 4 | 34 | 27 | 27 | 30 |
| 16 | 4 | 4 | 35 | 35 | 35 | 26 |
| 17 | 4 | 4 | 41 | 27 | 27 | 35 |
| 18 | 4 | 4 | 41 | 41 # | 28 | 28 |
| 19 | 3 | 3 | 41 | 41 | 41 | 24 |
| 20 | 3 | 3 | 39 | 39 | 39 | 9 |
| 21 | 3 | 3 | 41 | 41 | 41 | 16 |
| 22 | 3 | 3 | 39 | 39 | 39 | 11 |
| 23 | 3 | 3 | 29 | 29 | 29 | 37 |
| 24 | 3 | 3 | 34 | 34 | 34 | 37 |
| 25 | 3 | 3 | 36 | 36 | 36 | 26 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seitzer, P.; Jeanniard, A.; Ma, F.; Van Etten, J.L.; Facciotti, M.T.; Dunigan, D.D. Gene Gangs of the Chloroviruses: Conserved Clusters of Collinear Monocistronic Genes. Viruses 2018, 10, 576. https://0-doi-org.brum.beds.ac.uk/10.3390/v10100576
Seitzer P, Jeanniard A, Ma F, Van Etten JL, Facciotti MT, Dunigan DD. Gene Gangs of the Chloroviruses: Conserved Clusters of Collinear Monocistronic Genes. Viruses. 2018; 10(10):576. https://0-doi-org.brum.beds.ac.uk/10.3390/v10100576
Chicago/Turabian StyleSeitzer, Phillip, Adrien Jeanniard, Fangrui Ma, James L. Van Etten, Marc T. Facciotti, and David D. Dunigan. 2018. "Gene Gangs of the Chloroviruses: Conserved Clusters of Collinear Monocistronic Genes" Viruses 10, no. 10: 576. https://0-doi-org.brum.beds.ac.uk/10.3390/v10100576