Identification of a Novel Betacoronavirus (Merbecovirus) in Amur Hedgehogs from China

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. Detection of CoVs by RT-PCR and Sequencing
2.4. Viral Culture
2.5. Complete Genome Sequencing and Analysis of Ea-HedCoV HKU31
2.6. Estimation of Divergence Dates
2.7. Structural Modelling of Ea-HedCoV HKU31 Receptor-Binding Domain (RBD)
2.8. Recombination Analysis
2.9. Nucleotide Sequence Accession Numbers
3. Results
3.1. Detection of CoVs in Animals and Discovery of a Novel Species of Merbecovirus from Amur Hedgehogs
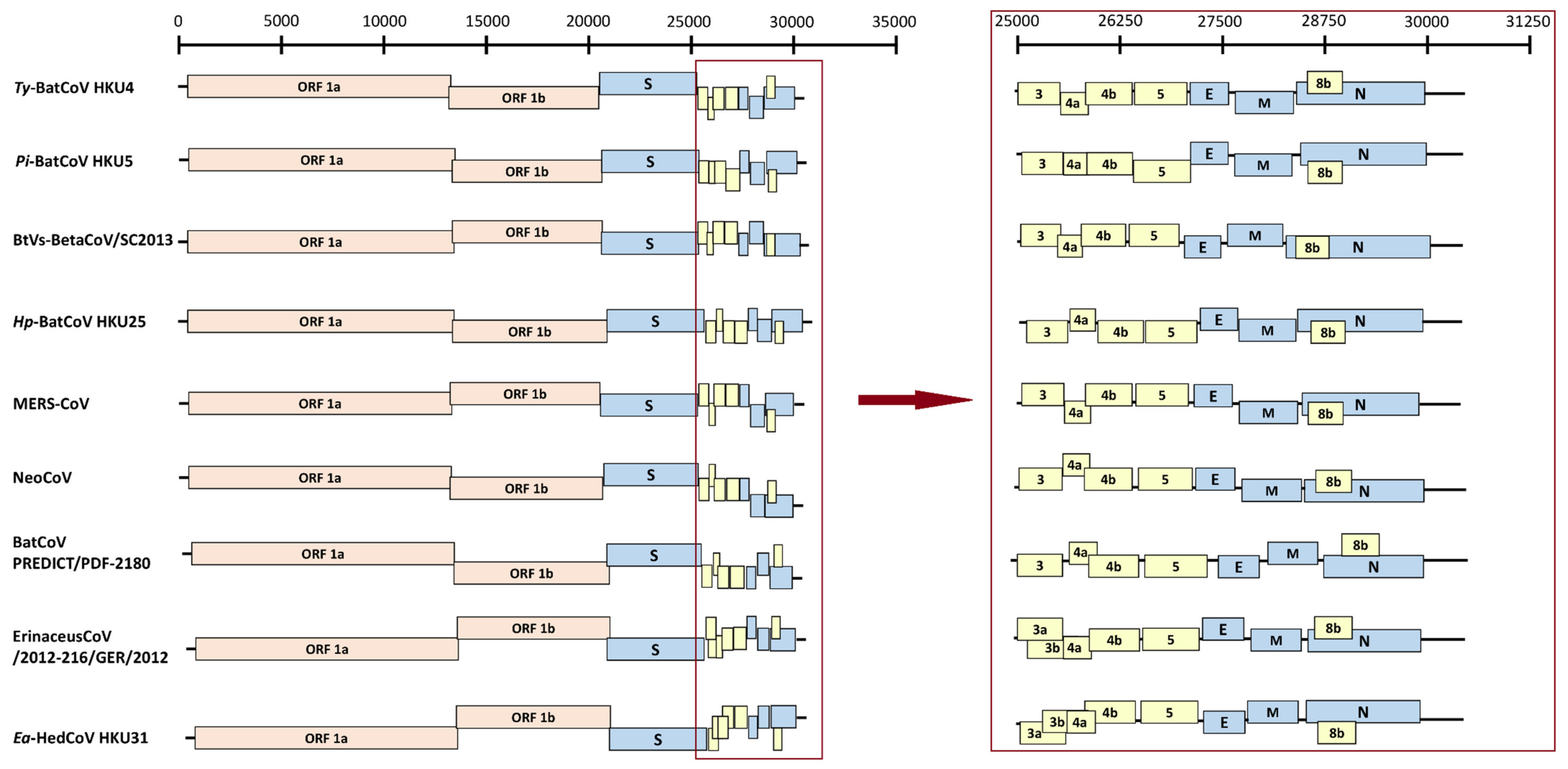
3.2. Genome Organization of Ea-HedCoV HKU31
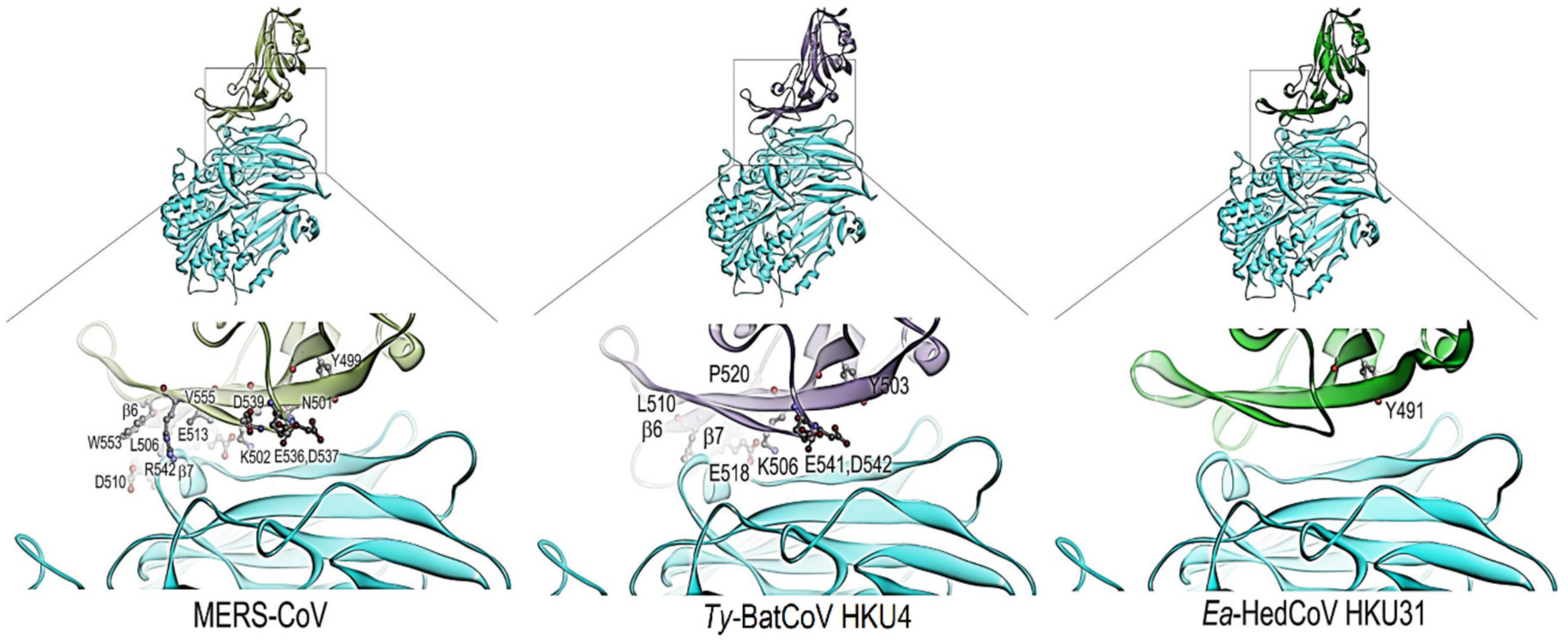
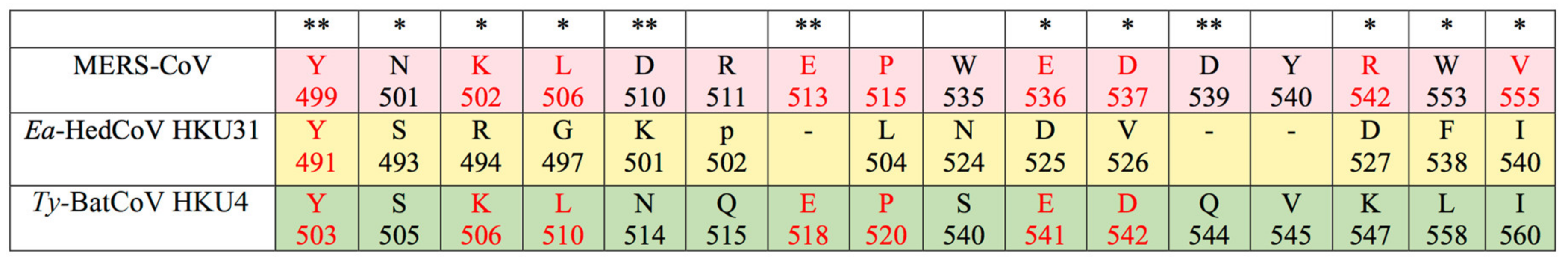
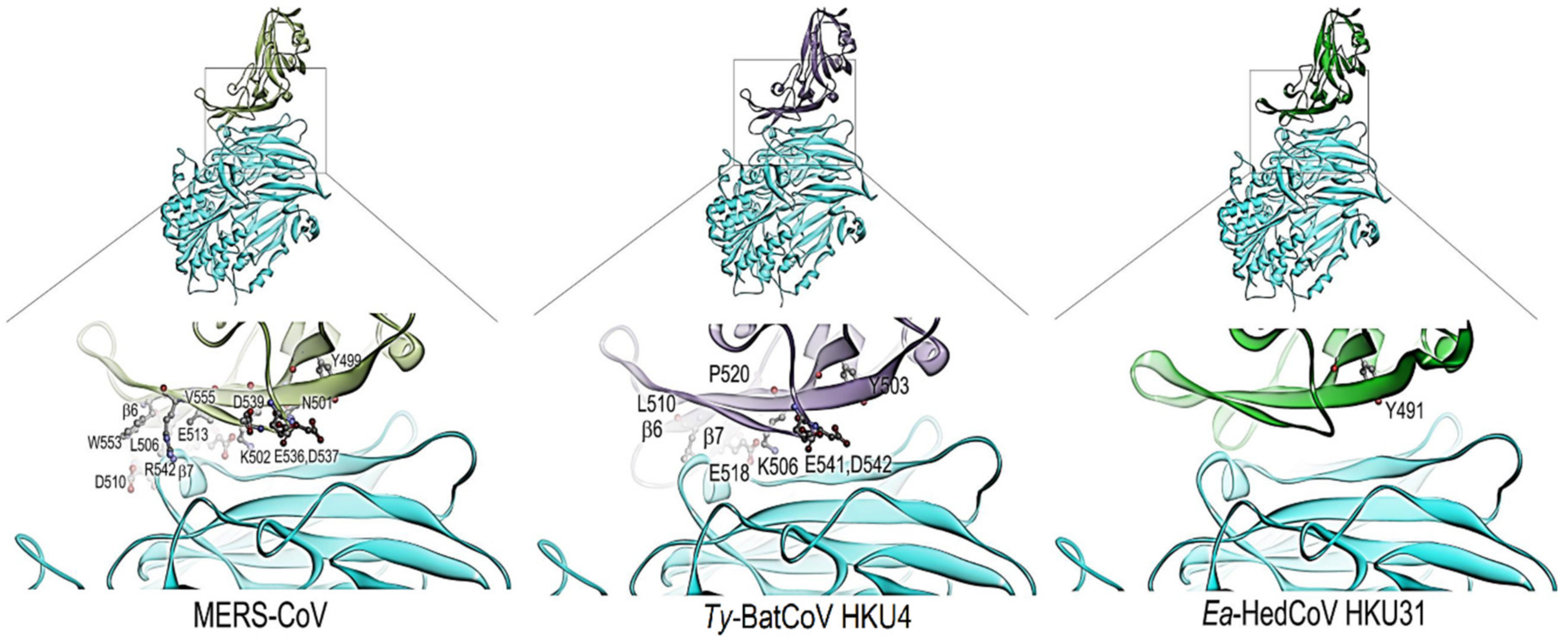
3.3. Sequence Analysis and Structural Modelling of the Ea-HedCoV HKU31 Spike Protein
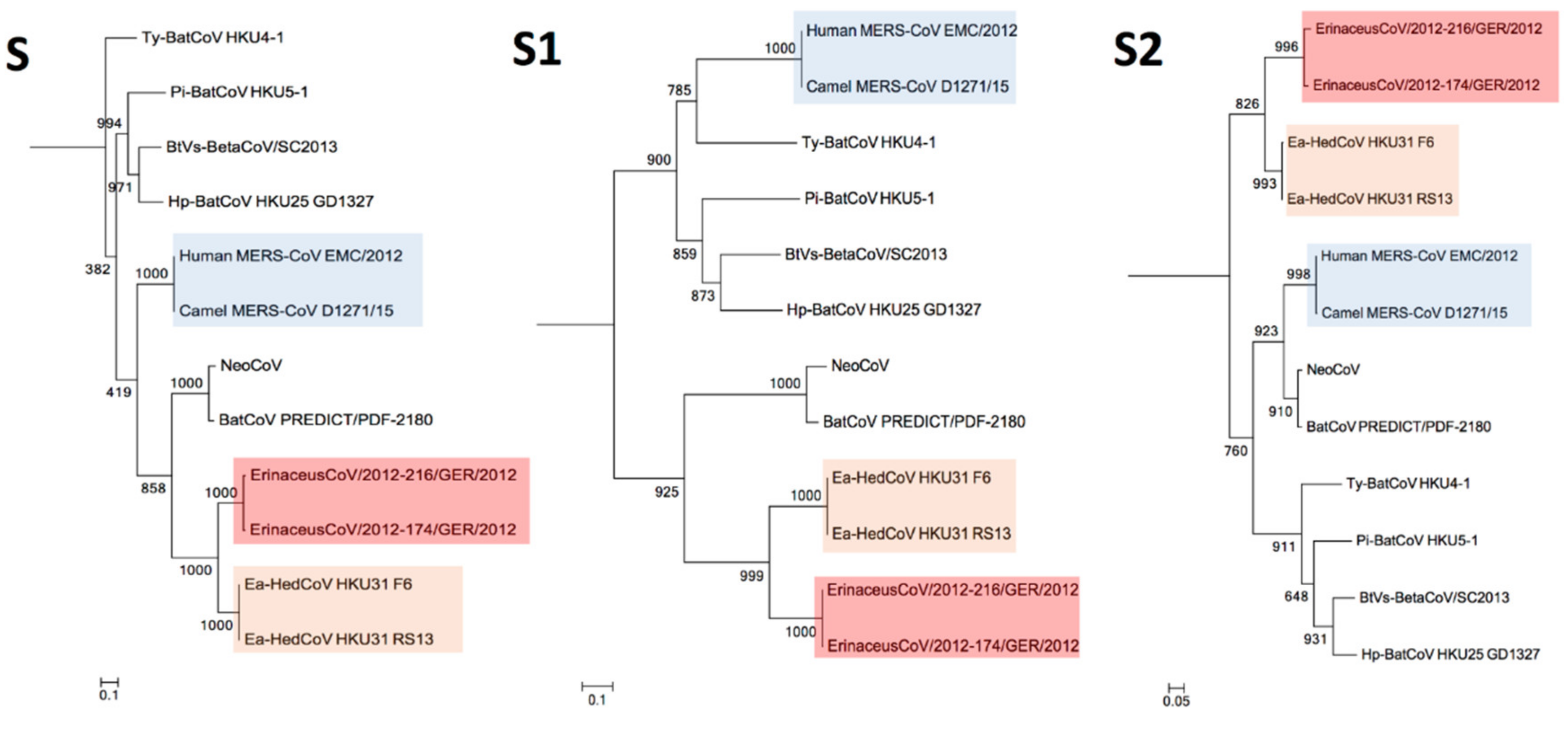
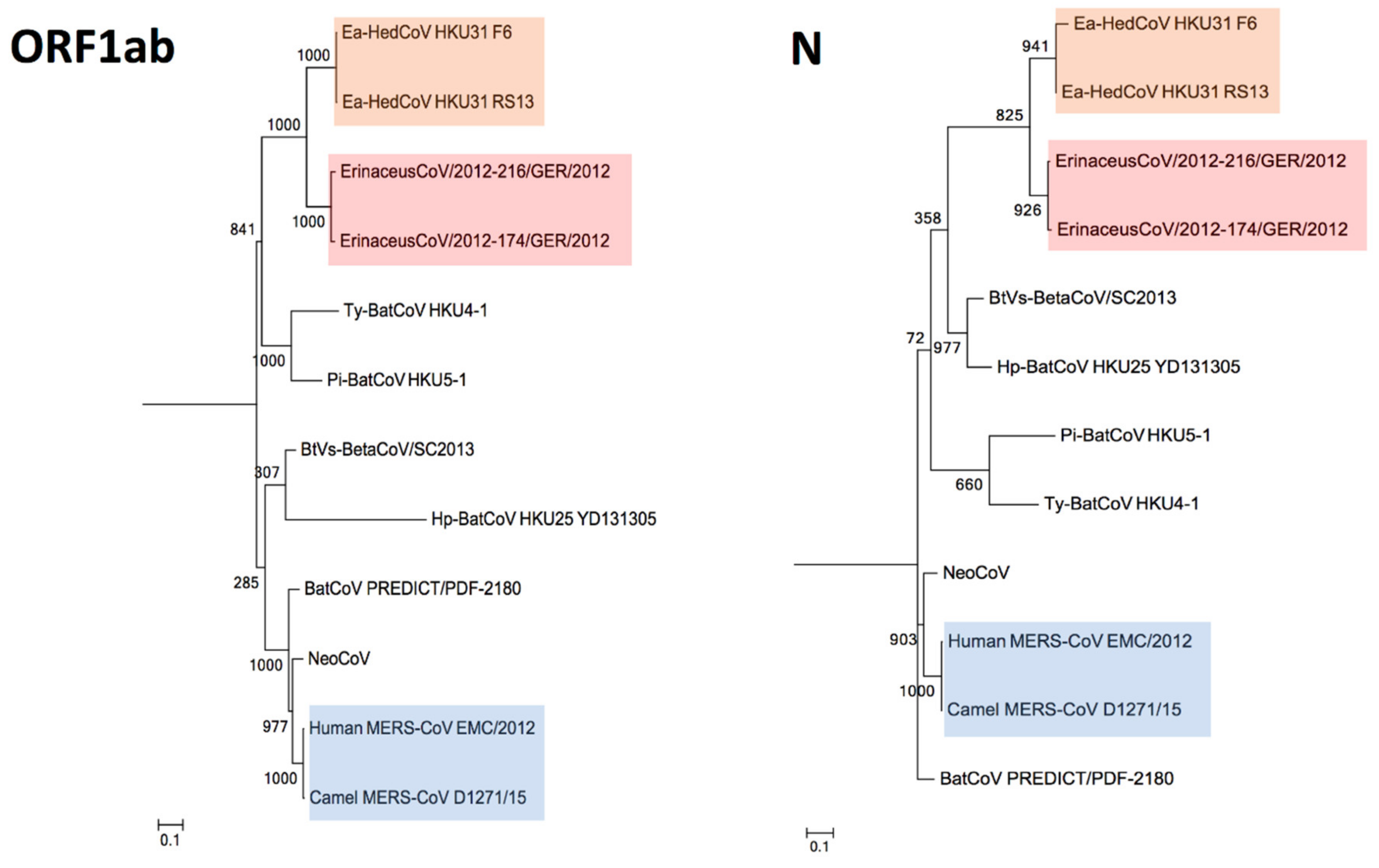
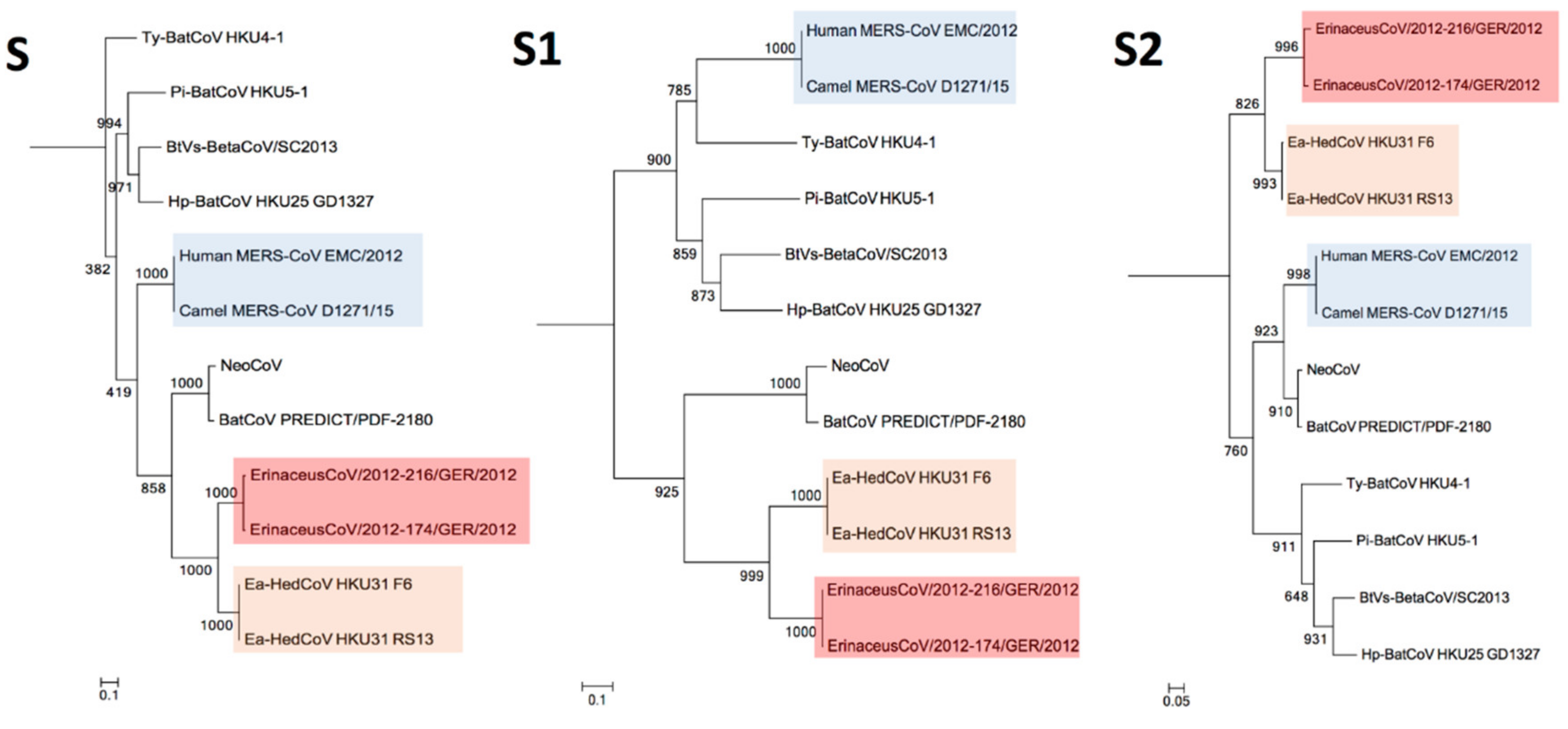
3.4. Phylogenetic Analysis
3.5. Estimation of Divergence Dates
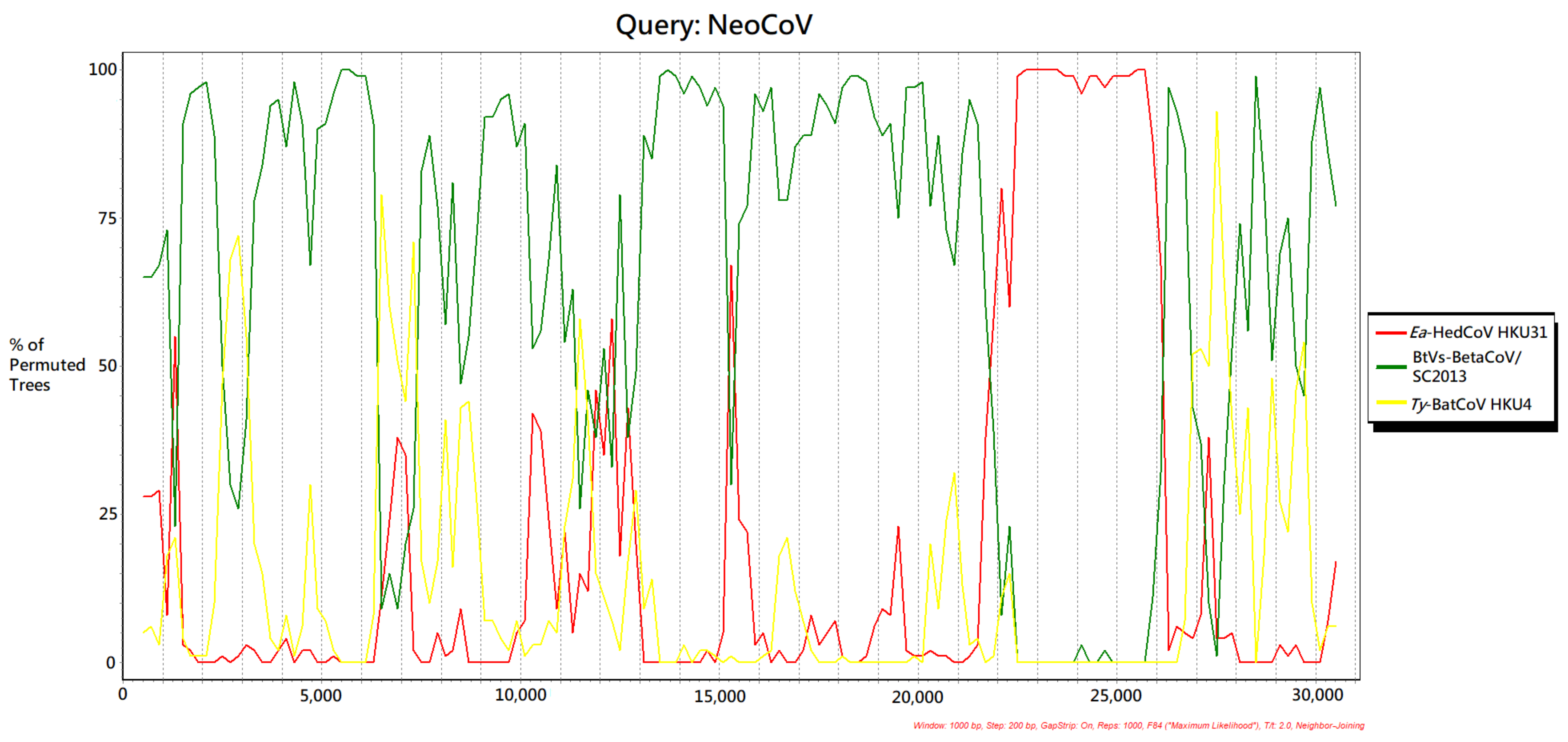
3.6. Recombination Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BetaCoV | Betacoronavirus |
| bp | Base pair |
| CoVs | Coronaviruses |
| HCoV | Human coronavirus |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| ORF | Open reading frame |
| PCR | Polymerase chain reaction |
| Pi | Pipistrellus |
| RdRp | RNA-dependent RNA polymerase |
| RT | Reverse transcription |
| S | Spike |
| SARSr-CoV | Severe Acute Respiratory Syndrome-related coronavirus |
| TRS | Transcription regulatory sequence |
| Ty | Tylonycteris |
| µL | Microliter |
References
- World Health Organization. Latest Disease Outbreak News: Middle East Respiratory Coronavirus (MERS-CoV); World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Reusken, C.B.; Haagmans, B.L.; Muller, M.A.; Gutierrez, C.; Godeke, G.J.; Meyer, B.; Muth, D.; Raj, V.S.; Vries, L.S.; Corman, V.M.; et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: A comparative serological study. Lancet Infect. Dis. 2013. [Google Scholar] [CrossRef]
- Haagmans, B.L.; Al Dhahiry, S.H.; Reusken, C.B.; Raj, V.S.; Galiano, M.; Myers, R.; Godeke, G.J.; Jonges, M.; Farag, E.; Diab, A.; et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet Infect. Dis. 2013. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Lau, S.K.P.; To, K.K.W.; Cheng, V.C.C.; Woo, P.C.Y.; Yuen, K.Y. Middle east respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing Sars-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Kallies, R.; Philipps, H.; Gopner, G.; Muller, M.A.; Eckerle, I.; Brunink, S.; Drosten, C.; Drexler, J.F. Characterization of a novel betacoronavirus related to middle East respiratory syndrome coronavirus in European hedgehogs. J. Virol. 2014, 88, 717–724. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Li, K.S.; Tsang, A.K.; Yuen, K.Y. Genetic relatedness of the novel human group c betacoronavirus to tylonycteris bat coronavirus HKU4 and pipistrellus bat coronavirus HKU5. Emerg. Microbes Infect. 2012, 1. [Google Scholar] [CrossRef]
- Corman, V.M.; Ithete, N.L.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the phylogenetic tree of Middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an african bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further evidence for bats as the evolutionary source of middle east respiratory syndrome coronavirus. Am. Soc. Microbiol. 2017, 8, e00373. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Zhang, L.; Luk, H.K.H.; Xiong, L.; Peng, X.; Li, K.S.M.; He, X.; Zhao, P.S.; Fan, R.Y.Y.; Wong, A.C.P.; et al. Receptor usage of a novel bat lineage c betacoronavirus reveals evolution of Middle East respiratory syndrome-related coronavirus spike proteins for human dipeptidyl peptidase 4 Binding. J. Infect. Dis. 2018, 218, 197–207. [Google Scholar] [CrossRef]
- Luo, C.M.; Wang, N.; Yang, X.L.; Liu, H.Z.; Zhang, W.; Li, B.; Hu, B.; Peng, C.; Geng, Q.B.; Zhu, G.J.; et al. Discovery of novel bat coronaviruses in South China that use the same receptor as Middle East respiratory syndrome coronavirus. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Lai, M.M.C.; Cavanagh, D. The molecular biology of coronaviruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 1997; pp. 1–100. [Google Scholar] [CrossRef]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. In Current Topics in Microbiology and Immunology; Springer-Verlag: Berlin, Germany, 2005; pp. 77–110. [Google Scholar] [CrossRef]
- De Groot, R.J.; Baric, R.; Enjuanes, L.; Gorbalenya, A.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.; Talbot, P.J.; Woo, P.C.; et al. Coronaviridae. In Virus Taxonomy, Classification and Nomenclature of Viruses; Ninth Report of the International Committee on Taxonomy of Viruses; International Union of Microbiological Societies, Virology Division; Microbiology Society: London, UK, 2011; pp. 806–828. [Google Scholar]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef]
- Woo, P.C.; Wang, M.; Lau, S.K.; Xu, H.; Poon, R.W.; Guo, R.; Wong, B.H.; Gao, K.; Tsoi, H.W.; Huang, Y.; et al. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 2007, 81, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Mihindukulasuriya, K.A.; Wu, G.; St Leger, J.; Nordhausen, R.W.; Wang, D. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J. Virol. 2008, 82, 5084–5088. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global epidemiology of bat coronaviruses. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Fouchier, R.A.M.; Hartwig, N.G.; Bestebroer, T.M.; Niemeyer, B.; de Jong, J.C.; Simon, J.H.; Osterhaus, A.D.M.E. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. USA 2004, 101, 6212–6216. [Google Scholar] [CrossRef] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Lai, S.T.; Poon, L.L.M.; Guan, Y.; Yam, L.Y.C.; Lim, W.; Nicholls, J.; Yee, W.K.S.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 2012, 3. [Google Scholar] [CrossRef]
- de Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Tsang, A.K.L.; Fan, R.Y.Y.; Luk, H.K.H.; Cai, J.-P.; Chan, K.-H.; Zheng, B.-J.; Wang, M.; et al. Discovery of a novel coronavirus, China rattus coronavirus HKU24, from Norway rats supports the murine origin of betacoronavirus 1 and has implications for the ancestor of betacoronavirus lineage, a. J. Virol. 2014, 89, 3076–3092. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Li, K.S.M.; Poon, R.W.S.; Wong, B.H.L.; Tsoi, H.-w.; Yip, B.C.K.; Huang, Y.; Chan, K.-h.; Yuen, K.-y. Molecular diversity of coronaviruses in bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.K.; Li, K.S.; Tsang, A.K.; Lam, C.S.; Ahmed, S.; Chen, H.; Chan, K.H.; Woo, P.C.; Yuen, K.Y. Genetic characterization of betacoronavirus lineage c viruses in bats reveals marked sequence divergence in the spike protein of pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: Implications for the origin of the novel Middle East respiratory syndrome coronavirus. J. Virol. 2013, 87, 8638–8650. [Google Scholar] [CrossRef]
- Yang, L.; Wu, Z.; Ren, X.; Yang, F.; Zhang, J.; He, G.; Dong, J.; Sun, L.; Zhu, Y.; Zhang, S.; et al. MERS-related betacoronavirus in Vespertilio superans bats, China. Emerg. Infect. Dis. 2014, 20, 1260–1262. [Google Scholar] [CrossRef]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Monchatre-Leroy, E.; Boue, F.; Boucher, J.M.; Renault, C.; Moutou, F.; Ar Gouilh, M.; Umhang, G. Identification of alpha and beta coronavirus in wildlife species in France: Bats, rodents, rabbits, and hedgehogs. Viruses 2017, 9, 364. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Zhang, H.J.; Fan, R.Y.; Zhang, A.J.; Chan, B.C.; Lam, C.S.; Yip, C.C.; Yuen, M.C.; et al. Identification of novel rosavirus species that infects diverse rodent species and causes multisystemic dissemination in mouse model. PLoS Pathog. 2016, 12, e1005911. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Wong, A.C.P.; Lau, T.C.K.; Woo, P.C.Y. Molecular Evolution of MERS Coronavirus: Dromedaries as a recent intermediate host or long-time animal reservoir? Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Woo, P.C.Y.; Yip, C.C.Y.; Fan, R.Y.Y.; Huang, Y.; Wang, M.; Guo, R.; Lam, C.S.F.; Tsang, A.K.L.; Lai, K.K.Y.; et al. Isolation and characterization of a novel betacoronavirus subgroup a coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J. Virol. 2012, 86, 5481–5496. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Fan, R.Y.Y.; Luk, H.K.H.; Zhu, L.; Fung, J.; Li, K.S.M.; Wong, E.Y.M.; Ahmed, S.S.; Chan, J.F.W.; Kok, R.K.H.; et al. Replication of MERS and SARS coronaviruses in bat cells offers insights to their ancestral origins. Emerg. Microbes Infect. 2018, 7, 209. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Birney, E.; Biswas, M.; Bucher, P.; Cerutti, L.; Corpet, F.; Croning, M.D.; et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001, 29, 37–40. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A. The Pfam protein families database. Nucleic Acids Res. 2002, 30, 276–280. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Lu, G.W.; Hu, Y.W.; Wang, Q.H.; Qi, J.X.; Gao, F.; Li, Y.; Zhang, Y.F.; Zhang, W.; Yuan, Y.; Bao, J.K.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500. [Google Scholar] [CrossRef]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Guo, D.; Fu, L.; Cui, Y.; Liu, X.; et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Qi, J.; Yuan, Y.; Xuan, Y.; Han, P.; Wan, Y.; Ji, W.; Li, Y.; Wu, Y.; Wang, J.; et al. Bat origins of MERS-CoV supported by bat coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe 2014, 16, 328–337. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Li, K.S.M.; Huang, Y.; Shek, C.T.; Tse, H.; Wang, M.; Choi, G.K.Y.; Xu, H.; Lam, C.S.F.; Guo, R.; et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related rhinolophus bat coronavirus in China reveal bats as a reservoir for Acute, self-limiting infection that allows recombination events. J. Virol. 2010, 84, 2808–2819. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Lau, S.K.; Feng, Y.; Chen, H.; Luk, H.K.; Yang, W.H.; Li, K.S.; Zhang, Y.Z.; Huang, Y.; Song, Z.Z.; Chow, W.N.; et al. Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. J. Virol. 2015, 89, 10532–10547. [Google Scholar] [CrossRef]
- Cotten, M.; Watson, S.J.; Zumla, A.I.; Makhdoom, H.Q.; Palser, A.L.; Ong, S.H.; Al Rabeeah, A.A.; Alhakeem, R.F.; Assiri, A.; Al-Tawfiq, J.A.; et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio 2014, 5. [Google Scholar] [CrossRef]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Cauchemez, S.; Fraser, C.; Van Kerkhove, M.D.; Donnelly, C.A.; Riley, S.; Rambaut, A.; Enouf, V.; van der Werf, S.; Ferguson, N.M. Middle East respiratory syndrome coronavirus: quantification of the extent of the epidemic, surveillance biases, and transmissibility. Lancet Infect. Dis. 2014, 14, 50–56. [Google Scholar] [CrossRef]
- Drosten, C.; Seilmaier, M.; Corman, V.M.; Hartmann, W.; Scheible, G.; Sack, S.; Guggemos, W.; Kallies, R.; Muth, D.; Junglen, S.; et al. Clinical features and virological analysis of a case of Middle East respiratory syndrome coronavirus infection. Lancet Infect. Dis. 2013, 13, 745–751. [Google Scholar] [CrossRef] [Green Version]
- De Benedictis, P.; Marciano, S.; Scaravelli, D.; Priori, P.; Zecchin, B.; Capua, I.; Monne, I.; Cattoli, G. Alpha and lineage C betaCoV infections in Italian bats. Virus Genes 2014, 48, 366–371. [Google Scholar] [CrossRef]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [PubMed]
- Annan, A.; Baldwin, H.J.; Corman, V.M.; Klose, S.M.; Owusu, M.; Nkrumah, E.E.; Badu, E.K.; Anti, P.; Agbenyega, O.; Meyer, B.; et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 2013, 19, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Falcon, A.; Vazquez-Moron, S.; Casas, I.; Aznar, C.; Ruiz, G.; Pozo, F.; Perez-Brena, P.; Juste, J.; Ibanez, C.; Garin, I.; et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch. Virol. 2011, 156, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelli, D.; Papetti, A.; Sabelli, C.; Rosti, E.; Moreno, A.; Boniotti, M.B. Detection of coronaviruses in bats of various species in Italy. Viruses 2013, 5, 2679–2689. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | Pairwise aa Sequence Identity with Ea-HedCoV HKU31 Strain F6 Sequence (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Coronavirus | Size (No. of Bases) | G+C Content | 3CLPro | RdRp | Hel | S | E | M | N |
| Merbecovirus | |||||||||
| Hp-BatCoV HKU25 | 30,497 | 0.42 | 79.4 | 90.0 | 92.5 | 57.1 | 76.8 | 78.4 | 67.2 |
| BtVs-BetaCoV/SC2013 | 30,423 | 0.43 | 78.4 | 90.7 | 92.6 | 57.3 | 79.3 | 79.8 | 68.9 |
| Human MERS-CoV ChinaGD01 | 30,114 | 0.41 | 77.8 | 89.4 | 91.3 | 57.5 | 73.2 | 79 | 66.7 |
| Human MERS-CoV England1 | 30,111 | 0.41 | 77.8 | 89.3 | 91.3 | 57.7 | 73.2 | 78.5 | 67.2 |
| Human MERS-CoV EMC/2012 | 30,119 | 0.41 | 77.8 | 89.4 | 91.1 | 57.7 | 73.2 | 79 | 67.2 |
| Camel MERS-CoV NRCE-HKU205 | 29,908 | 0.41 | 78.1 | 89.4 | 91.1 | 57.8 | 73.2 | 79 | 66.4 |
| Camel MERS-CoV Jeddah-1 | 29,851 | 0.41 | 77.8 | 89.4 | 91.3 | 57.7 | 73.2 | 79 | 67.2 |
| NeoCoV | 30,111 | 0.40 | 77.1 | 89.2 | 91.6 | 63.4 | 75.6 | 80.4 | 69.7 |
| Ty-BatCoV HKU4 | 30,286 | 0.38 | 71.2 | 87.6 | 90.8 | 57.5 | 64.6 | 77.6 | 63.8 |
| Pi-BatCoV HKU5 | 30,488 | 0.43 | 75.5 | 89.1 | 91 | 57.7 | 62.2 | 79.1 | 63.8 |
| BetaCoV/Erinaceus/VMC/DEU | 30,175 | 0.37 | 85.6 | 92.2 | 96.2 | 77.9 | 84.1 | 90.8 | 84.8 |
| Ea-HedCoV HKU31 RS13 | 29,951 | 0.38 | 100 | 100 | 100 | 99.9 | 100 | 100 | 96.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, S.K.P.; Luk, H.K.H.; Wong, A.C.P.; Fan, R.Y.Y.; Lam, C.S.F.; Li, K.S.M.; Ahmed, S.S.; Chow, F.W.N.; Cai, J.-P.; Zhu, X.; et al. Identification of a Novel Betacoronavirus (Merbecovirus) in Amur Hedgehogs from China. Viruses 2019, 11, 980. https://0-doi-org.brum.beds.ac.uk/10.3390/v11110980
Lau SKP, Luk HKH, Wong ACP, Fan RYY, Lam CSF, Li KSM, Ahmed SS, Chow FWN, Cai J-P, Zhu X, et al. Identification of a Novel Betacoronavirus (Merbecovirus) in Amur Hedgehogs from China. Viruses. 2019; 11(11):980. https://0-doi-org.brum.beds.ac.uk/10.3390/v11110980
Chicago/Turabian StyleLau, Susanna K. P., Hayes K. H. Luk, Antonio C. P. Wong, Rachel Y. Y. Fan, Carol S. F. Lam, Kenneth S. M. Li, Syed Shakeel Ahmed, Franklin W.N. Chow, Jian-Piao Cai, Xun Zhu, and et al. 2019. "Identification of a Novel Betacoronavirus (Merbecovirus) in Amur Hedgehogs from China" Viruses 11, no. 11: 980. https://0-doi-org.brum.beds.ac.uk/10.3390/v11110980