Interaction of the Mouse Polyomavirus Capsid Proteins with Importins Is Required for Efficient Import of Viral DNA into the Cell Nucleus



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Plasmid DNA and DNA Transfection
2.2. Viral Infection
2.3. Negative Staining
2.4. Transmission Electron Microscopy
2.5. Indirect In Situ Immunofluorescence
2.6. Co-Immunoprecipitation and Cross-Linking
2.7. PCR Detection of DNA Isolated from Immunocomplexes
2.8. Proximity Ligation Assay
2.9. Agarose Gel Electrophoresis
2.10. Virus Production, Isolation and Quantification
2.11. NLS Sequence Analysis
2.12. Viral Genome Quantification
2.13. SDS-PAGE and Western Blot Analysis
2.14. Introduction of Mutations into Individual Proteins VP1 and VP2
2.15. Antibodies
3. Results
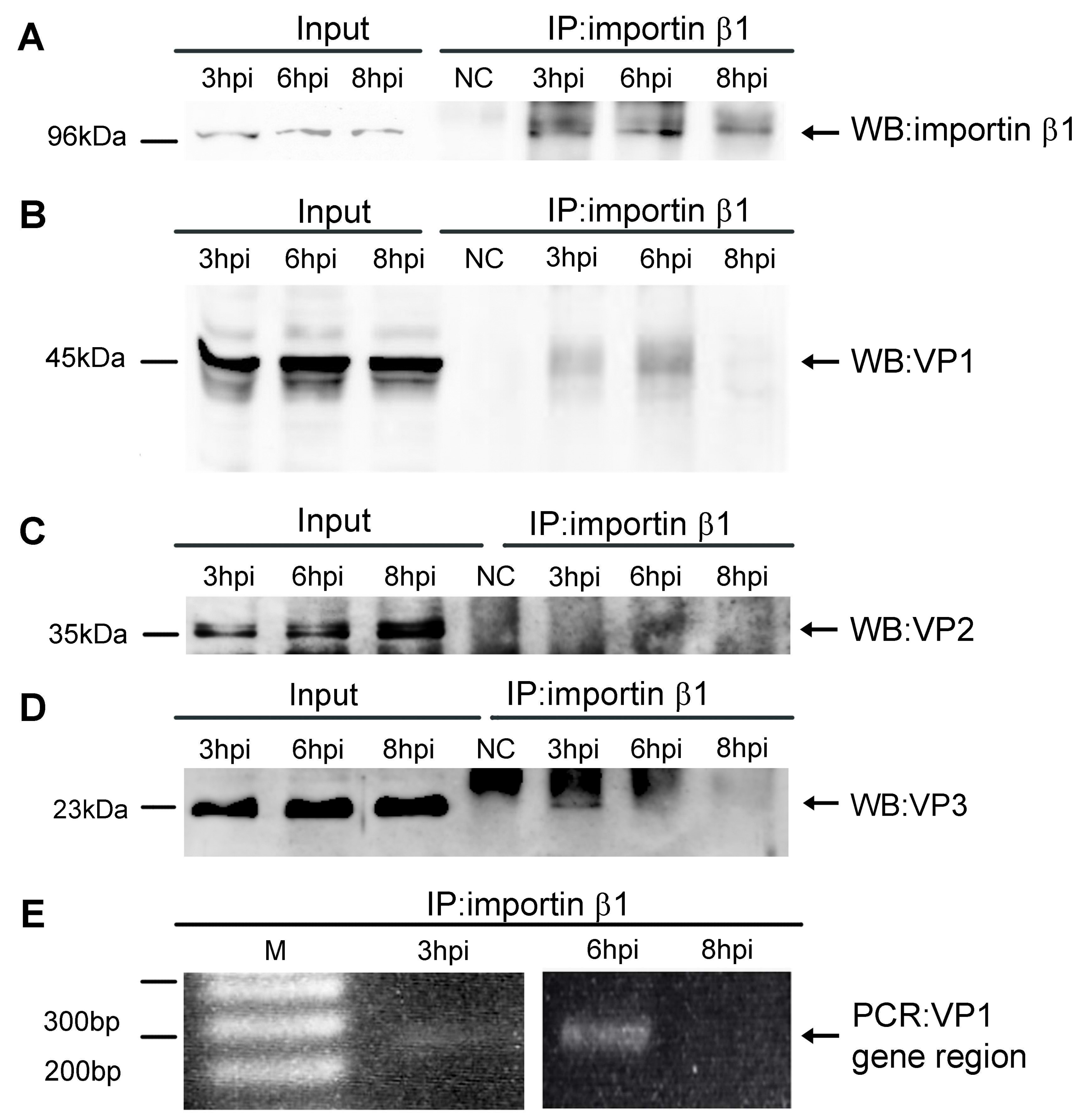
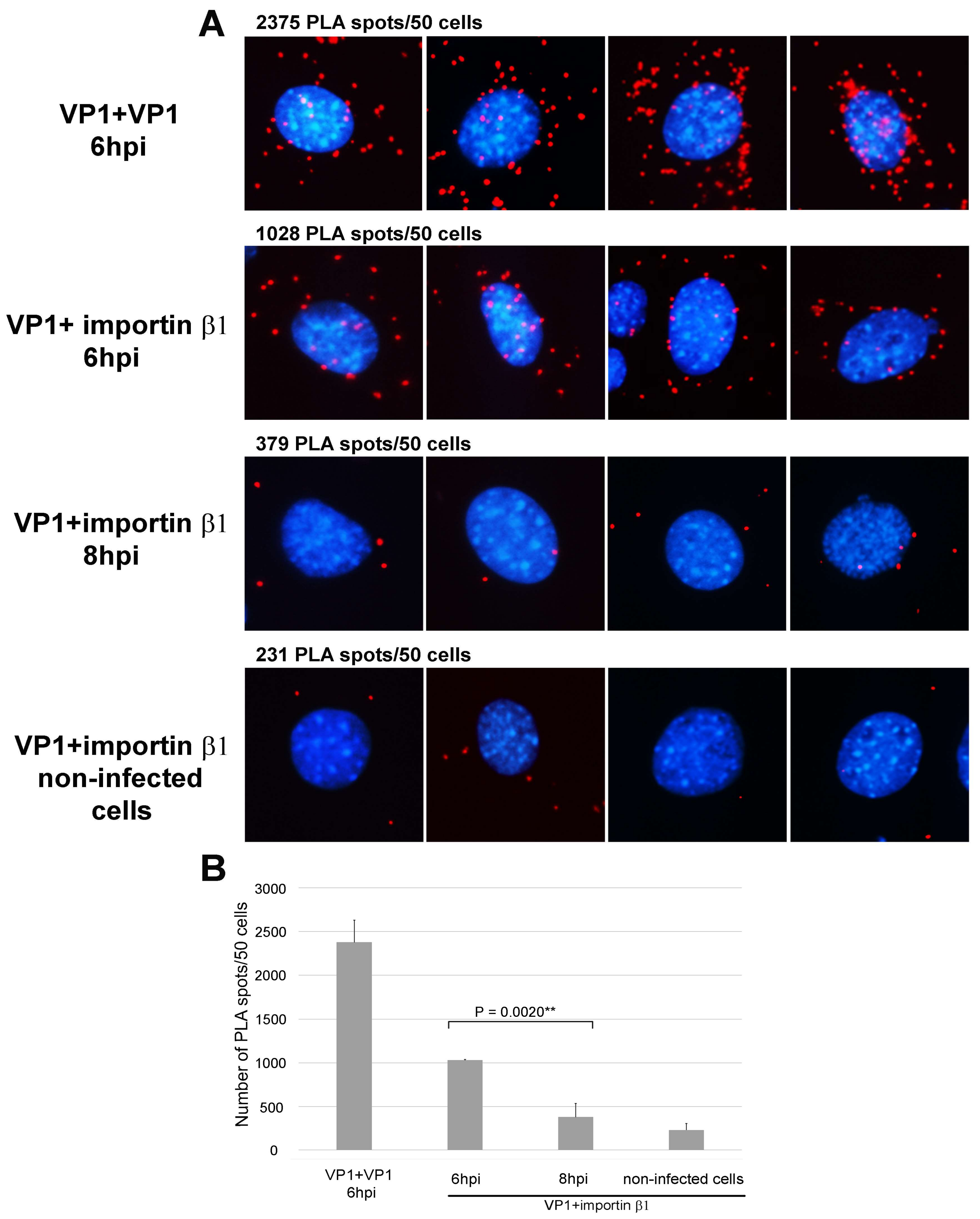
3.1. Importin β1 Binds Virions of MPyV at Early Times Post Infection
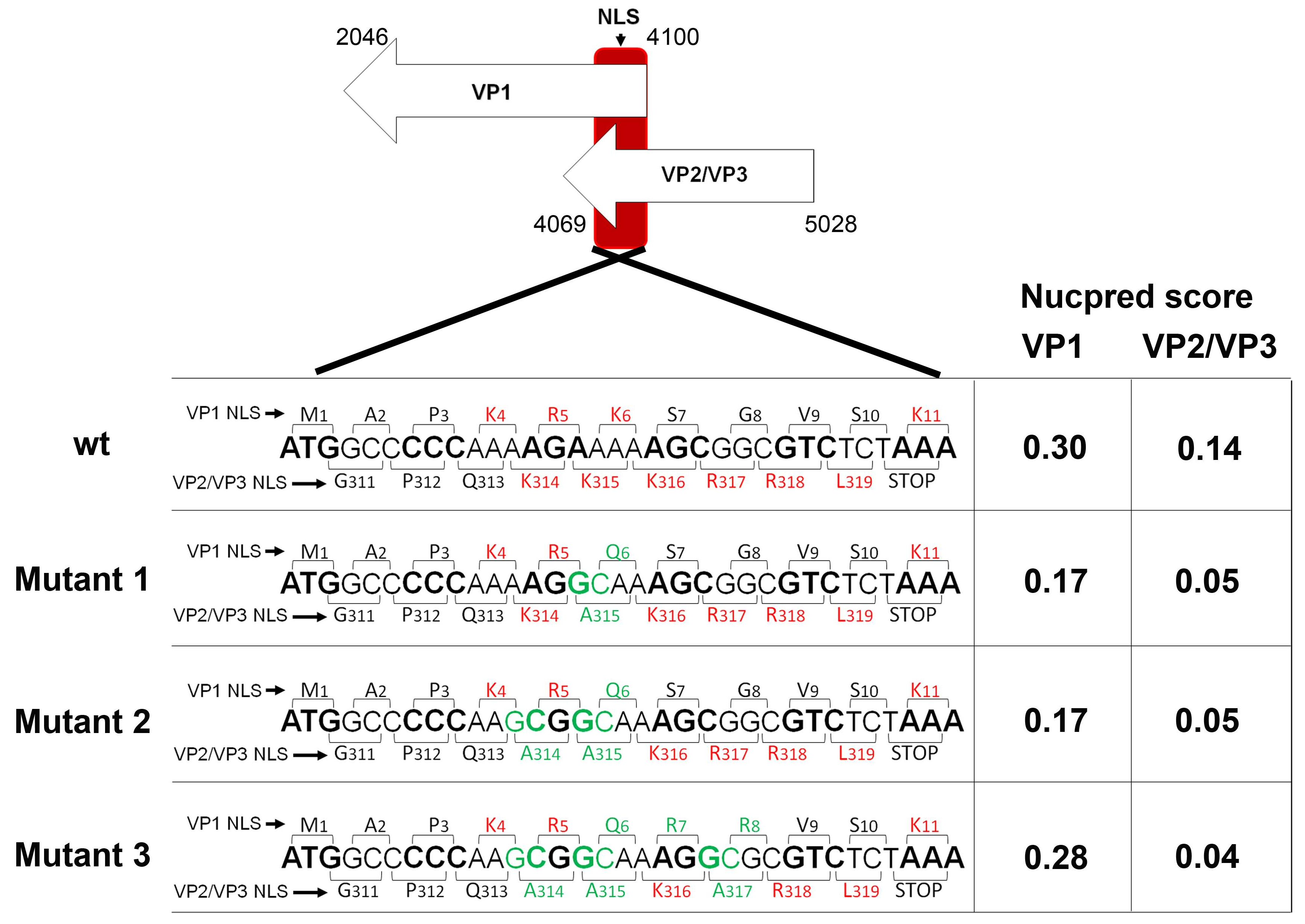
3.2. Mutagenesis Design of the Viruses Carrying Mutations in NLS Sequences of VP1 and VP2/VP3 Genes
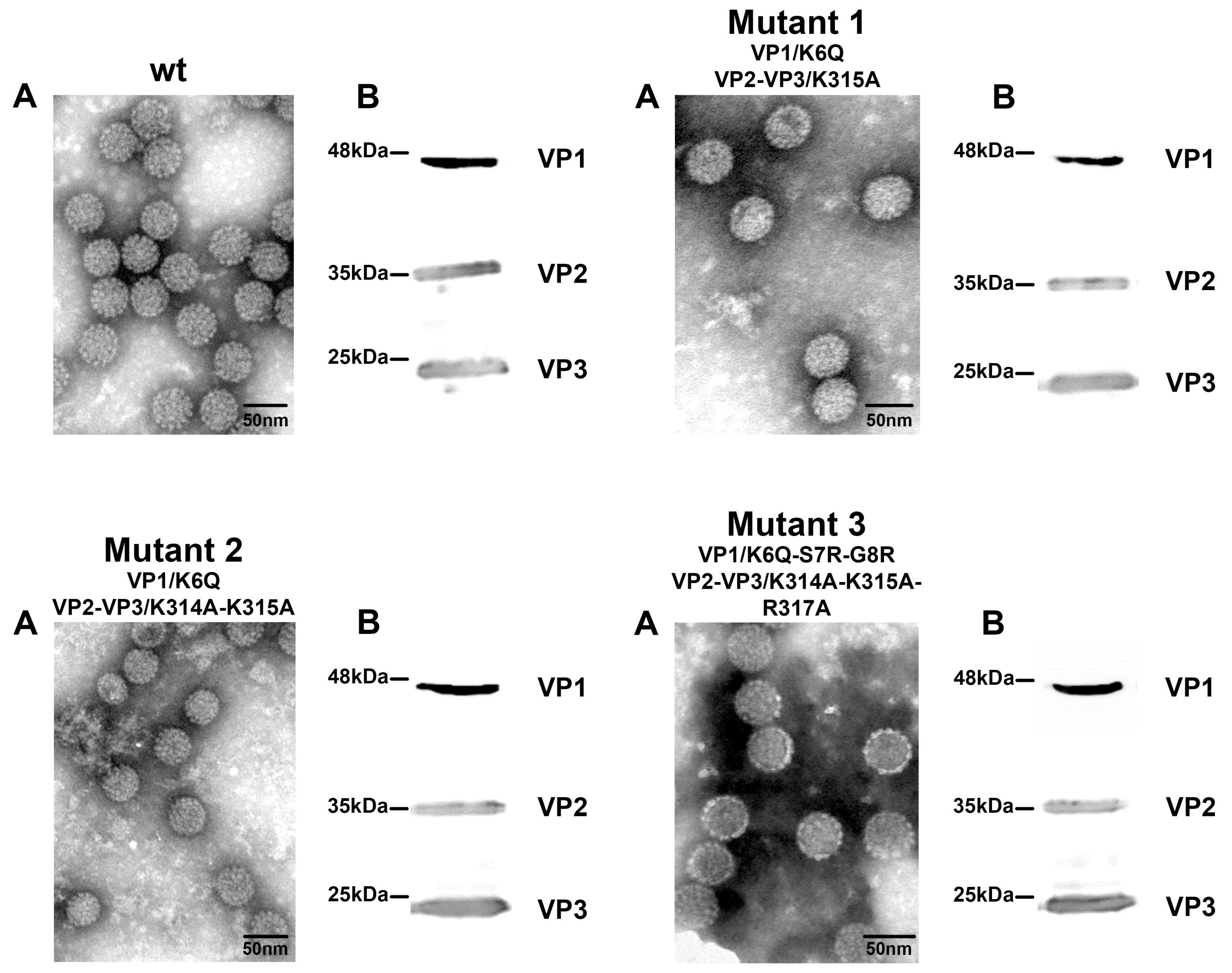
3.3. Mutant Virus Production and Characterization
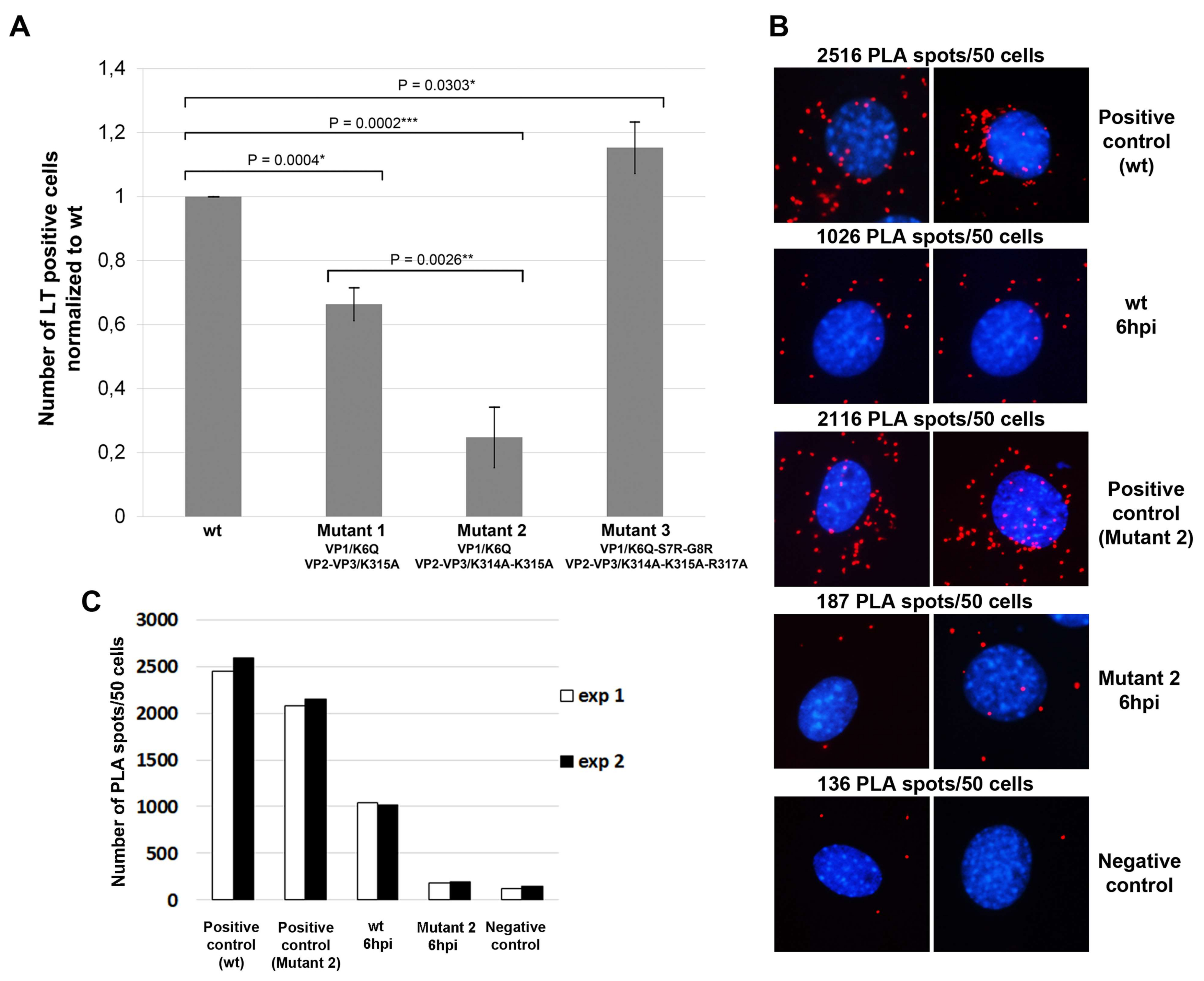
3.4. Amino Acid Substitutions in NLS of VP1 and VP2/VP3 Proteins Affect Virus Infection
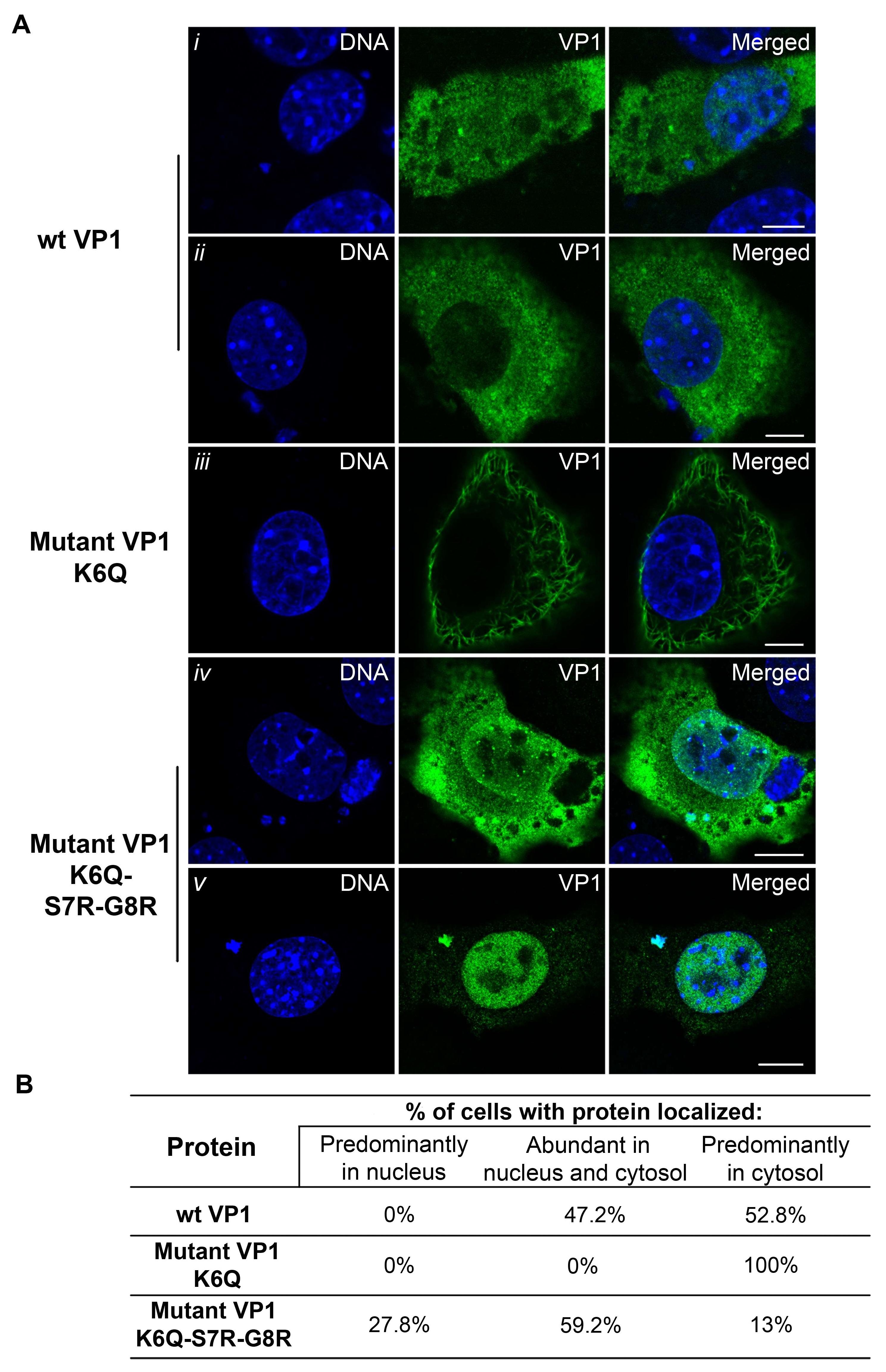
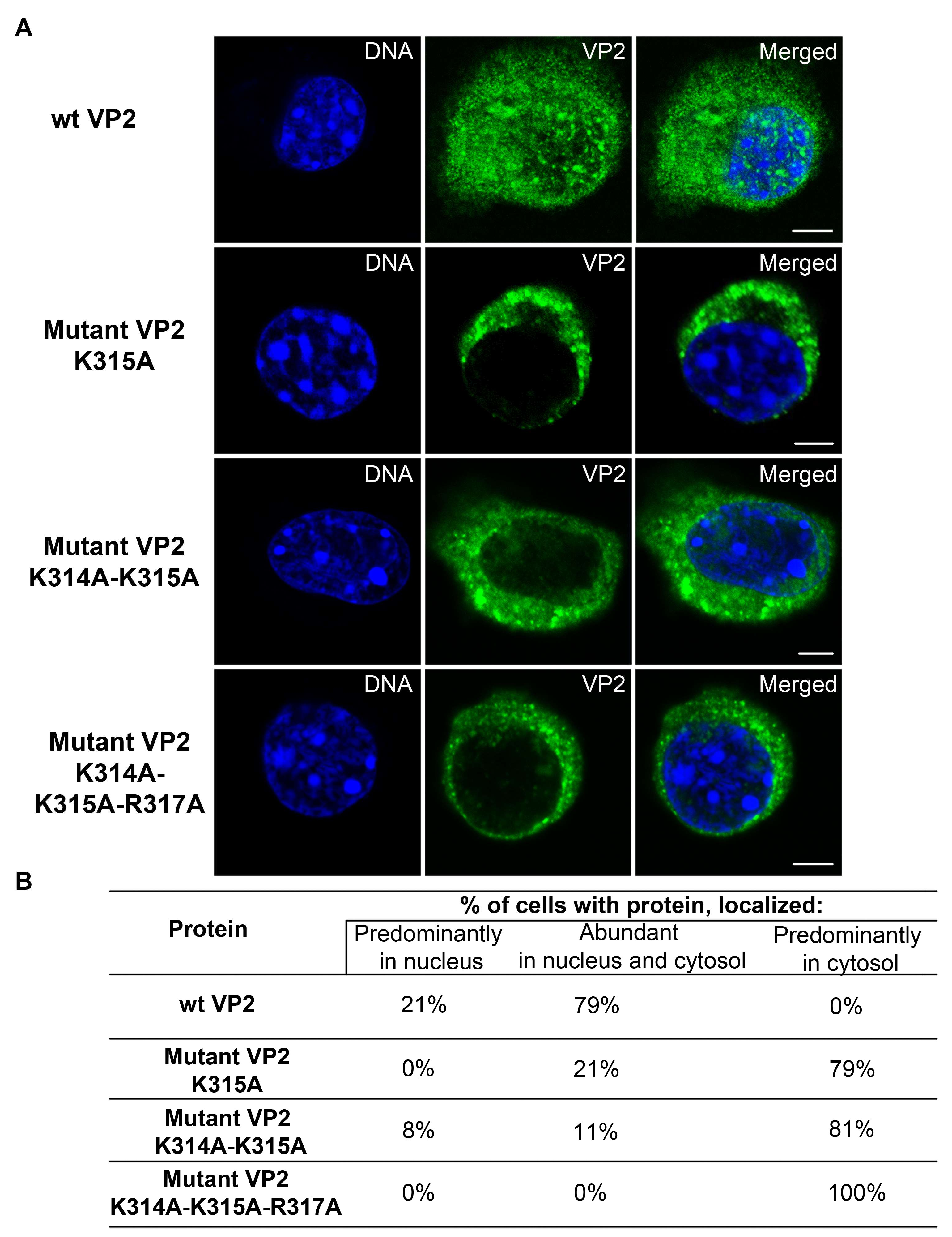
3.5. Mutations Introduced into the NLS of the MPyV Capsid Proteins Affect Their Transport into the Nucleus during Their Individual Expression
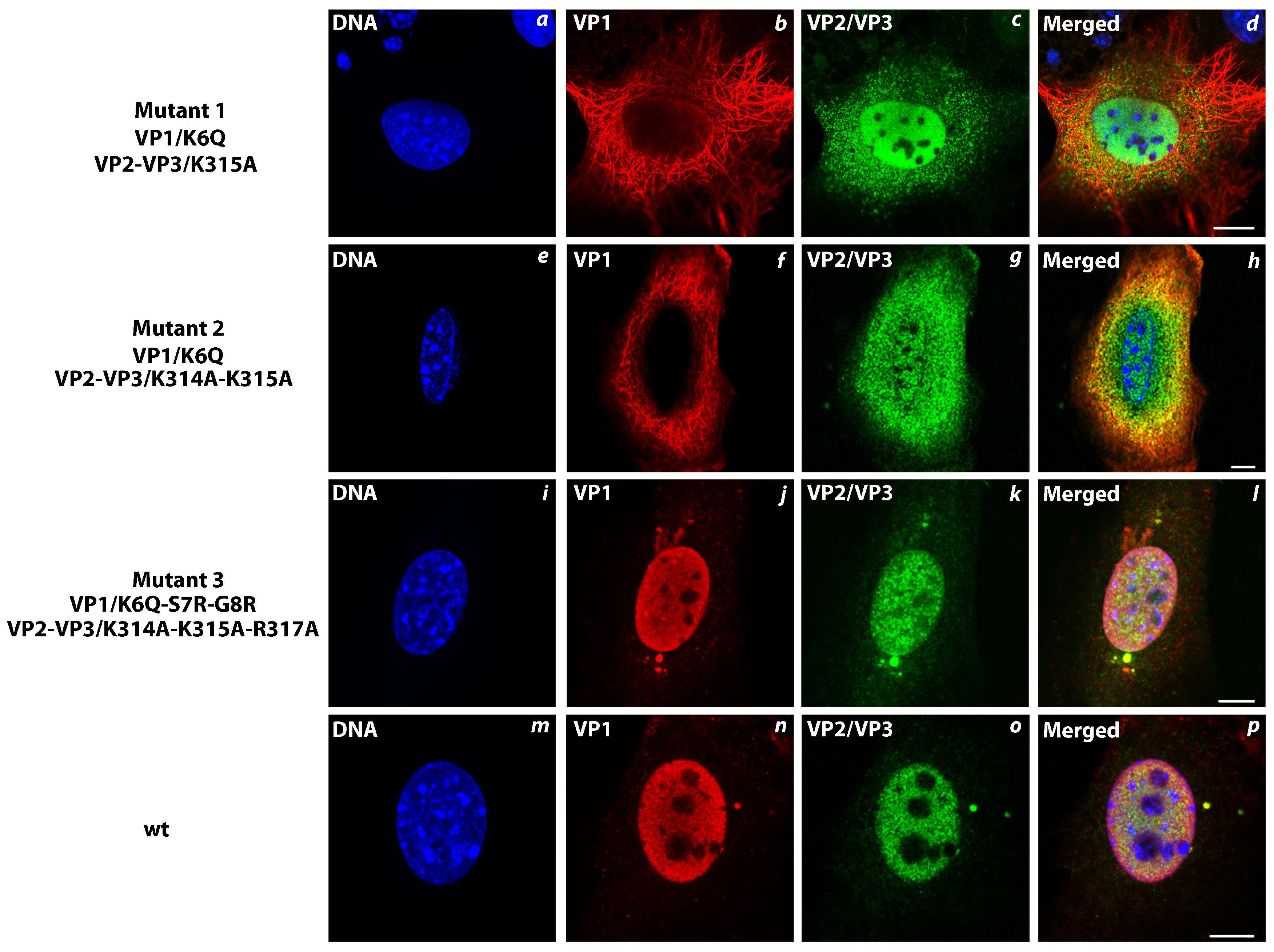
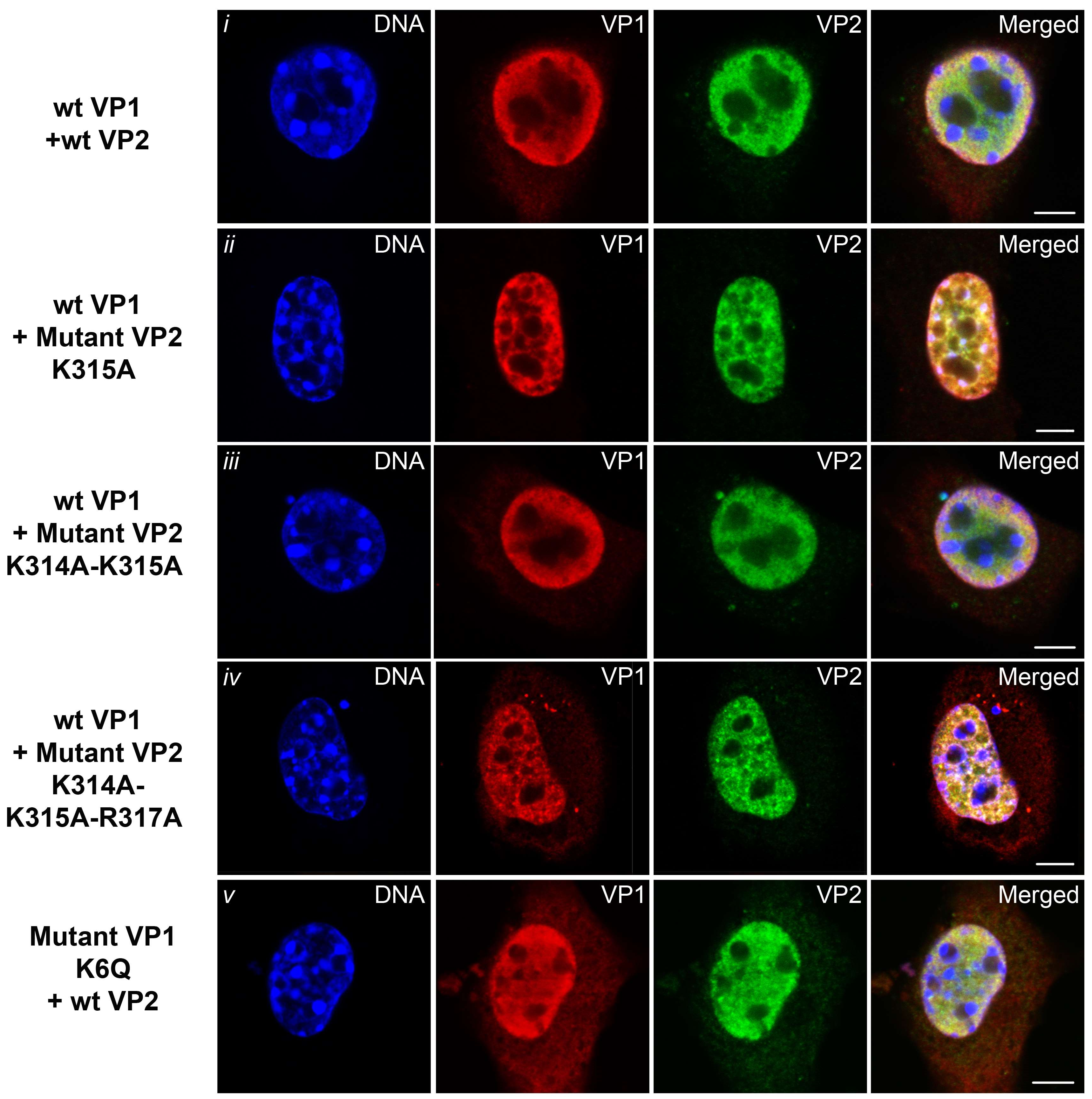
3.6. Transport of the Major and Minor Capsid Protein Complexes into the Cell Nucleus Is Efficient even when NLS of either VP1 or VP2 Proteins Is Abolished
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Buck, C.B.; Van Doorslaer, K.; Peretti, A.; Geoghegan, E.M.; Tisza, M.J.; An, P.; Katz, J.P.; Pipas, J.M.; McBride, A.A.; Camus, A.C.; et al. The ancient evolutionary history of Polyomaviruses. PLoS Pathog. 2016, 12, e1005574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klug, A. Structure of viruses of the papilloma-polyoma type. II. Comments on other work. J. Mol. Biol. 1965, 11, 424–431. [Google Scholar] [CrossRef]
- Chen, X.S.; Stehle, T.; Harrison, S.C. Interaction of Polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 1998, 17, 3233–3240. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.; Harrison, S. Interactions among the major and minor coat proteins of polyomavirus. J. Virol. 1994, 68, 3982–3989. [Google Scholar] [PubMed]
- Lin, W.; Hata, T.; Kasamatsu, H. Subcellular distribution of viral structural proteins during simian virus 40 infection. J. Virol. 1984, 50, 363–371. [Google Scholar] [PubMed]
- Forstova, J.; Krauzewicz, N.; Wallace, S.; Street, A.; Dilworth, S.; Beard, S.; Griffin, B. Cooperation of structural proteins during late events in the life cycle of polyomavirus. J. Virol. 1993, 67, 1405–1413. [Google Scholar] [PubMed]
- Delos, S.; Montross, L.; Moreland, R.; Garcea, R. Expression of the polyomavirus VP2 and VP3 proteins in insect cells: Coexpression with the major capsid protein VP1 alters VP2/VP3 subcellular localization. Virology 1993, 194, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A.J.; Bakayev, V.V.; Chumackov, P.M.; Georgiev, G.P. Minichromosome of simian virus 40: Presence of histone HI. Nucleic Acids Res. 1976, 3, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- You, J.; O’Hara, S.D.; Velupillai, P.; Castle, S.; Levery, S.; Garcea, R.L.; Benjamin, T. Ganglioside and non-ganglioside mediated host responses to the mouse polyomavirus. PLoS Pathog. 2015, 11, e1005175. [Google Scholar] [CrossRef] [PubMed]
- Richterová, Z.; Liebl, D.; Horák, M.; Palková, Z.; Stokrová, J.; Hozák, P.; Korb, J.; Forstová, J. Caveolae are involved in the trafficking of mouse polyomavirus virions and artificial VP1 pseudocapsids toward cell nuclei. J. Virol. 2001, 75, 10880–10891. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Kartenbeck, J.; Helenius, A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001, 3, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Drachenberg, C.B.; Papadimitriou, J.C.; Wali, R.; Cubitt, C.L.; Ramos, E. BK polyoma virus allograft nephropathy: Ultrastructural features from viral cell entry to lysis. Am. J. Transpl. 2003, 3, 1383–1392. [Google Scholar] [CrossRef]
- Liebl, D.; Difato, F.; Horníková, L.; Mannová, P.; Stokrová, J.; Forstová, J. Mouse polyomavirus enters early endosomes, requires their acidic PH for productive infection, and meets transferrin cargo in rab11-positive endosomes. J. Virol. 2006, 80, 4610–4622. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Cai, D.; Verhey, K.J.; Tsai, B. A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog. 2009, 5, e1000465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maul, G.G.; Rovera, G.; Vorbrodt, A.; Abramczuk, J. Membrane fusion as a mechanism of simian virus 40 entry into different cellular compartments. J. Virol. 1978, 28, 936–944. [Google Scholar] [PubMed]
- Mackay, R.L.; Consigli, R.A. Early events in polyoma virus infection: Attachment, penetration, and nuclear entry. J. Virol. 1976, 19, 620–636. [Google Scholar] [PubMed]
- Butin-Israeli, V.; Ben-nun-Shaul, O.; Kopatz, I.; Adam, S.A.; Shimi, T.; Goldman, R.D.; Oppenheim, A. Simian virus 40 induces lamin a/c fluctuations and nuclear envelope deformation during cell entry. Nucleus 2011, 2, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Li, P.P.; Qu, Q.; Jafri, Q.H.; Kasamatsu, H. Molecular dissection of nuclear entry-competent SV40 during infection. Virus Res. 2007, 124, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Sawa, H.; Suzuki, T.; Semba, S.; Henmi, C.; Okada, Y.; Tsuda, M.; Tanaka, S.; Atwood, W.J.; Nagashima, K. Nuclear entry mechanism of the human polyomavirus JC virus-like particle: Role of importins and the nuclear pore complex. J. Biol. Chem. 2004, 279, 27735–27742. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.M.; Zhao, L.; Bosard, C.; Imperiale, M.J. Role of a nuclear localization signal on the minor capsid proteins VP2 and VP3 in BKPyV nuclear entry. Virology 2015, 474, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, B.; Rainey, E.K.; Benjamin, T.; Baryshev, M.; Mkrtchian, S.; Tsai, B. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell 2005, 20, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Rainey-Barger, E.K.; Magnuson, B.; Tsai, B. A chaperone-activated nonenveloped virus perforates the physiologically relevant endoplasmic reticulum membrane. J. Virol. 2007, 81, 12996–13004. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Kuksin, D.; Norkin, L.C. Disassociation of the SV40 genome from capsid proteins prior to nuclear entry. Virol. J. 2012, 9, 158. [Google Scholar] [CrossRef] [PubMed]
- Huérfano, S.; Ryabchenko, B.; Španielová, H.; Forstová, J. Hydrophobic domains of mouse polyomavirus minor capsid proteins promote membrane association and virus exit from the ER. FEBS J. 2017, 284, 883–902. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Dosey, A.; Herbstman, J.F.; Ravindran, M.S.; Skiniotis, G.; Tsai, B. Erdj5 reductase cooperates with protein disulfide isomerase to promote simian virus 40 endoplasmic reticulum membrane translocation. J. Virol. 2015, 89, 8897–8908. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Malmström, J.; Pelkmans, L.; Haugstetter, J.; Ellgaard, L.; Grünewald, K.; Helenius, A. Simian virus 40 depends on ERprotein folding and quality control factors for entry into host cells. Cell 2007, 131, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsai, B. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog. 2011, 7, e1002037. [Google Scholar] [CrossRef] [PubMed]
- Chook, Y.M.; Blobel, G. Karyopherins and nuclear import. Curr. Opin. Struct. Biol. 2001, 11, 703–715. [Google Scholar] [CrossRef]
- Ishii, N.; Nakanishi, A.; Yamada, M.; Macalalad, M.H.; Kasamatsu, H. Functional complementation of nuclear targeting-defective mutants of simian virus 40 structural proteins. J. Virol. 1994, 68, 8209–8216. [Google Scholar] [PubMed]
- Chang, D.; Haynes, J.I.; Brady, J.N.; Consigli, R.A. Identification of a nuclear localization sequence in the polyomavirus capsid protein VP2. Virology 1992, 191, 978–983. [Google Scholar] [CrossRef]
- Chang, D.; Haynes, J.I.; Brady, J.N.; Consigli, R.A. Identification of amino acid sequences in the polyomavirus capsid proteins that serve as nuclear localization signals. Trans. Kans. Acad. Sci. 1993, 96, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Shum, D.; Morioka, H.; Otsuka, E.; Kasamatsu, H. Interaction of the VP3 nuclear localization signal with the importin α 2/β heterodimer directs nuclear entry of infecting simian virus 40. J. Virol. 2002, 76, 9368–9377. [Google Scholar] [CrossRef] [PubMed]
- Horníková, L.; Žíla, V.; Španielová, H.; Forstová, J. Mouse polyomavirus: Propagation, purification, quantification, and storage. Curr. Protoc. Microbiol. 2015, 38, 14F.1.1–14F.1.26. [Google Scholar] [CrossRef] [PubMed]
- Brameier, M.; Krings, A.; MacCallum, R.M. Nucpred-predicting nuclear localization of proteins. Bioinformatics 2007, 23, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Orlando, S.J.; Nabavi, M.; Gharakhanian, E. Rapid small-scale isolation of SV40 virions and SV40 DNA. J. Virol. Methods 2000, 90, 109–114. [Google Scholar] [CrossRef]
- Horníková, L.; Man, P.; Forstová, J. Blue native protein electrophoresis for studies of mouse polyomavirus morphogenesis and interactions between the major capsid protein VP1 and cellular proteins. J. Virol. Methods 2011, 178, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Huerfano, S.; Ryabchenko, B.; Forstová, J. Nucleofection of expression vectors induces a robust interferon response and inhibition of cell proliferation. DNA Cell Biol. 2013, 32, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Koos, B.; Andersson, L.; Clausson, C.M.; Grannas, K.; Klaesson, A.; Cane, G.; Söderberg, O. Analysis of protein interactions in situ by proximity ligation assays. Curr. Top. Microbiol. Immunol. 2014, 377, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Huang, C.Y. Use of in situ proximity ligation assays for systems analysis of signaling pathways. Curr. Protoc. Cell Biol. 2016, 71. [Google Scholar] [CrossRef]
- Chang, D.; Haynes, J.I.; Brady, J.N.; Consigli, R.A. The use of additive and subtractive approaches to examine the nuclear localization sequence of the polyomavirus major capsid protein VP1. Virology 1992, 189, 821–827. [Google Scholar] [CrossRef]
- Soeda, E.; Arrand, J.R.; Griffin, B.E. Polyoma virus DNA: Complete nucleotide sequence of the gene which codes for polyoma virus capsid protein VP1 and overlaps the VP2/VP3 genes. J. Virol. 1980, 33, 619–630. [Google Scholar] [PubMed]
- Huerfano, S.; Zíla, V.; Boura, E.; Spanielová, H.; Stokrová, J.; Forstová, J. Minor capsid proteins of mouse polyomavirus are inducers of apoptosis when produced individually but are only moderate contributors to cell death during the late phase of viral infection. FEBS J. 2010, 277, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Mannová, P.; Forstová, J. Mouse polyomavirus utilizes recycling endosomes for a traffic pathway independent of COPI vesicle transport. J. Virol. 2003, 77, 1672–1681. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fluck, M. Kinetic analysis of the steps of the polyomavirus lytic cycle. J. Virol. 2001, 75, 8368–8379. [Google Scholar] [CrossRef] [PubMed]
- Zila, V.; Difato, F.; Klimova, L.; Huerfano, S.; Forstova, J. Involvement of microtubular network and its motors in productive endocytic trafficking of mouse polyomavirus. PLoS ONE 2014, 9, e96922. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.H.; Suomalainen, M.; Andriasyan, V.; Kilcher, S.; Mercer, J.; Neef, A.; Luedtke, N.W.; Greber, U.F. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 2013, 14, 468–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flatt, J.W.; Greber, U.F. Misdelivery at the Nuclear Pore Complex-Stopping a Virus Dead in Its Tracks. Cells 2015, 28, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Hornikova, L.; Charles University, Prague, Czech Republic. The data when we analyzed by confocal microscopy the subcellular localization of the BK minor proteins. 2017. [Google Scholar]
- Schowalter, R.M.; Buck, C.B. The merkel cell polyomavirus minor capsid protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.A.; Stage, D.E.; Goldfarb, D.S. Evolution of the metazoan-specific importin α gene family. J. Mol. Evol. 2009, 68, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Pumroy, R.A.; Cingolani, G. Diversification of importin-α isoforms in cellular trafficking and disease states. Biochem. J. 2015, 466, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Köhler, M.; Speck, C.; Christiansen, M.; Bischoff, F.R.; Prehn, S.; Haller, H.; Görlich, D.; Hartmann, E. Evidence for distinct substrate specificities of importin α family members in nuclear protein import. Mol. Cell Biol. 1999, 19, 7782–7791. [Google Scholar] [CrossRef] [PubMed]
- Sankhala, R.S.; Lokareddy, R.K.; Begum, S.; Pumroy, R.A.; Gillilan, R.E.; Cingolani, G. Three-dimensional context rather than NLS amino acid sequence determines importin α subtype specificity for RCC1. Nat. Commun. 2017, 8, 979. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soldatova, I.; Prilepskaja, T.; Abrahamyan, L.; Forstová, J.; Huérfano, S. Interaction of the Mouse Polyomavirus Capsid Proteins with Importins Is Required for Efficient Import of Viral DNA into the Cell Nucleus. Viruses 2018, 10, 165. https://0-doi-org.brum.beds.ac.uk/10.3390/v10040165
Soldatova I, Prilepskaja T, Abrahamyan L, Forstová J, Huérfano S. Interaction of the Mouse Polyomavirus Capsid Proteins with Importins Is Required for Efficient Import of Viral DNA into the Cell Nucleus. Viruses. 2018; 10(4):165. https://0-doi-org.brum.beds.ac.uk/10.3390/v10040165
Chicago/Turabian StyleSoldatova, Irina, Terezie Prilepskaja, Levon Abrahamyan, Jitka Forstová, and Sandra Huérfano. 2018. "Interaction of the Mouse Polyomavirus Capsid Proteins with Importins Is Required for Efficient Import of Viral DNA into the Cell Nucleus" Viruses 10, no. 4: 165. https://0-doi-org.brum.beds.ac.uk/10.3390/v10040165