Human Fetal Astrocytes Infected with Zika Virus Exhibit Delayed Apoptosis and Resistance to Interferon: Implications for Persistence

 ,
, 
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Virus Strains and Cell Lines
2.3. Virus Infection
2.4. ZIKV Titration
2.5. Quantitative Reverse-Transcription PCR Assay
2.6. Enzyme-Linked Immunosorbent Assay
2.7. Cell Viability Assay
2.8. Immunofluorescence Staining and Imaging Analysis
2.9. Flow Cytometry
2.10. Virus Entry Inhibition Assays
2.11. Viral Persistence Assay
2.12. Statistical Analyses
2.13. RNAseq Libraries
2.14. Bioinformatics
2.15. Accession Number
3. Results
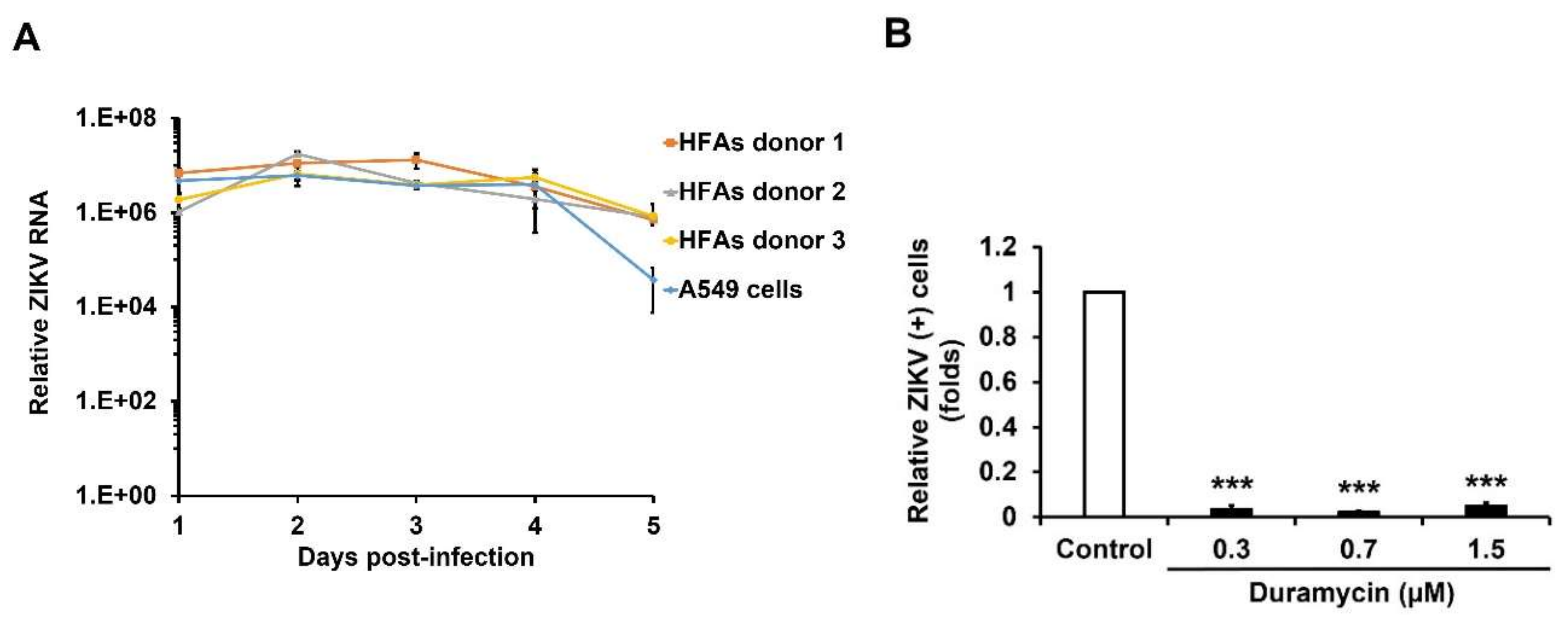
3.1. ZIKV Infects TIM/TAM-Expressing Cells
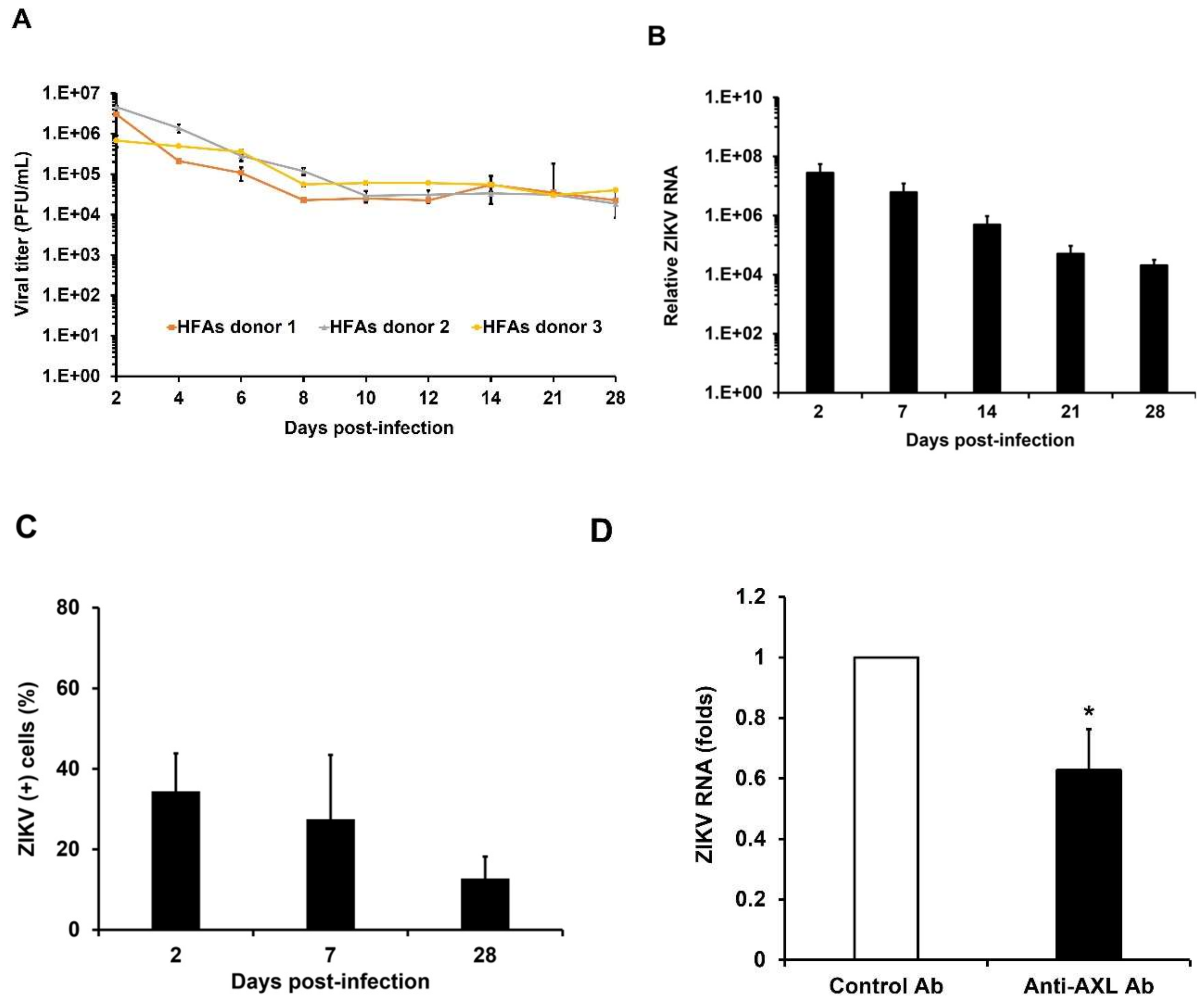
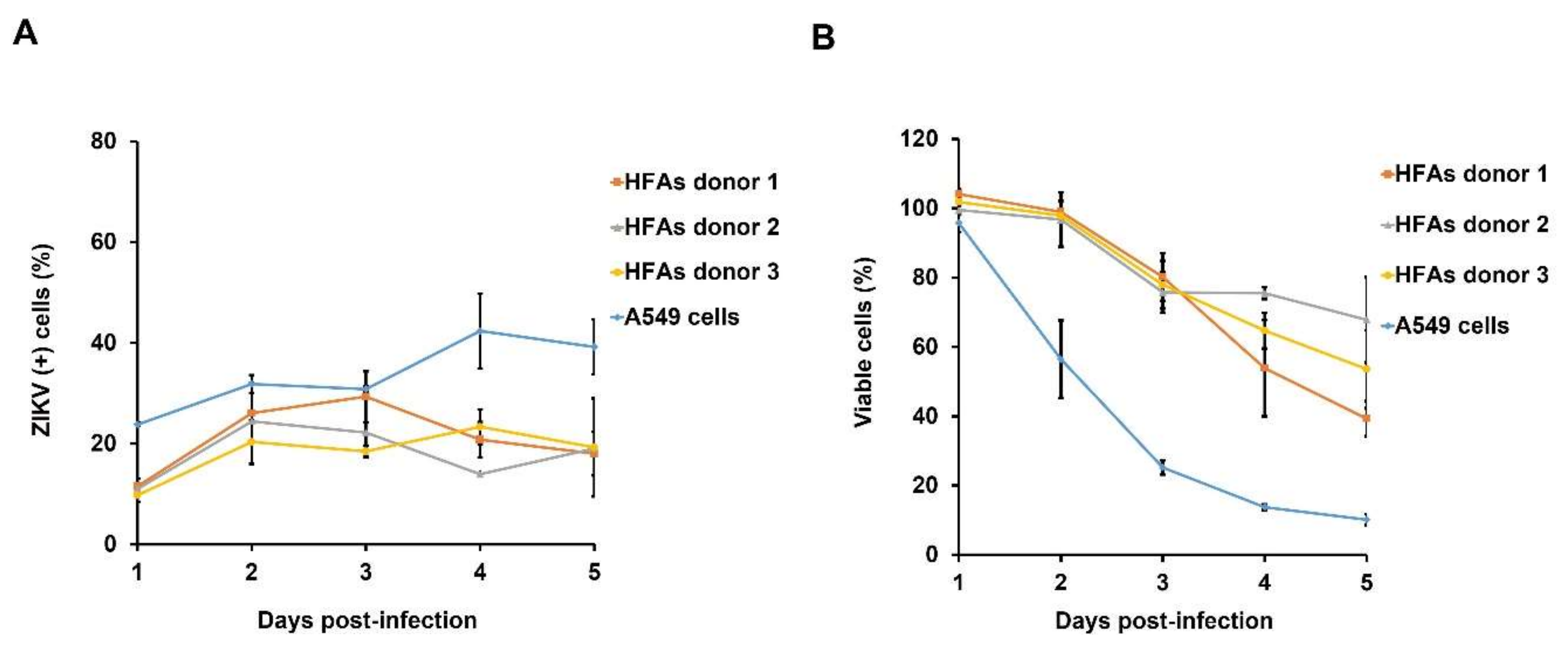
3.2. Kinetics of ZIKV Persistent Infection in HFAs
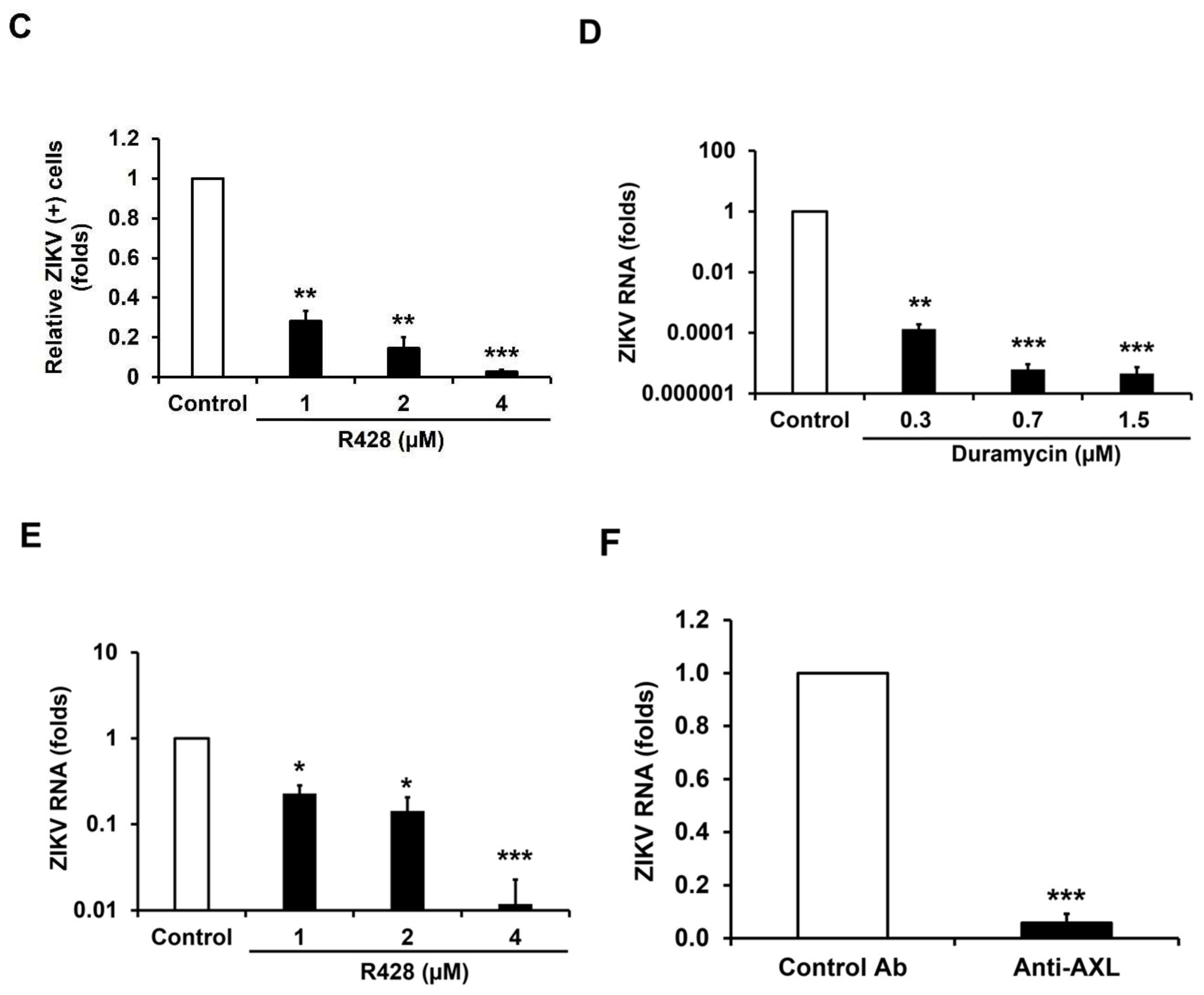
3.3. AXL Mediates Viral Spread During ZIKV Persistence
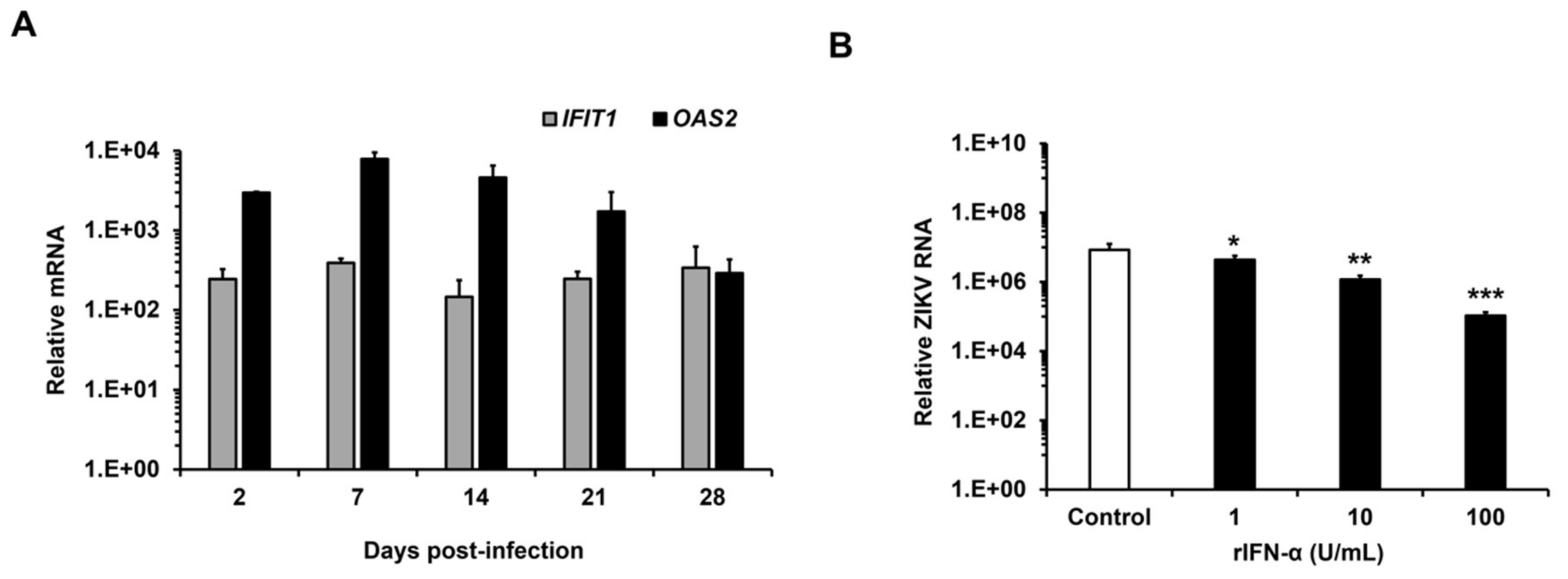
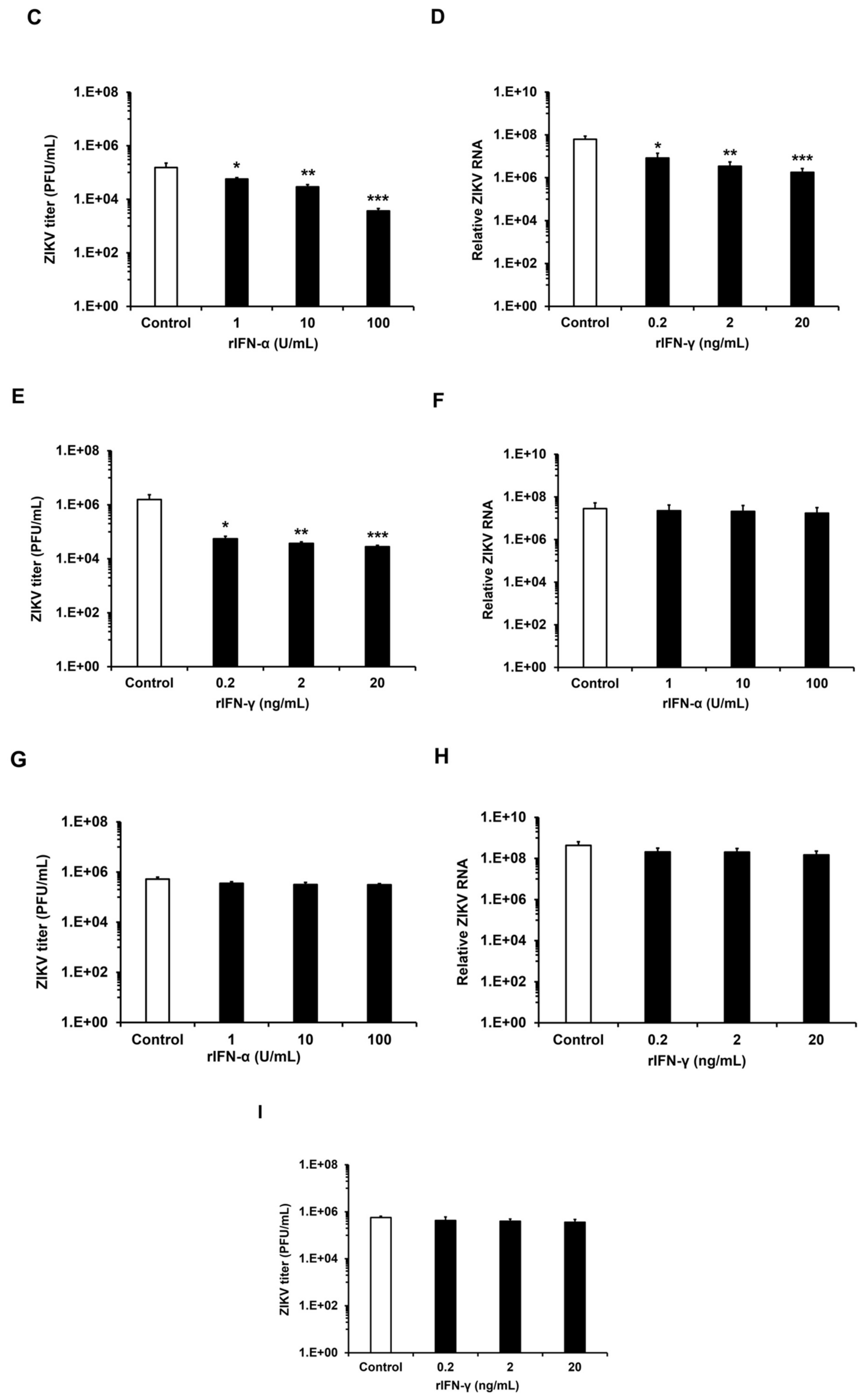
3.4. ZIKV Replicates in HFAs Despite a Robust Antiviral Response
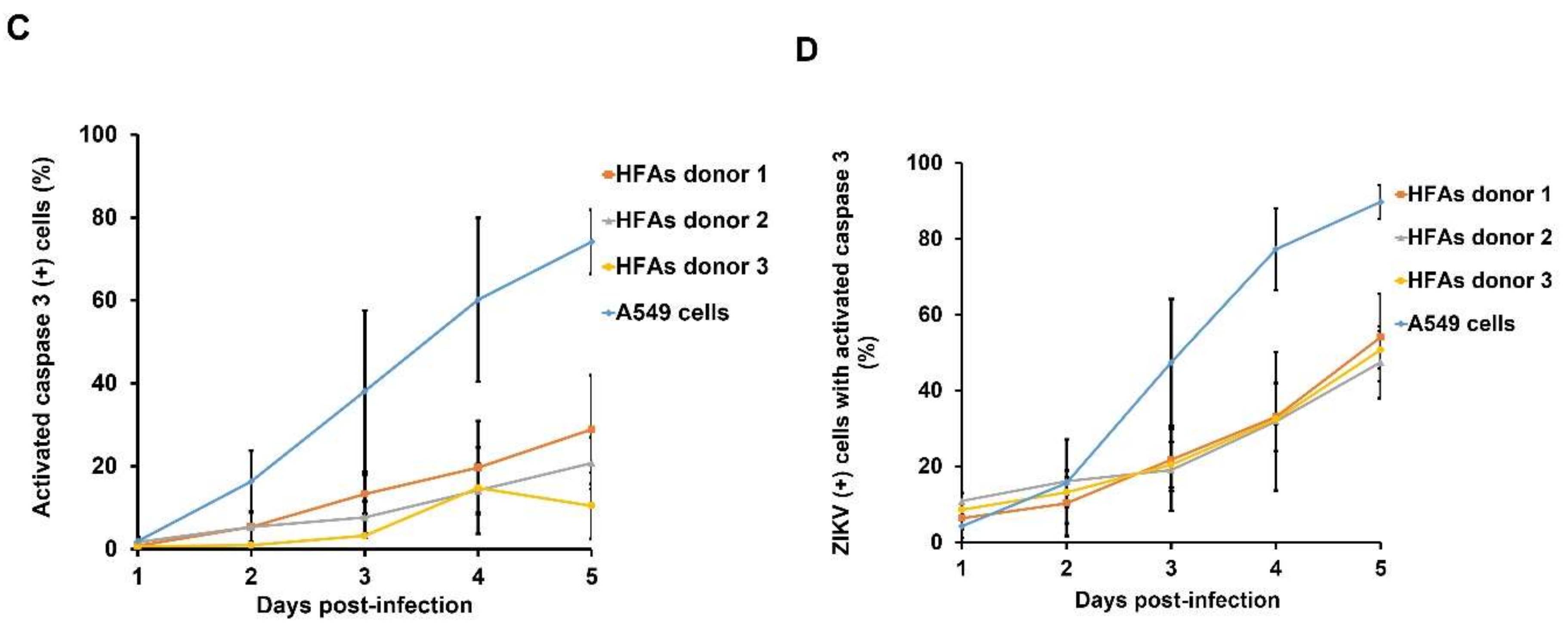
3.5. Resistance to Apoptosis Contributes to Persistent Infection of HFAs
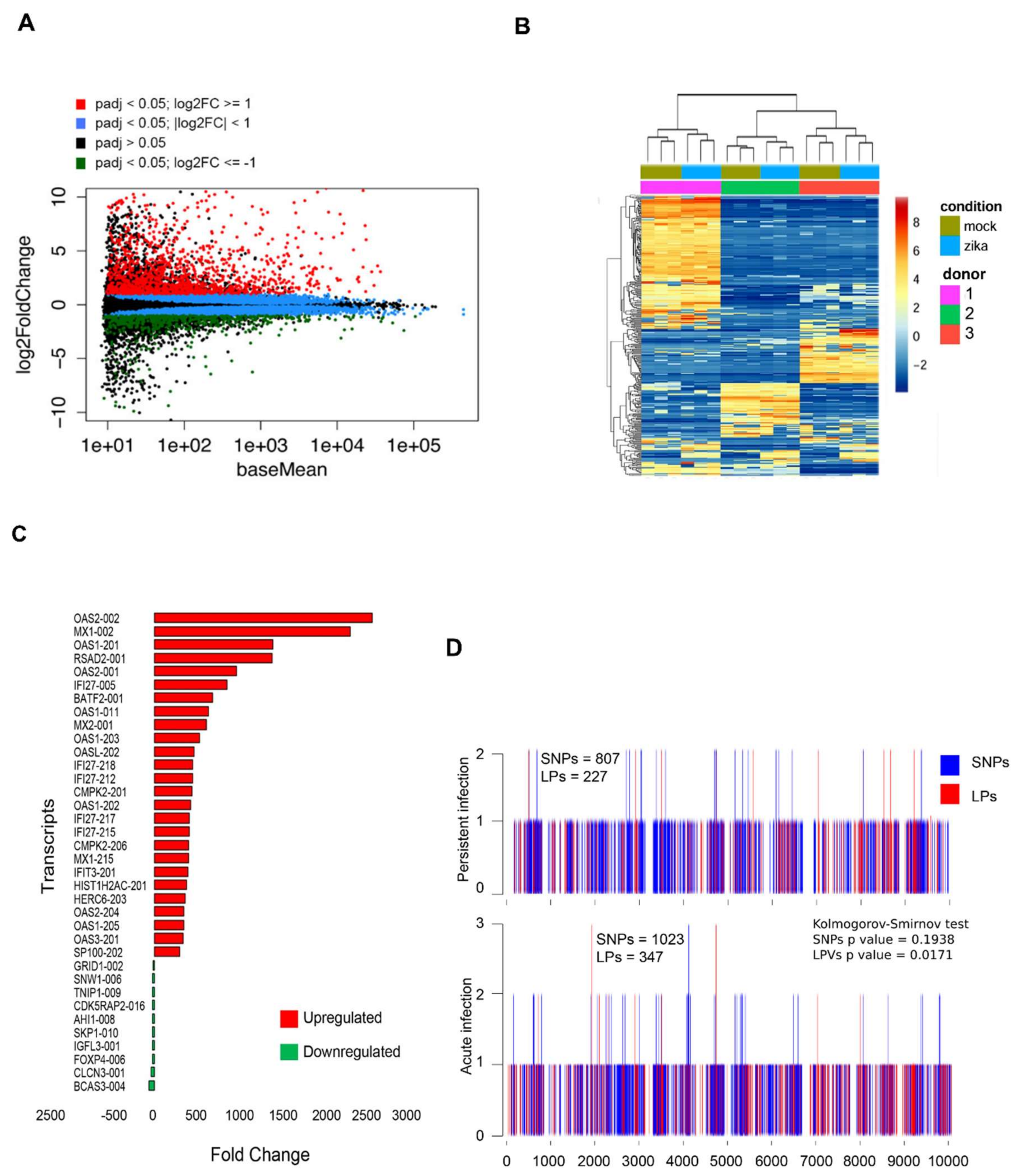
3.6. Persistent ZIKV Infection Has A Dramatic Effect on HFA Transcription
3.7. No Significant Mutations Are Accumulated in the ZIKV Genome After Persistence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Brasil, P.; Pereira, J.P., Jr.; Moreira, M.E.; Ribeiro Nogueira, R.M.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N. Engl. J. Med. 2016, 375, 2321–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franca, G.V.; Schuler-Faccini, L.; Oliveira, W.K.; Henriques, C.M.; Carmo, E.H.; Pedi, V.D.; Nunes, M.L.; Castro, M.C.; Serruya, S.; Silveira, M.F.; et al. Congenital Zika virus syndrome in Brazil: A case series of the first 1501 livebirths with complete investigation. Lancet 2016, 388, 891–897. [Google Scholar] [CrossRef]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanners, N.W.; Eitson, J.L.; Usui, N.; Richardson, R.B.; Wexler, E.M.; Konopka, G.; Schoggins, J.W. Western Zika Virus in Human Fetal Neural Progenitors Persists Long Term with Partial Cytopathic and Limited Immunogenic Effects. Cell Rep. 2016, 15, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Pol, A.N.; Mao, G.; Yang, Y.; Ornaghi, S.; Davis, J.N. Zika virus targeting in the developing brain. J. Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, R.; Mundt, F.; Gilthorpe, J.D.; Wolfel, S.; Gekara, N.O.; Kroger, A.; Overby, A.K. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflammation 2016, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Hollidge, B.; Daye, S.; Zeng, X.; Blancett, C.; Kuszpit, K.; Bocan, T.; Koehler, J.W.; Coyne, S.; Minogue, T.; et al. Neuropathogenesis of Zika Virus in a Highly Susceptible Immunocompetent Mouse Model after Antibody Blockade of Type, I. Interferon. PLoS Negl. Trop. Dis. 2017, 11, e0005296. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, T.J.; Pollen, A.A.; di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Mancia Leon, W.R.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Yang, Y.F.; Yang, Y.; Zou, P.; He, Y.; Shui, S.L.; Cui, Y.R.; Bai, R.; Liang, Y.J.; Hu, Y.; et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, K.; Meatherall, B.; Zarra, D.; Drebot, M.; MacDonald, J.; Pabbaraju, K.; Wong, S.; Webster, P.; Lindsay, R.; Tellier, R. First case of Zika virus infection in a returning Canadian traveler. Am. J. Trop. Med. Hyg. 2014, 91, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Lambert, A.J.; Holodniy, M.; Saavedra, S.; Signor Ldel, C. Phylogeny of Zika Virus in Western Hemisphere, 2015. Emerg. Infect. Dis. 2016, 22, 933–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, M.C.; Sourisseau, M.; Espino, M.M.; Gray, E.S.; Chambers, M.T.; Tortorella, D.; Evans, M.J. Rescue of the 1947 Zika Virus Prototype Strain with a Cytomegalovirus Promoter-Driven cDNA Clone. mSphere 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Callaway, J.B.; Smith, S.A.; Widman, D.G.; McKinnon, K.P.; Scholle, F.; Sempowski, G.D.; Dittmer, D.P.; Crowe, J.E., Jr.; de Silva, A.M.; Ting, J.P. Source and Purity of Dengue-Viral Preparations Impact Requirement for Enhancing Antibody to Induce Elevated IL-1beta Secretion: A Primary Human Monocyte Model. PLoS ONE 2015, 10, e0136708. [Google Scholar] [CrossRef] [PubMed]
- Vivithanaporn, P.; Maingat, F.; Lin, L.T.; Na, H.; Richardson, C.D.; Agrawal, B.; Cohen, E.A.; Jhamandas, J.H.; Power, C. Hepatitis C virus core protein induces neuroimmune activation and potentiates Human Immunodeficiency Virus-1 neurotoxicity. PLoS ONE 2010, 5, e12856. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Jovel, J.; Lopez-Orozco, J.; Limonta, D.; Airo, A.M.; Hou, S.; Stryapunina, I.; Fibke, C.; Moore, R.B.; Hobman, T.C. Human Sertoli cells support high levels of Zika virus replication and persistence. Sci. Rep. 2018, 8, 5477. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, H.; Bray, N.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-Seq incorporating quantification uncertainty. biorxiv 2016. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Charlebois, P.; Macalalad, A.; Henn, M.R.; Zody, M.C. V-Phaser 2: Variant inference for viral populations. BMC Genomics 2013, 14, 674. [Google Scholar] [CrossRef] [PubMed]
- McLendon, R.E.; Bigner, D.D. Immunohistochemistry of the glial fibrillary acidic protein: Basic and applied considerations. Brain Pathol. 1994, 4, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus entry receptors: An update. Viruses 2013, 6, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Bayless, N.L.; Greenberg, R.S.; Swigut, T.; Wysocka, J.; Blish, C.A. Zika Virus Infection Induces Cranial Neural Crest Cells to Produce Cytokines at Levels Detrimental for Neurogenesis. Cell Host Microbe 2016, 20, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jaaskelainen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Kari, V.; Karpiuk, O.; Tieg, B.; Kriegs, M.; Dikomey, E.; Krebber, H.; Begus-Nahrmann, Y.; Johnsen, S.A. A subset of histone H2B genes produces polyadenylated mRNAs under a variety of cellular conditions. PLoS ONE 2013, 8, e63745. [Google Scholar] [CrossRef] [PubMed]
- Sin, C.; Li, H.; Crawford, D.A. Transcriptional regulation by FOXP1, FOXP2, and FOXP4 dimerization. J. Mol. Neurosci. 2015, 55, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.S.S.; de Sousa, J.R.; Araujo, M.T.F.; Martins Filho, A.J.; de Alcantara, B.N.; Araujo, F.M.C.; Queiroz, M.G.L.; Cruz, A.C.R.; Vasconcelos, B.H.B.; Chiang, J.O.; et al. In situ immune response and mechanisms of cell damage in central nervous system of fatal cases microcephaly by Zika virus. Sci. Rep. 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams Waldorf, K.M.; Stencel-Baerenwald, J.E.; Kapur, R.P.; Studholme, C.; Boldenow, E.; Vornhagen, J.; Baldessari, A.; Dighe, M.K.; Thiel, J.; Merillat, S.; et al. Fetal brain lesions after subcutaneous inoculation of Zika virus in a pregnant nonhuman primate. Nat. Med. 2016, 22, 1256–1259. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Maestre, A.M.; Fernandez-Sesma, A.; Garcia-Sastre, A. Antagonism of type I interferon by flaviviruses. Biochem. Biophys. Res. Commun. 2017, 492, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.S.; Aguiar, R.S.; Amorim, M.M.; Arruda, M.B.; Melo, F.O.; Ribeiro, S.T.; Batista, A.G.; Ferreira, T.; Dos Santos, M.P.; Sampaio, V.V.; et al. Congenital Zika Virus Infection: Beyond Neonatal Microcephaly. JAMA Neurol. 2016, 73, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.F.; Salick, M.R.; Wiskow, O.; Ho, D.J.; Worringer, K.A.; Ihry, R.J.; Kommineni, S.; Bilican, B.; Klim, J.R.; Hill, E.J.; et al. Genetic Ablation of AXL Does Not Protect Human Neural Progenitor Cells and Cerebral Organoids from Zika Virus Infection. Cell Stem Cell 2016, 19, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Massey, A.; Shives, K.D.; Stenglein, M.D.; Ebel, G.D.; Beckham, J.D. West Nile Virus Population Structure, Injury, and Interferon-Stimulated Gene Expression in the Brain From a Fatal Case of Encephalitis. Open Forum Infect. Dis. 2016, 3, ofv182. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Preusser, M.; Laggner, U.; Garzuly, F.; Holzmann, H.; Heinz, F.X.; Budka, H. Inflammatory response in human tick-borne encephalitis: Analysis of postmortem brain tissue. J. Neurovirol. 2006, 12, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Limonta, D.; Capo, V.; Torres, G.; Perez, A.B.; Guzman, M.G. Apoptosis in tissues from fatal dengue shock syndrome. J. Clin. Virol. 2007, 40, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Ferraris, P.; Wichit, S.; Diop, F.; Talignani, L.; Pompon, J.; Garcia, D.; Liegeois, F.; Sall, A.A.; Yssel, H.; et al. African and Asian Zika virus strains differentially induce early antiviral responses in primary human astrocytes. Infect. Genet. Evol. 2017, 49, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goo, L.; VanBlargan, L.A.; Dowd, K.A.; Diamond, M.S.; Pierson, T.C. A single mutation in the envelope protein modulates flavivirus antigenicity, stability, and pathogenesis. PLoS Pathog. 2017, 13, e1006178. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.L.; Fayzulin, R.; Dewsbury, N.; Bourne, N.; Mason, P.W. Mutations in West Nile virus nonstructural proteins that facilitate replicon persistence in vitro attenuate virus replication in vitro and in vivo. Virology 2007, 364, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Fulton, B.O.; Sachs, D.; Schwarz, M.C.; Palese, P.; Evans, M.J. Transposon Mutagenesis of the Zika Virus Genome Highlights Regions Essential for RNA Replication and Restricted for Immune Evasion. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liao, H.M.; Liu, H.; Tsai, S.; Zhang, J.; Hung, G.C.; Chin, P.J.; Gao, Y.; Lo, S.C. Comparative genomics, infectivity and cytopathogenicity of Zika viruses produced by acutely and persistently infected human hematopoietic cell lines. PLoS ONE 2018, 13, e0203331. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limonta, D.; Jovel, J.; Kumar, A.; Airo, A.M.; Hou, S.; Saito, L.; Branton, W.; Ka-Shu Wong, G.; Mason, A.; Power, C.; et al. Human Fetal Astrocytes Infected with Zika Virus Exhibit Delayed Apoptosis and Resistance to Interferon: Implications for Persistence. Viruses 2018, 10, 646. https://0-doi-org.brum.beds.ac.uk/10.3390/v10110646
Limonta D, Jovel J, Kumar A, Airo AM, Hou S, Saito L, Branton W, Ka-Shu Wong G, Mason A, Power C, et al. Human Fetal Astrocytes Infected with Zika Virus Exhibit Delayed Apoptosis and Resistance to Interferon: Implications for Persistence. Viruses. 2018; 10(11):646. https://0-doi-org.brum.beds.ac.uk/10.3390/v10110646
Chicago/Turabian StyleLimonta, Daniel, Juan Jovel, Anil Kumar, Adriana M. Airo, Shangmei Hou, Leina Saito, William Branton, Gane Ka-Shu Wong, Andrew Mason, Christopher Power, and et al. 2018. "Human Fetal Astrocytes Infected with Zika Virus Exhibit Delayed Apoptosis and Resistance to Interferon: Implications for Persistence" Viruses 10, no. 11: 646. https://0-doi-org.brum.beds.ac.uk/10.3390/v10110646