Phototracking Vaccinia Virus Transport Reveals Dynamics of Cytoplasmic Dispersal and a Requirement for A36R and F12L for Exit from the Site of Wrapping


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Constructs
2.2. Infection
2.3. Plaque Assays
2.4. Fluorescent Virus Generation
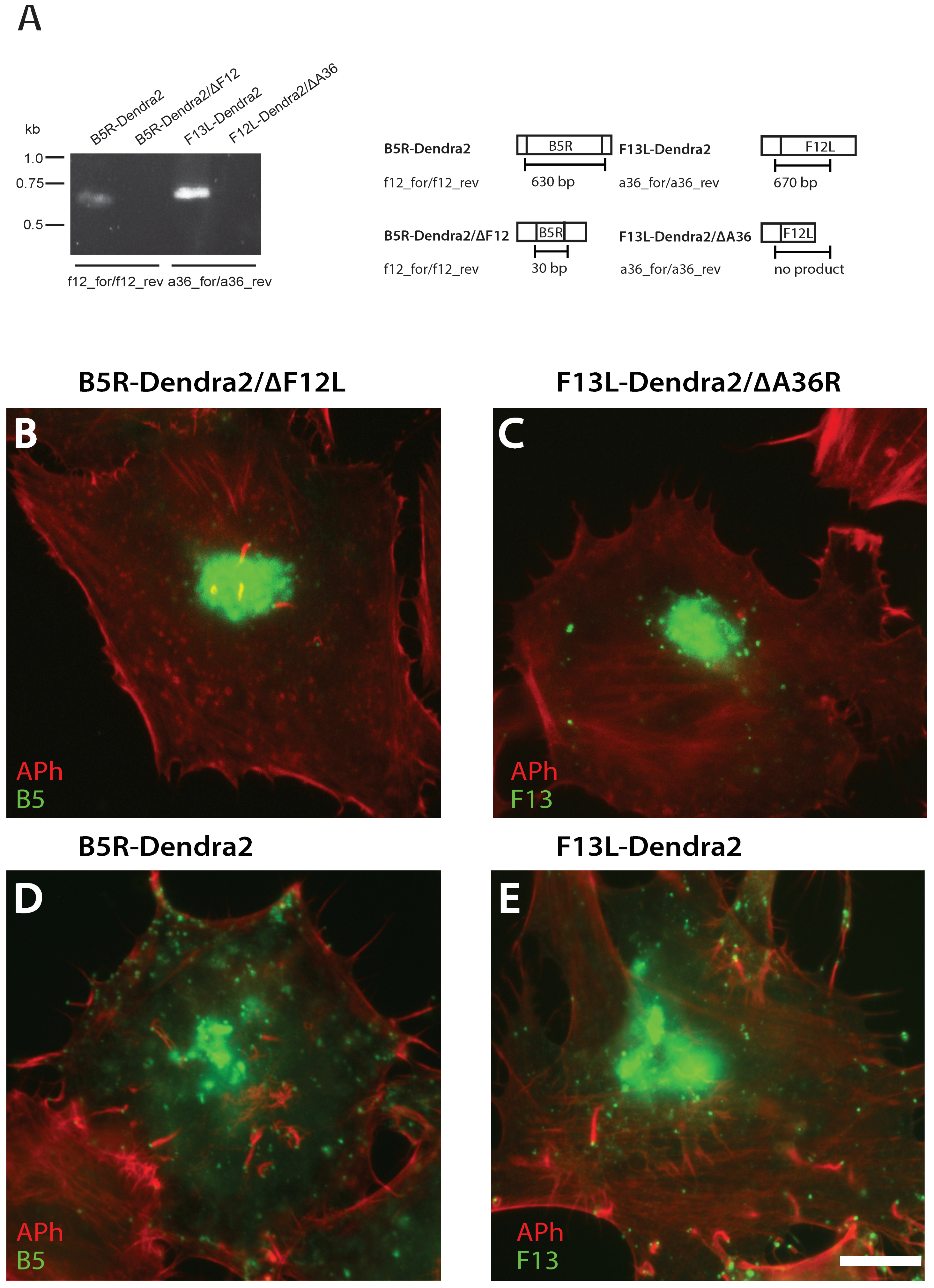
2.5. Validating B5R-Dendra2/∆F12L and F13L-Dendra2/∆A36R Null Mutants
2.6. Immunofluorescence Assays
2.7. Confocal Microscopy
3. Results
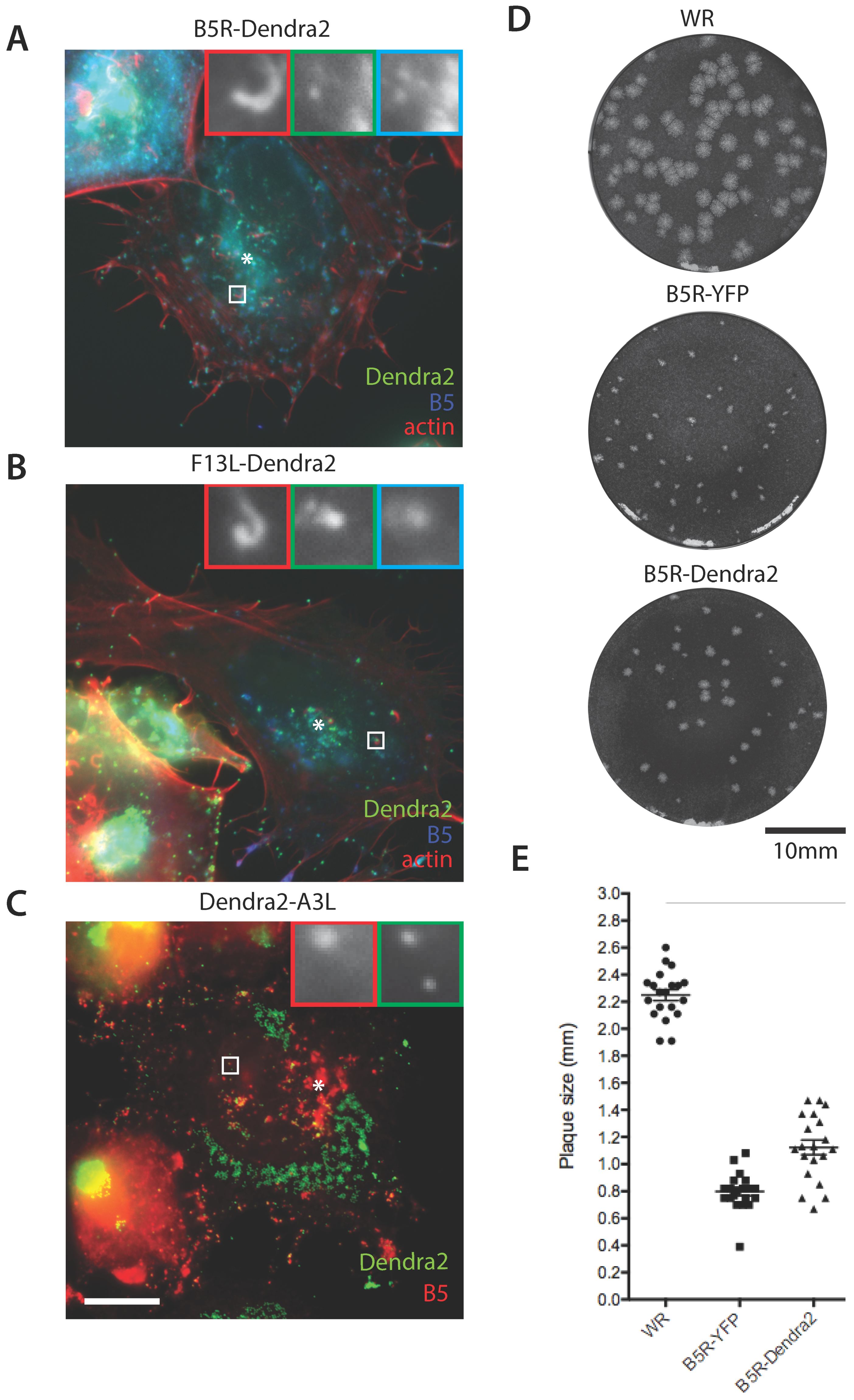
3.1. Construction of Photoconvertible VACV
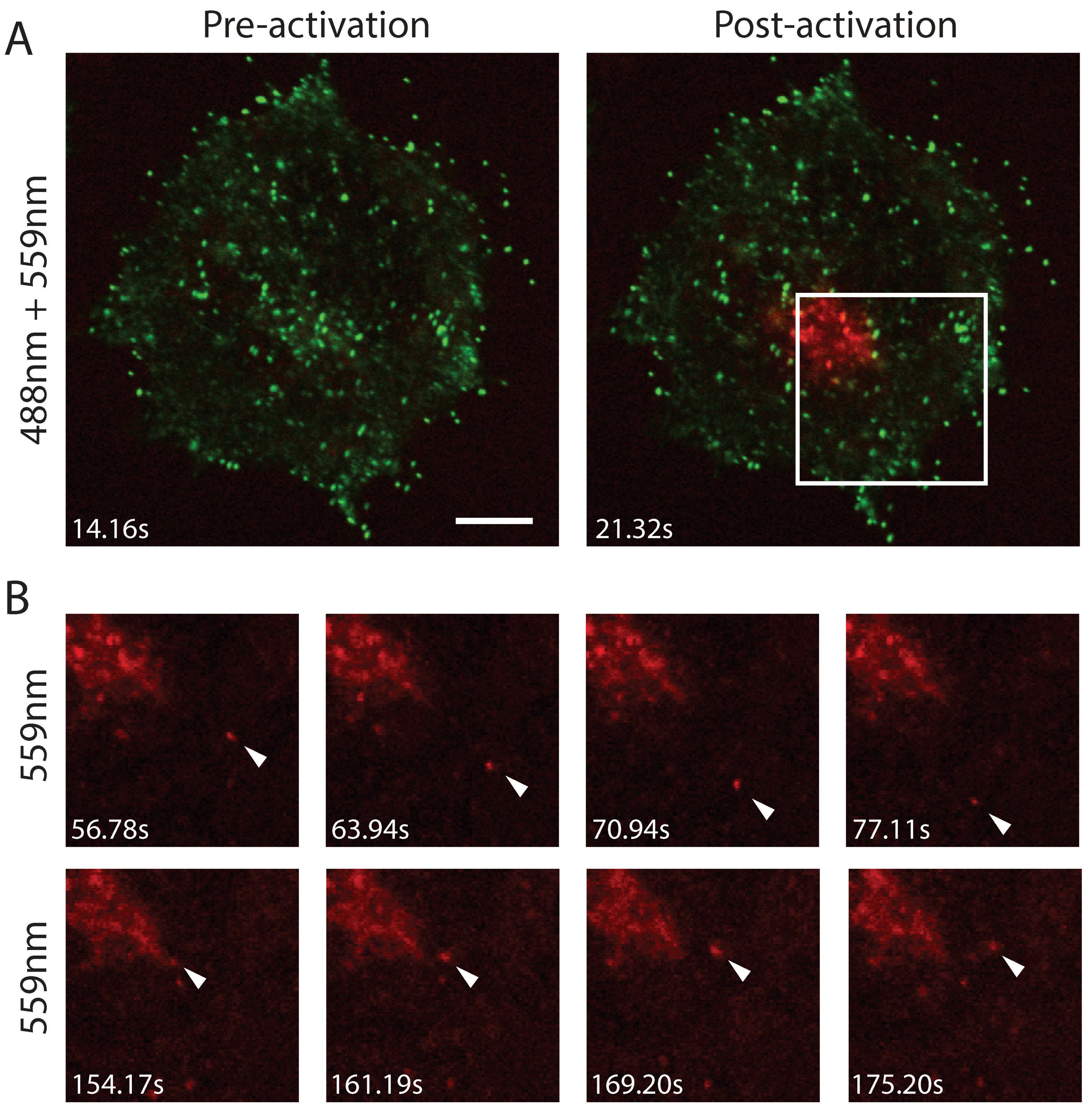
3.2. Efficient Photoconversion of Dendra2 Viruses
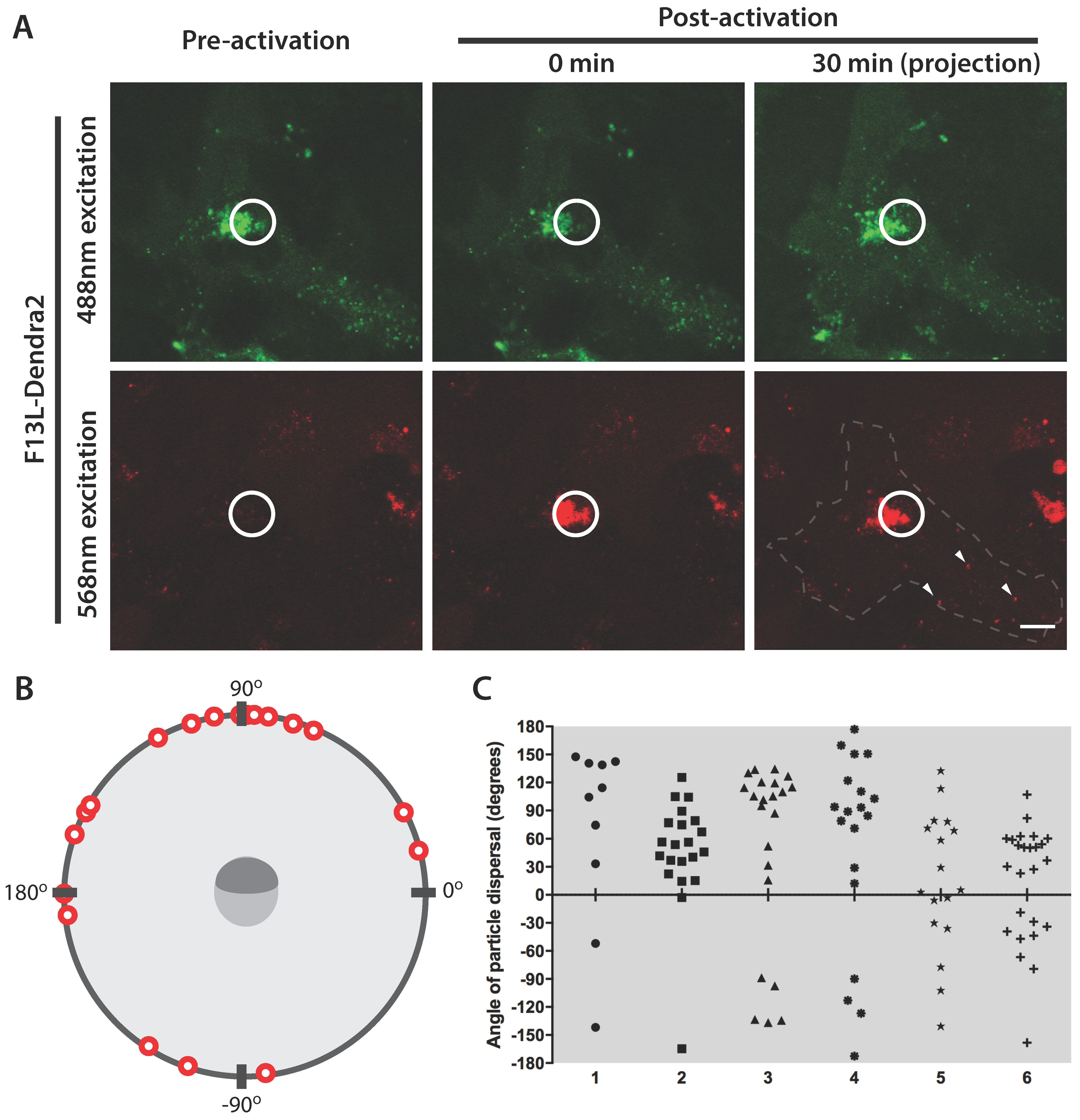
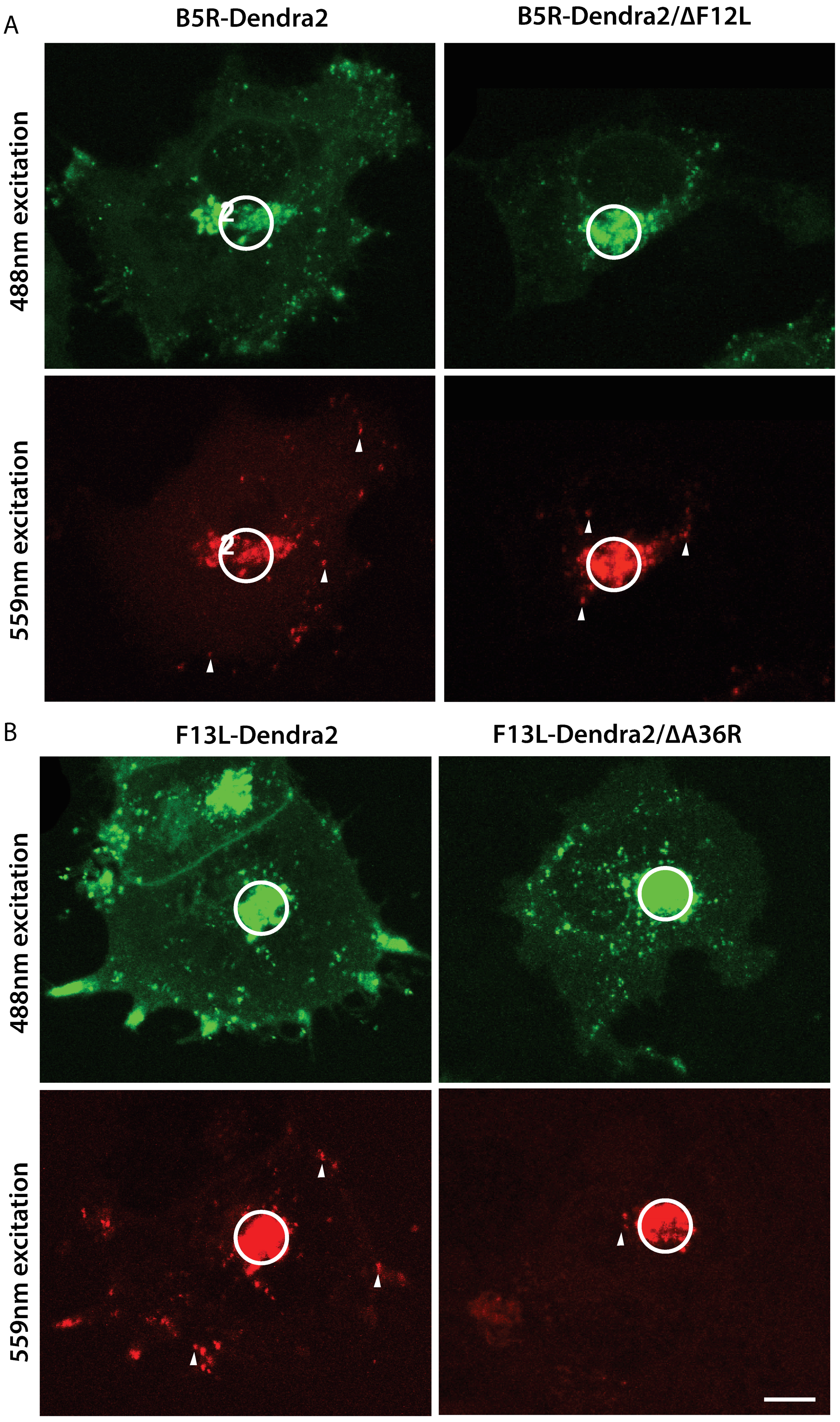
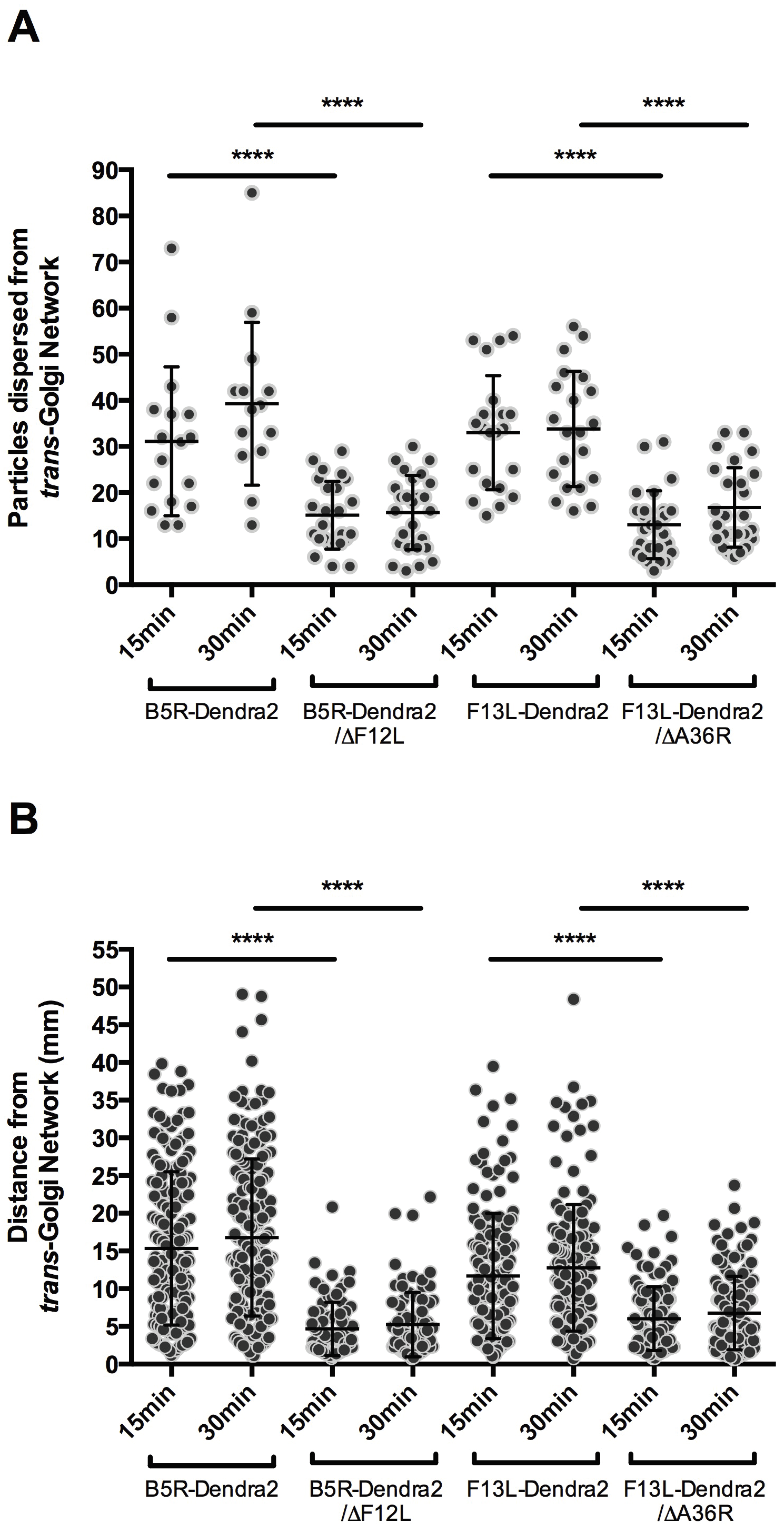
3.3. F12L and A36R Mutants Reveal a Role for Kinesin-1 in WV Exit from the TGN
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dodding, M.P.; Way, M. Coupling viruses to dynein and kinesin-1. EMBO J. 2011, 30, 3527–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, M.J.; Lakadamyali, M.; Brandenburg, B.; Zhuang, X. Single-virus tracking in live cells. Cold Spring Harb. Protoc. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Verkhusha, V.V.; Staroverov, D.B.; Souslova, E.A.; Lukyanov, S.; Lukyanov, K.A. Photoswitchable cyan fluorescent protein for protein tracking. Nat. Biotechnol. 2004, 22, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Wiedenmann, J.; Ivanchenko, S.; Oswald, F.; Schmitt, F.; Rocker, C.; Salih, A.; Spindler, K.D.; Nienhaus, G.U. Eosfp, a fluorescent marker protein with uv-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15905–15910. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Lukyanov, S.; Lukyanov, K.A. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat. Protoc. 2007, 2, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.H.; McCaffery, J.M.; Chan, D.C. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis 2012, 50, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.; Mathur, N.; Mathur, J. Simultaneous live-imaging of peroxisomes and the er in plant cells suggests contiguity but no luminal continuity between the two organelles. Front. Physiol. 2013, 4, 196. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.; Way, M. The role of signalling and the cytoskeleton during vaccinia virus egress. Virus Res. 2015, 209, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Condit, R.C.; Moussatche, N.; Traktman, P. In a nutshell: Structure and assembly of the vaccinia virion. Adv. Virus Res. 2006, 66, 31–124. [Google Scholar] [PubMed]
- Schepis, A.; Stauber, T.; Krijnse Locker, J. Kinesin-1 plays multiple roles during the vaccinia virus life cycle. Cell. Microbiol. 2007, 9, 1960–1973. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, C.M.; Hollinshead, M.; Smith, G.L. The vaccinia virus A27L protein is needed for the microtubule-dependent transport of intracellular mature virus particles. J. Gen. Virol. 2000, 81, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, K.L.; Smith, G.L. Vaccinia virus morphogenesis and dissemination. Trends Microbiol. 2008, 16, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Rietdorf, J.; Ploubidou, A.; Reckmann, I.; Holmstrom, A.; Frischknecht, F.; Zettl, M.; Zimmermann, T.; Way, M. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol. 2001, 3, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, B.M.; Moss, B. Vaccinia virus A36R membrane protein provides a direct link between intracellular enveloped virions and the microtubule motor kinesin. J. Virol. 2004, 78, 2486–2493. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.P.; Mitter, R.; Humphries, A.C.; Way, M. A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J. 2011, 30, 4523–4538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.N.D.; Carpentier, D.C.J.; Ewles, H.A.; Lee, S.A.; Smith, G.L. Vaccinia virus proteins a36 and f12/e2 show strong preferences for different kinesin light chain isoforms. Traffic 2017, 18, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.; Newsome, T.P.; Collinson, L.; Edwards, C.; Way, M. An E2-F12 complex is required for intracellular enveloped virus morphogenesis during vaccinia infection. Cell. Microbiol. 2009, 11, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, D.C.; Gao, W.N.; Ewles, H.; Morgan, G.W.; Smith, G.L. Vaccinia virus protein complex F12/E2 interacts with kinesin light chain isoform 2 to engage the kinesin-1 motor complex. PLoS Pathog. 2015, 11, e1004723. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.C.; Ward, B.M. The vaccinia virus protein F12 associates with intracellular enveloped virus through an interaction with A36. J. Virol. 2009, 83, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Martinez, E.; Roberts, K.L.; Hollinshead, M.; Smith, G.L. Vaccinia virus intracellular enveloped virions move to the cell periphery on microtubules in the absence of the A36R protein. J. Gen. Virol. 2005, 86, 2961–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, D.C.J.; Hollinshead, M.S.; Ewles, H.A.; Lee, S.A.; Smith, G.L. Tagging of the vaccinia virus protein F13 with mcherry causes aberrant virion morphogenesis. J. Gen. Virol. 2017, 98, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Newsome, T.P.; Marzook, N.B. Viruses that ride on the coat-tails of actin nucleation. Semin. Cell Dev. Biol. 2015, 46, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.A.; Harriman, P.D.; Wall, J.D. Escherichia coli mutants deficient in guanine-xanthine phosphoribosyltransferase. J. Bacteriol. 1976, 126, 1141–1148. [Google Scholar] [PubMed]
- Campbell, R.E.; Tour, O.; Palmer, A.E.; Steinbach, P.A.; Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 7877–7882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkner, F.G.; Moss, B. Escherichia coli gpt gene provides dominant selection for vaccinia virus open reading frame expression vectors. J. Virol. 1988, 62, 1849–1854. [Google Scholar] [PubMed]
- Frischknecht, F.; Cudmore, S.; Moreau, V.; Reckmann, I.; Rottger, S.; Way, M. Tyrosine phosphorylation is required for actin-based motility of vaccinia but not listeria or shigella. Curr. Biol. 1999, 9, 89–92. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Sisler, J.R.; Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques 1997, 23, 1094–1097. [Google Scholar] [PubMed]
- Marzook, N.B.; Procter, D.J.; Lynn, H.; Yamamoto, Y.; Horsington, J.; Newsome, T.P. Methodology for the efficient generation of fluorescently tagged vaccinia virus proteins. J. Vis. Exp. 2014, 83, e51151. [Google Scholar] [CrossRef] [PubMed]
- Engelstad, M.; Smith, G.L. The vaccinia virus 42-kda envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 1993, 194, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Wolffe, E.J.; Isaacs, S.N.; Moss, B. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J. Virol. 1993, 67, 4732–4741. [Google Scholar] [PubMed]
- Zhang, W.H.; Wilcock, D.; Smith, G.L. Vaccinia virus F12L protein is required for actin tail formation, normal plaque size, and virulence. J. Virol. 2000, 74, 11654–11662. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J.E.; Smith, G.L. Vaccinia virus gene A36R encodes a Mr 43-50 K protein on the surface of extracellular enveloped virus. Virology 1994, 204, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, S.N. Vaccinia virus and Poxvirology: Methods and Protocols; Humana Press Inc.: Totowa, NJ, USA, 2004; Volume 269. [Google Scholar]
- Geada, M.M.; Galindo, I.; Lorenzo, M.M.; Perdiguero, B.; Blasco, R. Movements of vaccinia virus intracellular enveloped virions with GFP tagged to the F13L envelope protein. J. Gen. Virol. 2001, 82, 2747–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, Y.; Cordeiro, J.V.; Schleich, S.; Newsome, T.P.; Way, M. The release of vaccinia from infected cells requires rhoa-mdia modulation of cortical actin. Cell Host Microbe 2007, 1, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Koizumi, K.; Macrae-Crerar, A.; Gallagher, K.L. Assessing the utility of photoswitchable fluorescent proteins for tracking intercellular protein movement in the arabidopsis root. PLoS ONE 2011, 6, e27536. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.Y.; Hope, T.J. Mobility of human immunodeficiency virus type 1 pr55gag in living cells. J. Virol. 2006, 80, 8796–8806. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.M.; Gretton, S.N.; McLauchlan, J.; Targett-Adams, P. Mobility analysis of an NS5A-GFP fusion protein in cells actively replicating hepatitis C virus subgenomic RNA. J. Gen. Virol. 2007, 88, 470–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardo, L.; Hatch, S.C.; Chen, J.; Nikolaitchik, O.; Burdick, R.C.; Chen, D.; Westlake, C.J.; Lockett, S.; Pathak, V.K.; Hu, W.S. Dynamics of HIV-1 RNA near the plasma membrane during virus assembly. J. Virol. 2015, 89, 10832–10840. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, G.; Ried, M.U.; Endress, T.; Buning, H.; Hallek, M.; Brauchle, C. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science 2001, 294, 1929–1932. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Pomeranz, L.; Gross, S.P.; Enquist, L.W. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. USA 2004, 101, 16034–16039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willard, M. Rapid directional translocations in virus replication. J. Virol. 2002, 76, 5220–5232. [Google Scholar] [CrossRef] [PubMed]
- Dohner, K.; Wolfstein, A.; Prank, U.; Echeverri, C.; Dujardin, D.; Vallee, R.; Sodeik, B. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 2002, 13, 2795–2809. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welte, M.A. Bidirectional transport along microtubules. Curr. Biol. 2004, 14, R525–R537. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010, 6, e1000991. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; Beerli, C.; Pereira, P.M.; Scherer, K.M.; Samolej, J.; Bleck, C.K.; Mercer, J.; Henriques, R. Virusmapper: Open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci. Rep. 2016, 6, 29132. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| a3_1 | AAGTCGACCGTTGACGCCGAGCAATGC |
| a3_2 | TTGGATCCCGAGAATGAATAAGTACTAAAGG |
| a3_3 | AAGGCGGCCGCGAAGCCGTGGTCAATAGCG |
| a3_4 | TTAAGCTTCGAGAATGAATAAGTACTAAAGG |
| a3_5 | AAAGATCTGTCGACCGTTGACGCCGAGCAATGC |
| a3_6 | GTTAATTCCCGGGGTGTTCATTATTTATATTCGTAGTTTTTAC |
| a3_7 | GTAAAAACTACGAATATAAATAATGAACACCCCGGGAATTAAC |
| den_1 | TTGCGGCCGCCCCACACCTGGCTGGGCAGGGG |
| den_2 | AAAGATCTACCATGAACACCCCGGGAATTAAC |
| den_3 | AAGCGGCCGCCAACACCCCGGGAATTAACCTG |
| den_4 | TTGGATCCTTACCACACCTGGCTGGGCAG |
| b5_1 | GTTCCATAAATTGCTACCG |
| b5_2 | AAAGGATCCTATACCATTAAGTGTATCCATCACC |
| f13_1 | AAAGATCTACCATGTGGCCATTTGCATCG |
| f13_2 | TTGCGGCCGCCAATTTTTAACGATTTACTGTG |
| Name | Sequence |
|---|---|
| f12_for | GAATATCCTGCTCTGATAGCAG |
| f12_rev | GCTAGGGTTATTTGGATGGATGCG |
| a36_for | ATGATGCTGGTACCTATCACG |
| a36_rev | CACGAACAGGGAGATATAGCAC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lynn, H.; Howell, L.M.; Diefenbach, R.J.; Newsome, T.P. Phototracking Vaccinia Virus Transport Reveals Dynamics of Cytoplasmic Dispersal and a Requirement for A36R and F12L for Exit from the Site of Wrapping. Viruses 2018, 10, 390. https://0-doi-org.brum.beds.ac.uk/10.3390/v10080390
Lynn H, Howell LM, Diefenbach RJ, Newsome TP. Phototracking Vaccinia Virus Transport Reveals Dynamics of Cytoplasmic Dispersal and a Requirement for A36R and F12L for Exit from the Site of Wrapping. Viruses. 2018; 10(8):390. https://0-doi-org.brum.beds.ac.uk/10.3390/v10080390
Chicago/Turabian StyleLynn, Helena, Liam M. Howell, Russell J. Diefenbach, and Timothy P. Newsome. 2018. "Phototracking Vaccinia Virus Transport Reveals Dynamics of Cytoplasmic Dispersal and a Requirement for A36R and F12L for Exit from the Site of Wrapping" Viruses 10, no. 8: 390. https://0-doi-org.brum.beds.ac.uk/10.3390/v10080390