Simultaneous Detection of Different Zika Virus Lineages via Molecular Computation in a Point-of-Care Assay

 , and
, and 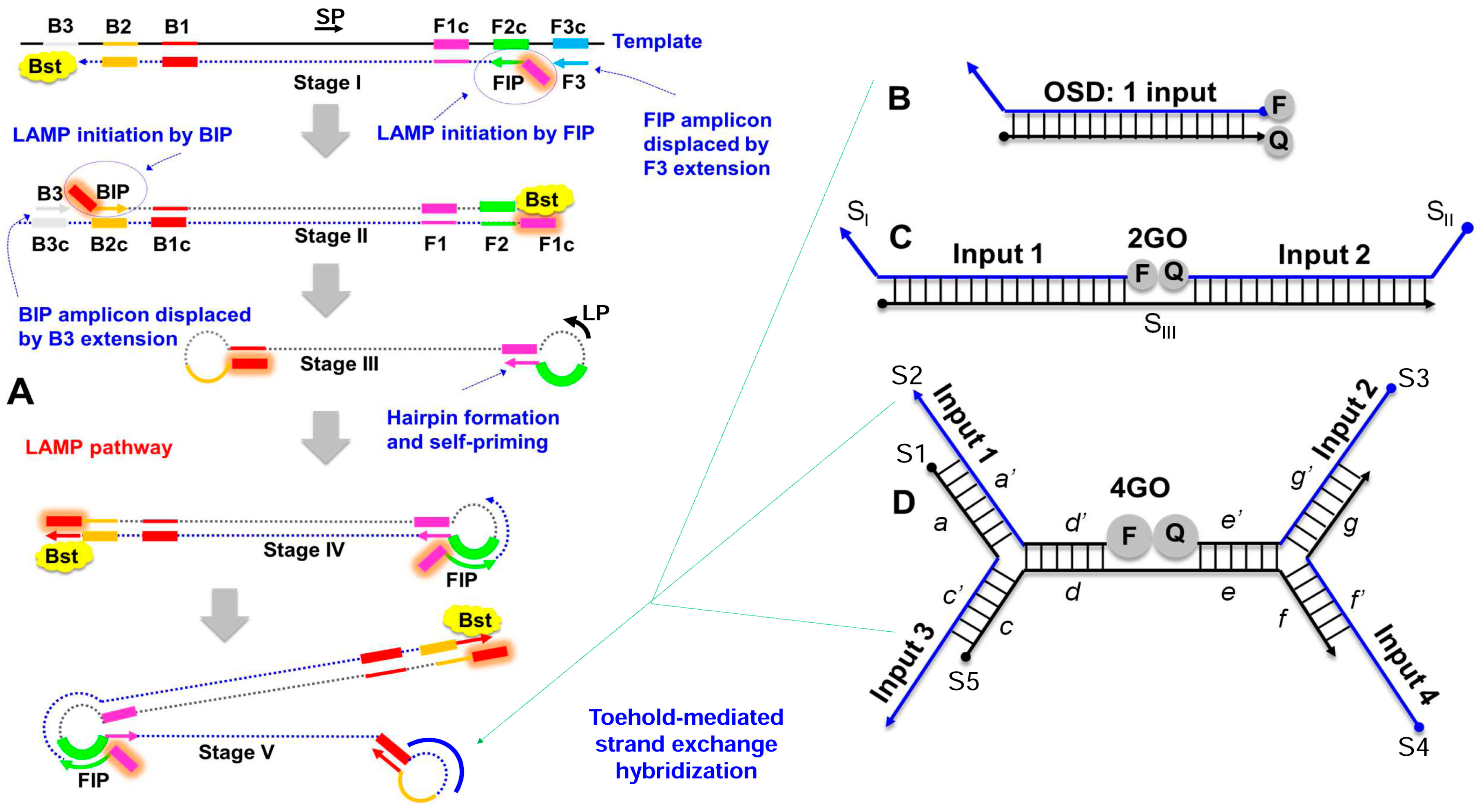{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cloning of gBlocks and PCR Amplification of Transcription Templates
2.3. In Vitro Transcription
2.4. Denaturing Polyacrylamide Gel Electrophoresis and RNA Gel Purification
2.5. LAMP Primer and Strand Exchange Probe Design
2.6. Assembly of Strand Exchange Probes
2.7. RT-LAMP Assay
2.8. TaqMan qRT-PCR Analysis
2.9. Analysis of ZIKV Virions
2.10. Rearing and Analysis of ZIKV-Infected Mosquitoes
3. Results
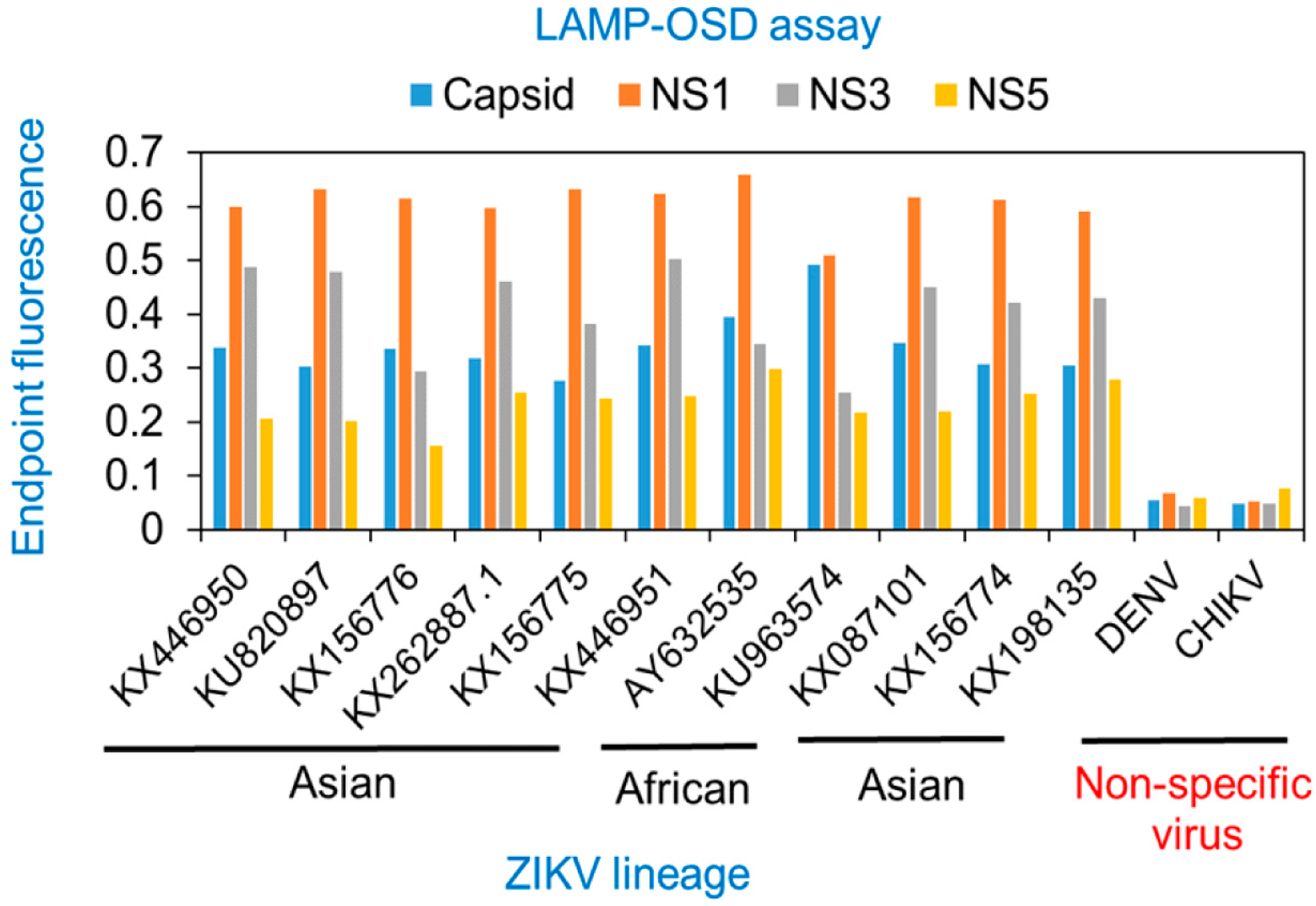
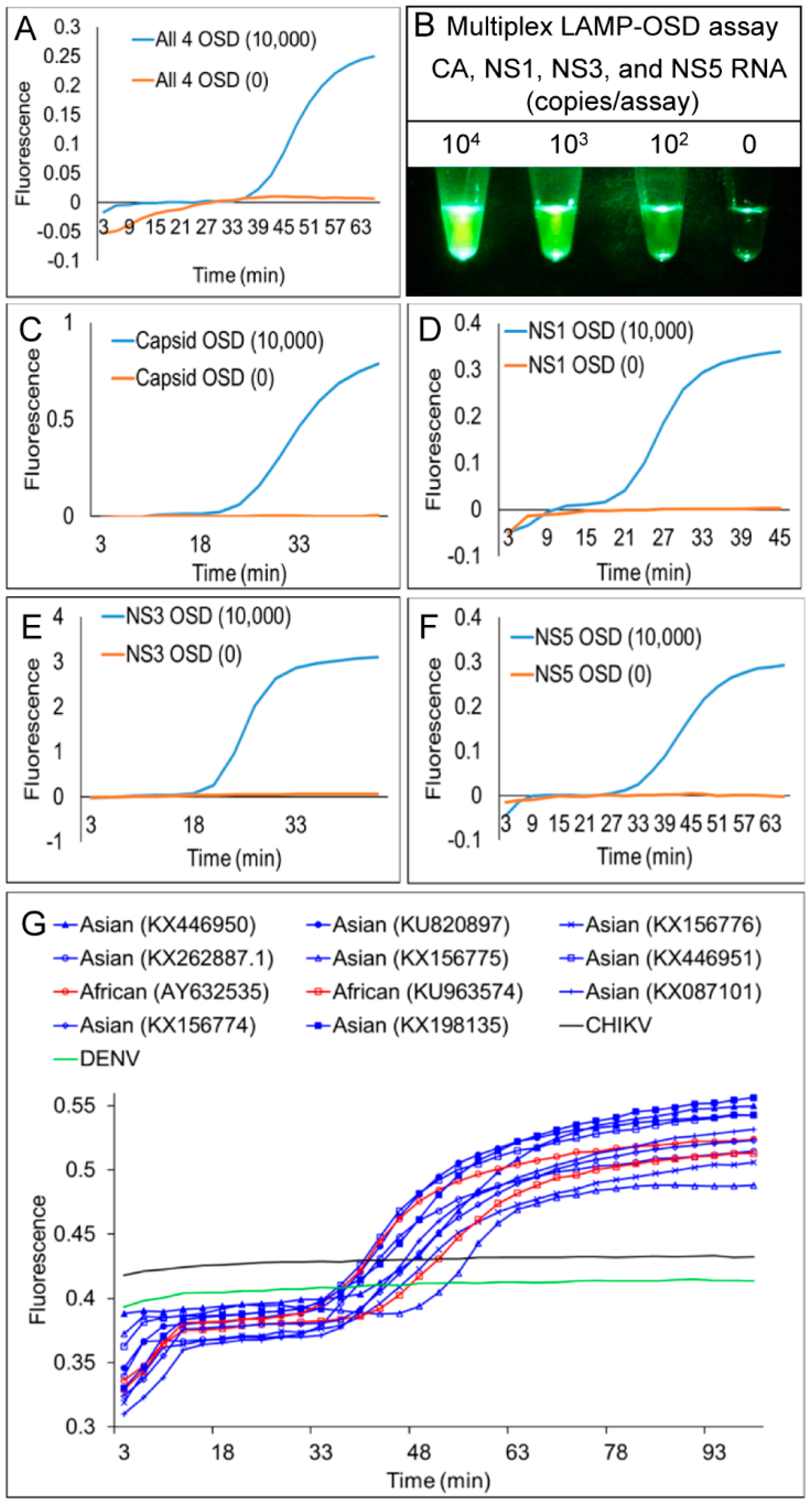
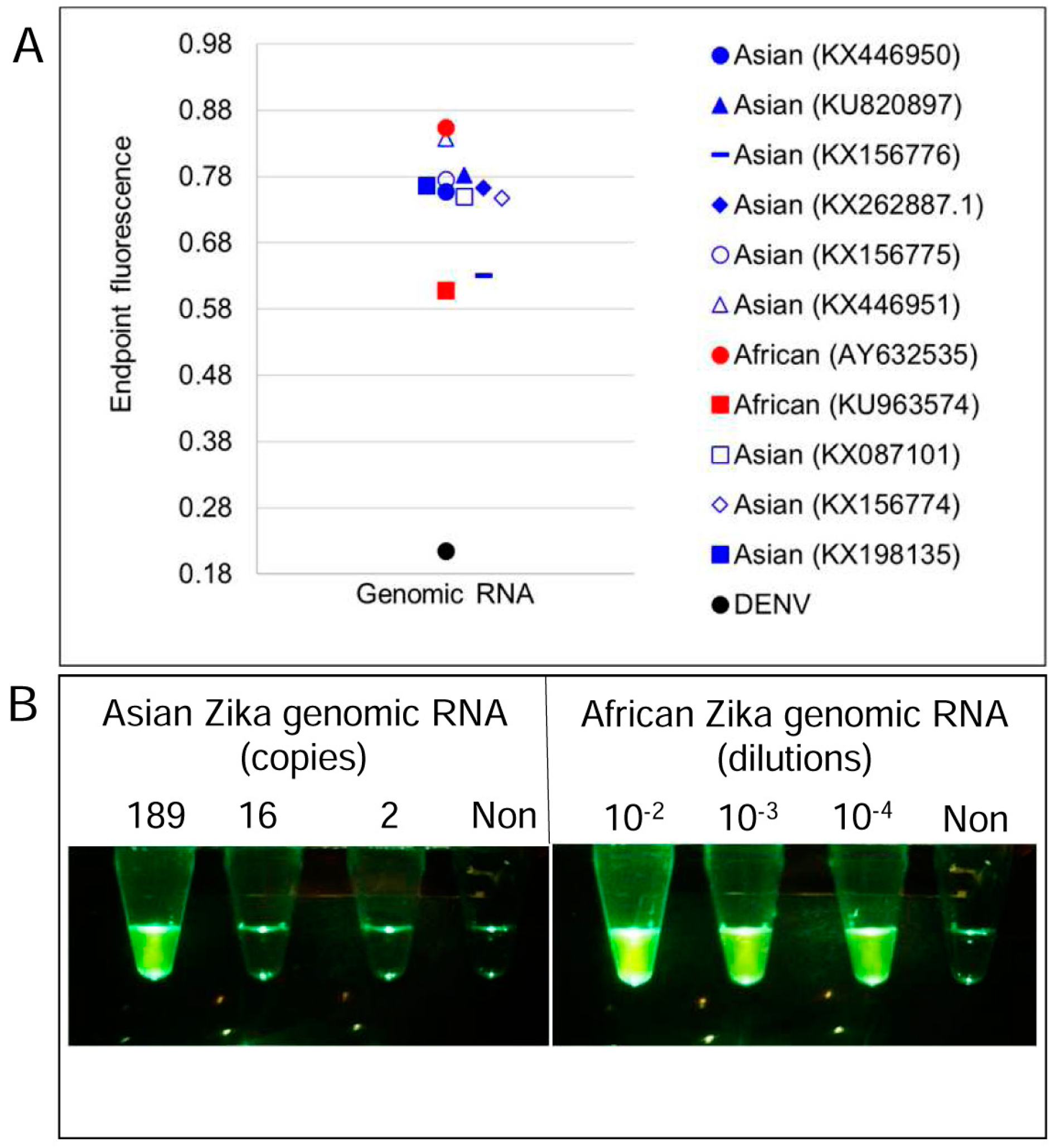
3.1. Degenerate LAMP-OSD Assays for Detection of Asian and African Lineage ZIKV
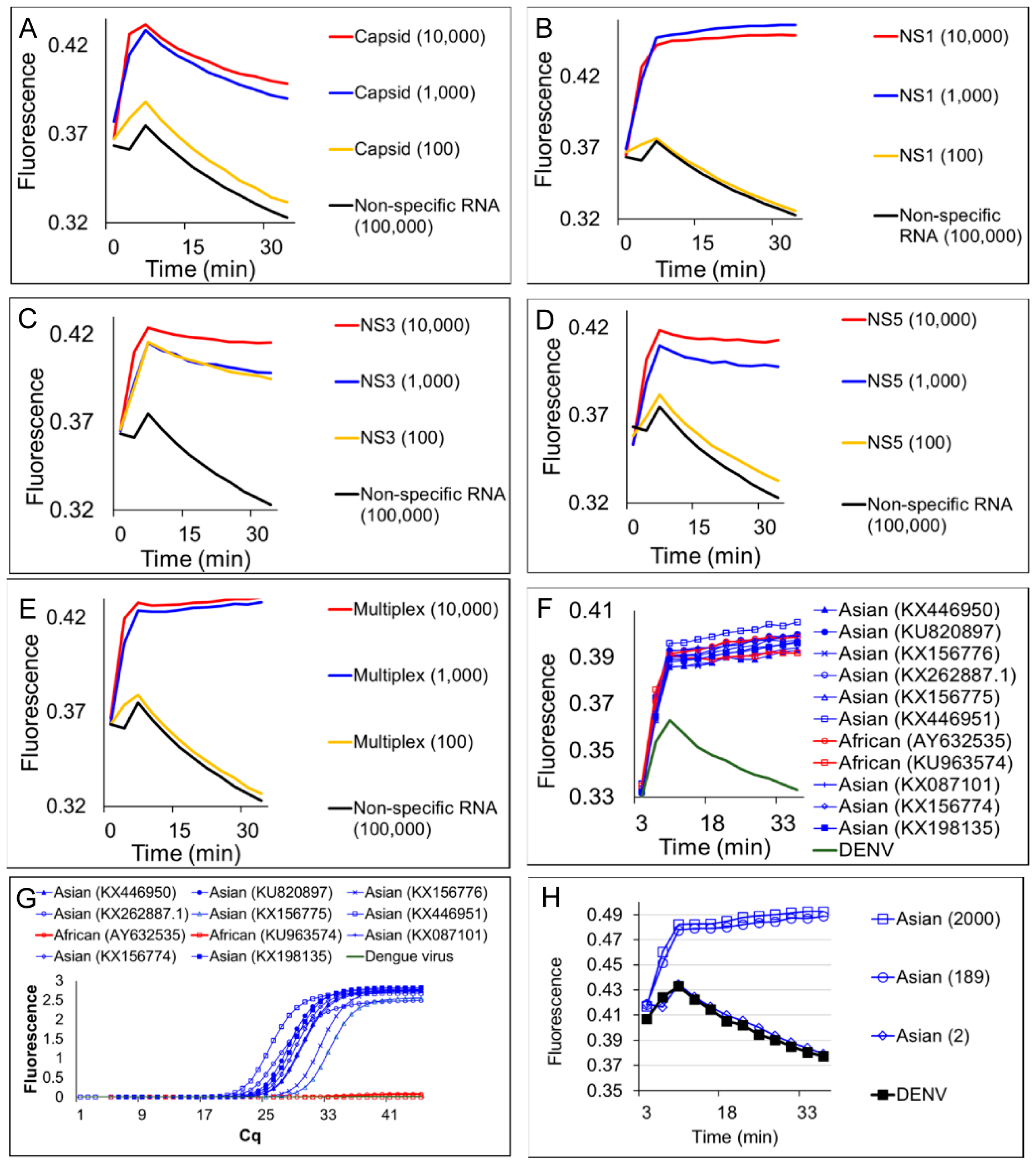
3.2. Multiplex LAMP with Degenerate Primers and OSD Probes
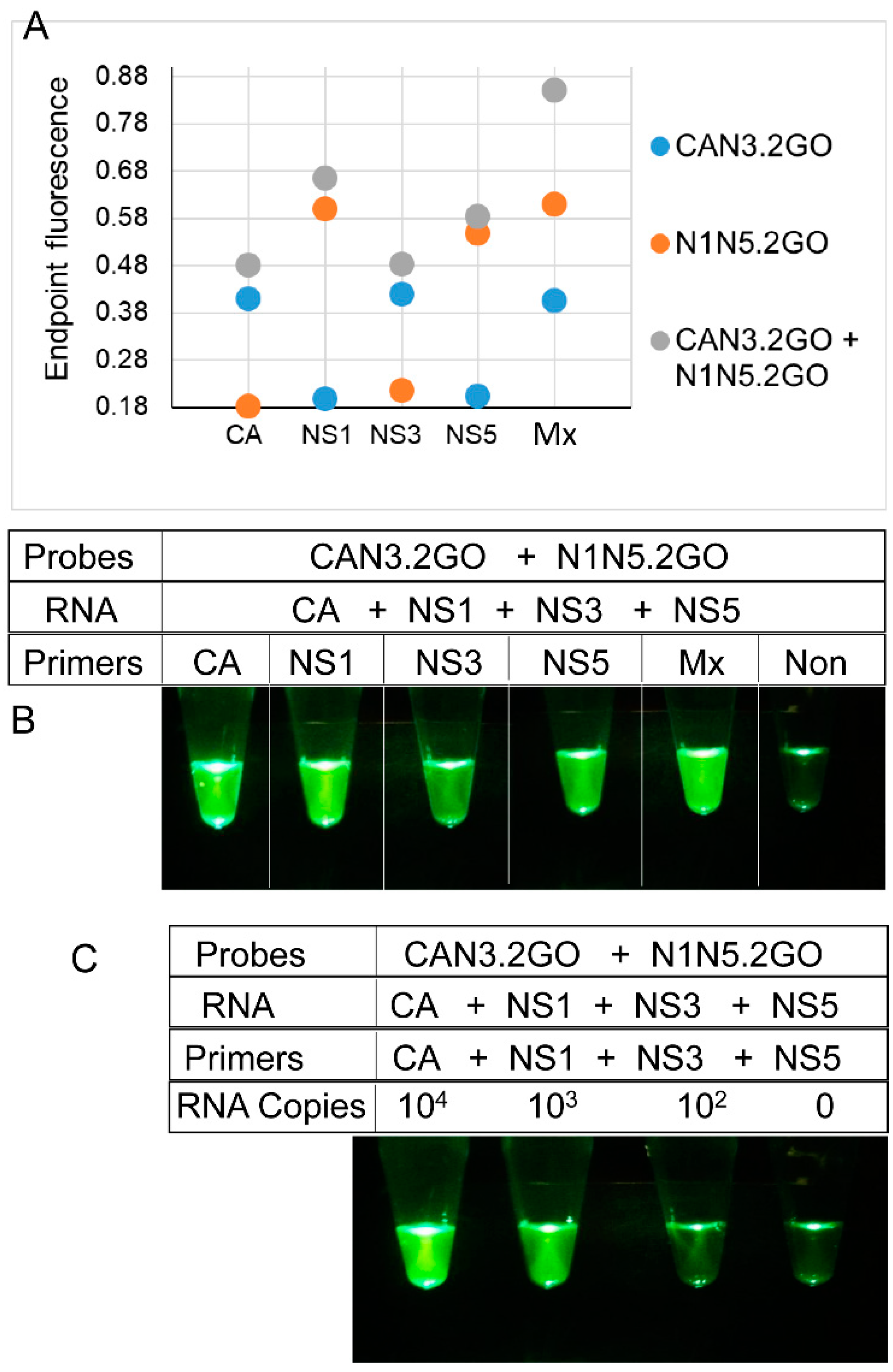
3.3. Multiplex LAMP with Degenerate Primers and Four-Input Logic-Processing Probes
3.4. Multiplex LAMP with Degenerate Primers and Two-Input Logic-Processing Probes
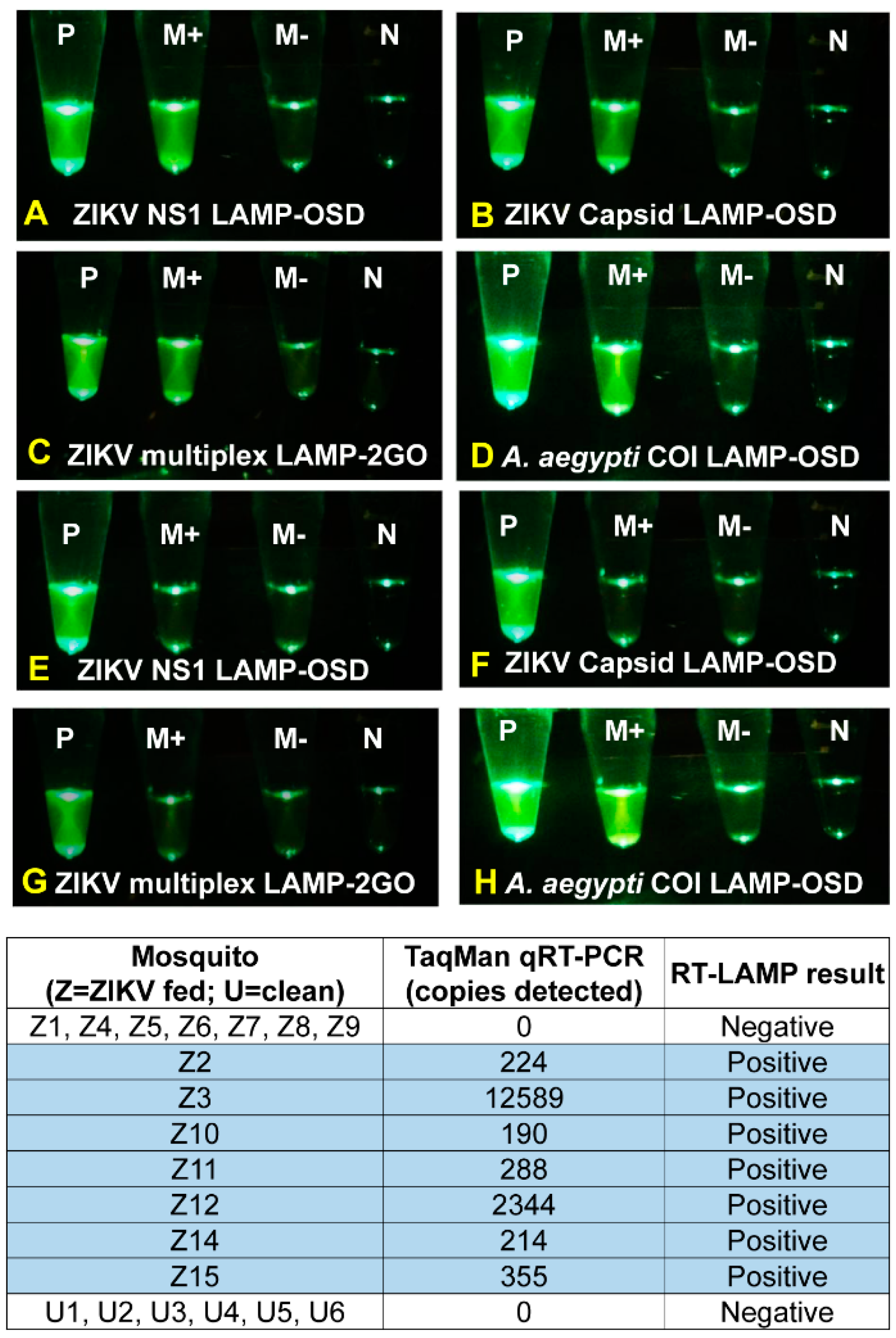
3.5. Identification of ZIKV-Infected Mosquitoes using Cellphones and Smart Molecular Diagnostics
5. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bruen, D.; Delaney, C.; Florea, L.; Diamond, D. Glucose sensing for diabetes monitoring: Recent developments. Sensors 2017, 17, 1866. [Google Scholar] [CrossRef] [PubMed]
- Gnoth, C.; Johnson, S. Strips of hope: Accuracy of home pregnancy tests and new developments. Geburtshilfe Frauenheilkd. 2014, 74, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Kozel, T.R.; Burnham-Marusich, A.R. Point-of-care testing for infectious diseases: Past, present, and future. J. Clin. Microbiol. 2017, 55, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Tiwari, S.; Jayant, R.D.; Vashist, A.; Nikkhah-Moshaie, R.; El-Hage, N.; Nair, M. Electrochemical biosensors for early stage zika diagnostics. Trends Biotechnol. 2017, 35, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Yndart, A.; Kumar, S.; Jayant, R.D.; Vashist, A.; Brown, A.N.; Li, C.Z.; Nair, M. A sensitive electrochemical immunosensor for label-free detection of zika-virus protein. Sci. Rep. 2018, 8, 9700. [Google Scholar] [CrossRef] [PubMed]
- Craw, P.; Balachandran, W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: A critical review. Lab Chip 2012, 12, 2469–2486. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Gullberg, M.; Dahl, F.; Szuhai, K.; Raap, A.K. Real-time monitoring of rolling-circles amplification using a modified molecular beacon design. Nucleic Acids Res. 2002, 30, e66. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Boil. 2006, 4, e204. [Google Scholar] [CrossRef] [PubMed]
- Kubota, R.; Jenkins, D.M. Real-time duplex applications of loop-mediated amplification (LAMP) by assimilating probes. Int. J. Mol. Sci. 2015, 16, 4786–4799. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.Y.; Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 2011, 3, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.S.; Bhadra, S.; Li, B.; Wu, Y.R.; Milligan, J.N.; Ellington, A.D. Robust strand exchange reactions for the sequence-specific, real-time detection of nucleic acid amplicons. Anal. Chem. 2015, 87, 3314–3320. [Google Scholar] [CrossRef]
- Jiang, Y.S.; Riedel, T.E.; Popoola, J.A.; Morrow, B.R.; Cai, S.; Ellington, A.D.; Bhadra, S. Portable platform for rapid in-field identification of human fecal pollution in water. Water Res. 2017, 131, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Riedel, T.E.; Saldaña, M.A.; Hegde, S.; Pederson, N.; Hughes, G.L.; Ellington, A.D. Direct nucleic acid analysis of mosquitoes for high fidelity species identification and detection of Wolbachia using a cellphone. PLoS Negl. Trop. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Jiang, Y.S.; Kumar, M.R.; Johnson, R.F.; Hensley, L.E.; Ellington, A.D. Real-time sequence-validated loop-mediated isothermal amplification assays for detection of Middle East respiratory syndrome coronavirus (MERS-CoV). PLoS ONE 2015, 10, e0123126. [Google Scholar] [CrossRef]
- Jung, C.; Ellington, A.D. Diagnostic applications of nucleic acid circuits. Acc. Chem. Res. 2014, 47, 1825–1835. [Google Scholar] [CrossRef]
- Du, Y.; Hughes, R.A.; Bhadra, S.; Jiang, Y.S.; Ellington, A.D.; Li, B. A sweet spot for molecular diagnostics: Coupling isothermal amplification and strand exchange circuits to glucometers. Sci. Rep. 2015, 5, 11039. [Google Scholar] [CrossRef]
- Li, B.; Ellington, A.D.; Chen, X. Rational, modular adaptation of enzyme-free DNA circuits to multiple detection methods. Nucleic Acids Res. 2011, 39, e110. [Google Scholar] [CrossRef]
- Allen, P.B.; Arshad, S.A.; Li, B.; Chen, X.; Ellington, A.D. DNA circuits as amplifiers for the detection of nucleic acids on a paperfluidic platform. Lab Chip 2012, 12, 2951–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardee, K.; Green, A.A.; Takahashi, M.K.; Braff, D.; Lambert, G.; Lee, J.W.; Ferrante, T.; Ma, D.; Donghia, N.; Fan, M.; et al. Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell 2016, 165, 1255–1266. [Google Scholar] [CrossRef]
- Qian, L.; Winfree, E. Scaling up digital circuit computation with DNA strand displacement cascades. Science 2011, 332, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Winfree, E.; Bruck, J. Neural network computation with DNA strand displacement cascades. Nature 2011, 475, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Turberfield, A.J.; Yurke, B.; Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science 2007, 318, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.S.; Stacy, A.; Whiteley, M.; Ellington, A.D.; Bhadra, S. Amplicon competition enables end-point quantitation of nucleic acids following isothermal amplification. ChemBioChem 2017. [Google Scholar] [CrossRef]
- Faria, N.R.; Quick, J.; Claro, I.M.; Theze, J.; de Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; da Costa, A.C.; et al. Establishment and cryptic transmission of Zika virus in brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Rasche, A.; Baronti, C.; Aldabbagh, S.; Cadar, D.; Reusken, C.B.; Pas, S.D.; Goorhuis, A.; Schinkel, J.; Molenkamp, R.; et al. Assay optimization for molecular detection of Zika virus. Bull. World Health Organ. 2016, 94, 880–892. [Google Scholar] [CrossRef]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, S.; Kellogg, D.E.; McKinney, N.; Spasic, D.; Goda, L.; Levenson, C.; Sninsky, J.J. Effects of primer-template mismatches on the polymerase chain reaction: Human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 1990, 18, 999–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petruska, J.; Goodman, M.F.; Boosalis, M.S.; Sowers, L.C.; Cheong, C.; Tinoco, I., Jr. Comparison between DNA melting thermodynamics and DNA polymerase fidelity. Proc. Natl. Acad. Sci. USA 1988, 85, 6252–6256. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, G.H.; Gardner, S.N. Predicting the sensitivity and specificity of published real-time PCR assays. Ann. Clin. Microbiol. Antimicrob. 2008, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Chan, J.F.; Tee, K.M.; Choi, G.K.; Lau, S.K.; Woo, P.C.; Tse, H.; Yuen, K.Y. Comparative genomic analysis of pre-epidemic and epidemic Zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerg. Microbes Infect. 2016, 5, e22. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schaffer, A.A.; Brister, J.R. Virus variation resource—Improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Zadeh, J.N.; Steenberg, C.D.; Bois, J.S.; Wolfe, B.R.; Pierce, M.B.; Khan, A.R.; Dirks, R.M.; Pierce, N.A. Nupack: Analysis and design of nucleic acid systems. J. Comput. Chem. 2011, 32, 170–173. [Google Scholar] [CrossRef]
- Waggoner, J.J.; Pinsky, B.A. Zika virus: Diagnostics for an emerging pandemic threat. J. Clin. Microbiol. 2016, 54, 860–867. [Google Scholar] [CrossRef]
- Muller, J.A.; Harms, M.; Schubert, A.; Jansen, S.; Michel, D.; Mertens, T.; Schmidt-Chanasit, J.; Munch, J. Inactivation and environmental stability of Zika virus. Emerg. Infect. Dis. 2016, 22, 1685–1687. [Google Scholar] [CrossRef] [PubMed]
- Faye, O.; Freire, C.C.; Iamarino, A.; de Oliveira, J.V.; Diallo, M.; Zanotto, P.M.; Sall, A.A. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl. Trop. Dis. 2014, 8, e2636. [Google Scholar] [CrossRef]
- Gandelman, O.; Jackson, R.; Kiddle, G.; Tisi, L. Loop-mediated amplification accelerated by stem primers. Int. J. Mol. Sci. 2011, 12, 9108–9124. [Google Scholar] [CrossRef]
- Faria, N.R.; Azevedo, R.; Kraemer, M.U.G.; Souza, R.; Cunha, M.S.; Hill, S.C.; Theze, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciota, A.T.; Bialosuknia, S.M.; Zink, S.D.; Brecher, M.; Ehrbar, D.J.; Morrissette, M.N.; Kramer, L.D. Effects of Zika virus strain and Aedes mosquito species on vector competence. Emerg. Infect. Dis. 2017, 23, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Chouin-Carneiro, T.; Vega-Rua, A.; Vazeille, M.; Yebakima, A.; Girod, R.; Goindin, D.; Dupont-Rouzeyrol, M.; Lourenco-de-Oliveira, R.; Failloux, A.B. Differential susceptibilities of Aedes aegypti and Aedes albopictus from the Americas to Zika virus. PLoS Negl. Trop. Dis. 2016, 10, e0004543. [Google Scholar] [CrossRef]
- Weger-Lucarelli, J.; Ruckert, C.; Chotiwan, N.; Nguyen, C.; Garcia Luna, S.M.; Fauver, J.R.; Foy, B.D.; Perera, R.; Black, W.C.; Kading, R.C.; et al. Vector competence of American mosquitoes for three strains of Zika virus. PLoS Negl. Trop. Dis. 2016, 10, e0005101. [Google Scholar] [CrossRef]
- Diagne, C.T.; Diallo, D.; Faye, O.; Ba, Y.; Faye, O.; Gaye, A.; Dia, I.; Faye, O.; Weaver, S.C.; Sall, A.A.; et al. Potential of selected Senegalese Aedes spp. Mosquitoes (diptera: Culicidae) to transmit Zika virus. BMC Infect. Dis. 2015, 15, 492. [Google Scholar] [CrossRef]
- Roundy, C.M.; Azar, S.R.; Rossi, S.L.; Huang, J.H.; Leal, G.; Yun, R.; Fernandez-Salas, I.; Vitek, C.J.; Paploski, I.A.; Kitron, U.; et al. Variation in Aedes aegypti mosquito competence for Zika virus transmission. Emerg. Infect. Dis. 2017, 23, 625–632. [Google Scholar] [CrossRef]
- Nunes, M.R.; Vianez, J.L., Jr.; Nunes, K.N.; da Silva, S.P.; Lima, C.P.; Guzman, H.; Martins, L.C.; Carvalho, V.L.; Tesh, R.B.; Vasconcelos, P.F. Analysis of a reverse transcription loop-mediated isothermal amplification (RT-LAMP) for yellow fever diagnostic. J. Virol. Methods 2015, 226, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.L.; Leung, C.S.; Chan, K.H.; Lee, J.H.; Yuen, K.Y.; Guan, Y.; Peiris, J.S. Detection of human influenza a viruses by loop-mediated isothermal amplification. J. Clin. Microbiol. 2005, 43, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Mamba, T.S.; Mbae, C.K.; Kinyua, J.; Mulinge, E.; Mburugu, G.N.; Njiru, Z.K. Lateral flow Loop-Mediated isothermal amplification test with stem primers: Detection of cryptosporidium species in Kenyan children presenting with diarrhea. J. Trop. Med. 2018, 2018, 7659730. [Google Scholar] [CrossRef] [PubMed]
- Blaser, S.; Diem, H.; von Felten, A.; Gueuning, M.; Andreou, M.; Boonham, N.; Tomlinson, J.; Muller, P.; Utzinger, J.; Frey, J.E.; et al. From laboratory to point of entry: Development and implementation of a loop-mediated isothermal amplification (LAMP)-based genetic identification system to prevent introduction of quarantine insect species. Pest Manag. Sci. 2018, 74, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Zhou, Q.; Zhao, K.; Chi, Y.; Liu, B.; Min, X.; Shi, Z.; Zou, B.; Cui, L. Detection of influenza viruses by coupling multiplex reverse-transcription loop-mediated isothermal amplification with cascade invasive reaction using nanoparticles as a sensor. Int. J. Nanomed. 2017, 12, 2645–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.S.; Yan, Y.H.; Zhang, D.Y. Modular probes for enriching and detecting complex nucleic acid sequences. Nat. Chem. 2017, 9, 1222–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, S.; Yue, S.; Zhang, S. Hybridization chain reaction: A versatile molecular tool for biosensing, bioimaging, and biomedicine. Chem. Soc. Rev. 2017, 46, 4281–4298. [Google Scholar] [CrossRef]
- Francois, P.; Tangomo, M.; Hibbs, J.; Bonetti, E.J.; Boehme, C.C.; Notomi, T.; Perkins, M.D.; Schrenzel, J. Robustness of a loop-mediated isothermal amplification reaction for diagnostic applications. FEMS Immunol. Med. Microbiol. 2011, 62, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Ebbinghaus, P.; von Samson-Himmelstjerna, G.; Krucken, J. Direct loop-mediated isothermal amplification from Plasmodium chabaudi infected blood samples: Inability to discriminate genomic and cDNA sequences. Exp. Parasitol. 2012, 131, 40–44. [Google Scholar] [CrossRef]
- Cook, J.; Aydin-Schmidt, B.; Gonzalez, I.J.; Bell, D.; Edlund, E.; Nassor, M.H.; Msellem, M.; Ali, A.; Abass, A.K.; Martensson, A.; et al. Loop-mediated isothermal amplification (LAMP) for point-of-care detection of asymptomatic low-density malaria parasite carriers in Zanzibar. Malar. J. 2015, 14, 43. [Google Scholar] [CrossRef]
- Nakiyingi, L.; Nakanwagi, P.; Briggs, J.; Agaba, T.; Mubiru, F.; Mugenyi, M.; Ssengooba, W.; Joloba, M.L.; Manabe, Y.C. Performance of loop-mediated isothermal amplification assay in the diagnosis of pulmonary tuberculosis in a high prevalence TB/HIV rural setting in Uganda. BMC Infect. Dis. 2018, 18, 87. [Google Scholar] [CrossRef]
- Xing, W.; Yu, X.; Feng, J.; Sun, K.; Fu, W.; Wang, Y.; Zou, M.; Xia, W.; Luo, Z.; He, H.; et al. Field evaluation of a recombinase polymerase amplification assay for the diagnosis of Schistosoma japonicum infection in Hunan province of China. BMC Infect. Dis. 2017, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Escadafal, C.; Faye, O.; Sall, A.A.; Faye, O.; Weidmann, M.; Strohmeier, O.; von Stetten, F.; Drexler, J.; Eberhard, M.; Niedrig, M.; et al. Rapid molecular assays for the detection of yellow fever virus in low-resource settings. PLoS Negl. Trop. Dis. 2014, 8, e2730. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase polymerase amplification for diagnostic applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Nolte, F.S.; Gauld, L.; Barrett, S.B. Direct comparison of Alere i and cobas Liat Influenza A and B tests for rapid detection of influenza virus infection. J. Clin. Microbiol. 2016, 54, 2763–2766. [Google Scholar] [CrossRef] [PubMed]
- LaBarre, P.; Hawkins, K.R.; Gerlach, J.; Wilmoth, J.; Beddoe, A.; Singleton, J.; Boyle, D.; Weigl, B. A simple, inexpensive device for nucleic acid amplification without electricity-toward instrument-free molecular diagnostics in low-resource settings. PLoS ONE 2011, 6, e19738. [Google Scholar] [CrossRef]
- Powell, M.L.; Bowler, F.R.; Martinez, A.J.; Greenwood, C.J.; Armes, N.; Piepenburg, O. New Fpg probe chemistry for direct detection of recombinase polymerase amplification on lateral flow strips. Anal. Biochem. 2018, 543, 108–115. [Google Scholar] [CrossRef]
- Maples, B.K.; Holmberg, R.C.; Miller, A.P.; Provins, J.W.; Roth, R.B.; Mandell, J.G. Nicking and Extension Amplification Reaction for the Exponential Amplification of Nucleic Acids. U.S. Patent 20090017453A1, 15 January 2009. [Google Scholar]
- Carter, C.; Akrami, K.; Hall, D.; Smith, D.; Aronoff-Spencer, E. Lyophilized visually readable loop-mediated isothermal reverse transcriptase nucleic acid amplification test for detection Ebola Zaire RNA. J. Virol. Methods 2017, 244, 32–38. [Google Scholar] [CrossRef]
- Chen, H.W.; Ching, W.M. Evaluation of the stability of lyophilized loop-mediated isothermal amplification reagents for the detection of Coxiella burnetii. Heliyon 2017, 3, e00415. [Google Scholar] [CrossRef]
- Wong, Y.P.; Othman, S.; Lau, Y.L.; Radu, S.; Chee, H.Y. Loop-mediated isothermal amplification (LAMP): A versatile technique for detection of micro-organisms. J. Appl. Microbiol. 2018, 124, 626–643. [Google Scholar] [CrossRef]
- Howson, E.L.A.; Armson, B.; Madi, M.; Kasanga, C.J.; Kandusi, S.; Sallu, R.; Chepkwony, E.; Siddle, A.; Martin, P.; Wood, J.; et al. Evaluation of two lyophilized molecular assays to rapidly detect foot-and-mouth disease virus directly from clinical samples in field settings. Transbound. Emerg. Dis. 2017, 64, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, N.W.; Gaye, M.; Diallo, M.A.; Goldman, I.F.; Ljolje, D.; Deme, A.B.; Badiane, A.; Ndiaye, Y.D.; Barnwell, J.W.; Udhayakumar, V.; et al. Evaluation of the Illumigene malaria LAMP: A robust molecular diagnostic tool for malaria parasites. Sci. Rep. 2016, 6, 36808. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhadra, S.; Saldaña, M.A.; Han, H.G.; Hughes, G.L.; Ellington, A.D. Simultaneous Detection of Different Zika Virus Lineages via Molecular Computation in a Point-of-Care Assay. Viruses 2018, 10, 714. https://0-doi-org.brum.beds.ac.uk/10.3390/v10120714
Bhadra S, Saldaña MA, Han HG, Hughes GL, Ellington AD. Simultaneous Detection of Different Zika Virus Lineages via Molecular Computation in a Point-of-Care Assay. Viruses. 2018; 10(12):714. https://0-doi-org.brum.beds.ac.uk/10.3390/v10120714
Chicago/Turabian StyleBhadra, Sanchita, Miguel A. Saldaña, Hannah Grace Han, Grant L. Hughes, and Andrew D. Ellington. 2018. "Simultaneous Detection of Different Zika Virus Lineages via Molecular Computation in a Point-of-Care Assay" Viruses 10, no. 12: 714. https://0-doi-org.brum.beds.ac.uk/10.3390/v10120714