Activity of Selected Nucleoside Analogue ProTides against Zika Virus in Human Neural Stem Cells

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
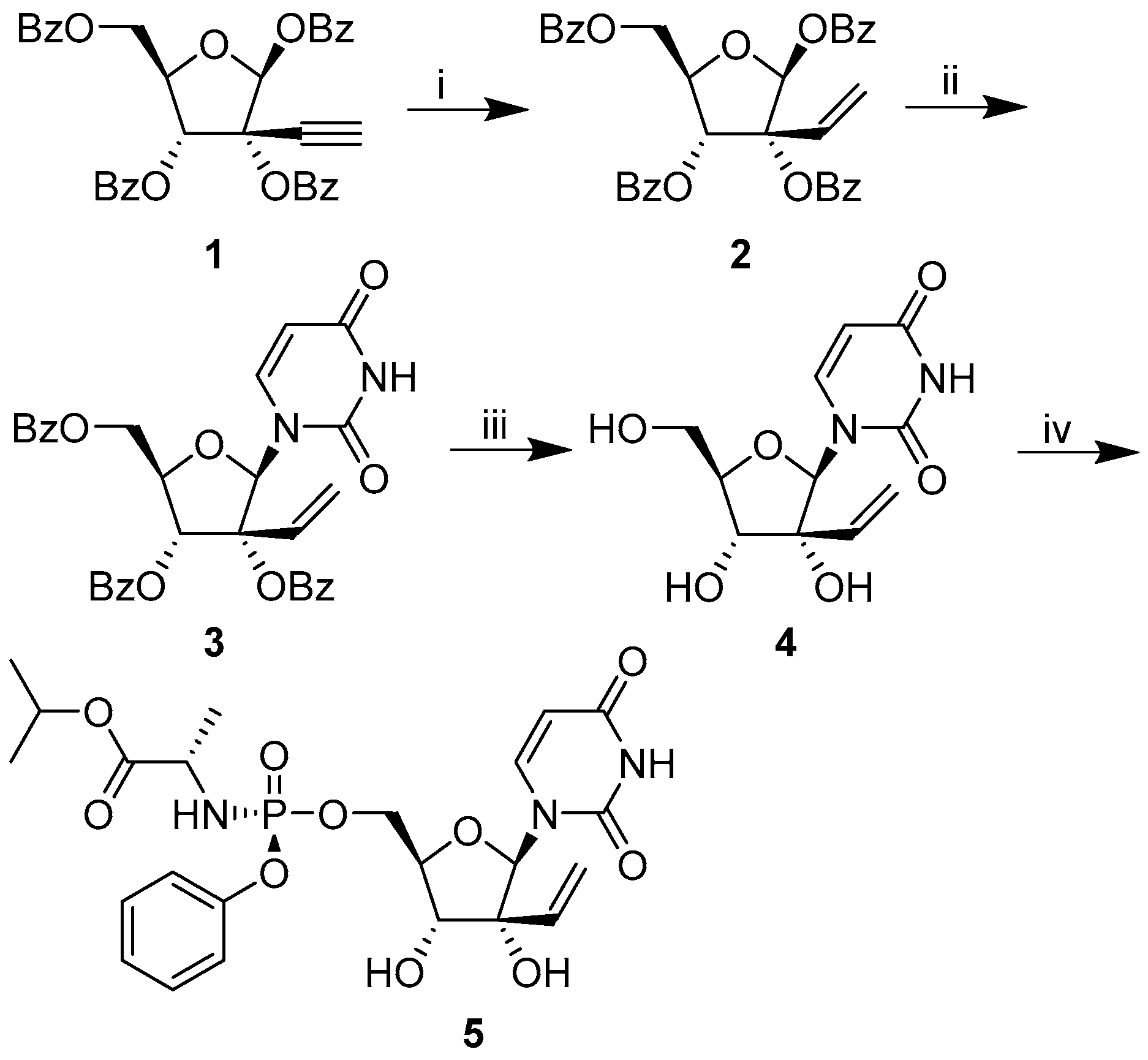
2.1. Chemistry
2.1.1. General Synthetic Experimental
2′-C-Ethynyluridine Aryloxy Phosphoramidate
5-((Benzoyloxy)methyl)-3-vinyltetrahydrofuran-2,3,4-triyl tribenzoate 2
5-((Benzoyloxy)Methyl)-2-(2,4-Dioxo-3,4-Hihydropyrimidin-1(2H)-yl)-3-Vinyltetrahydrofuran-3,4-diyl Benzoate 3
1-(3,4-dihydroxy-5-(hydromethyl)-3-vinyltetrahydrofuran2-yl)pyrimidine-2,4(1H,3H)-dione 4
isopropyl(((5-(2,4-dioxo-3,4-dihydropyridimin-1(2H)-yl)-3,4-dihydroxy-3-inyltetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)-L-alaninate 5
(5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxy-4-methyltetrahydrofuran-2-yl)methyl (2-(methylthio)ethyl) (2-(1H-indol-3-yl)ethyl)phosphoramidate (2′-C-methyluridine methylthioethyl tryptamine ProTide)
2.2. Cell Culture
2.3. Viruses
2.4. CellTiter-Glo Activity Assay
2.5. Bright-Field Microscopy
2.6. Plaque Assay
2.7. ZIKV RdRP Expression and Purification
2.8. Single Nucleotide Incorporation Assay
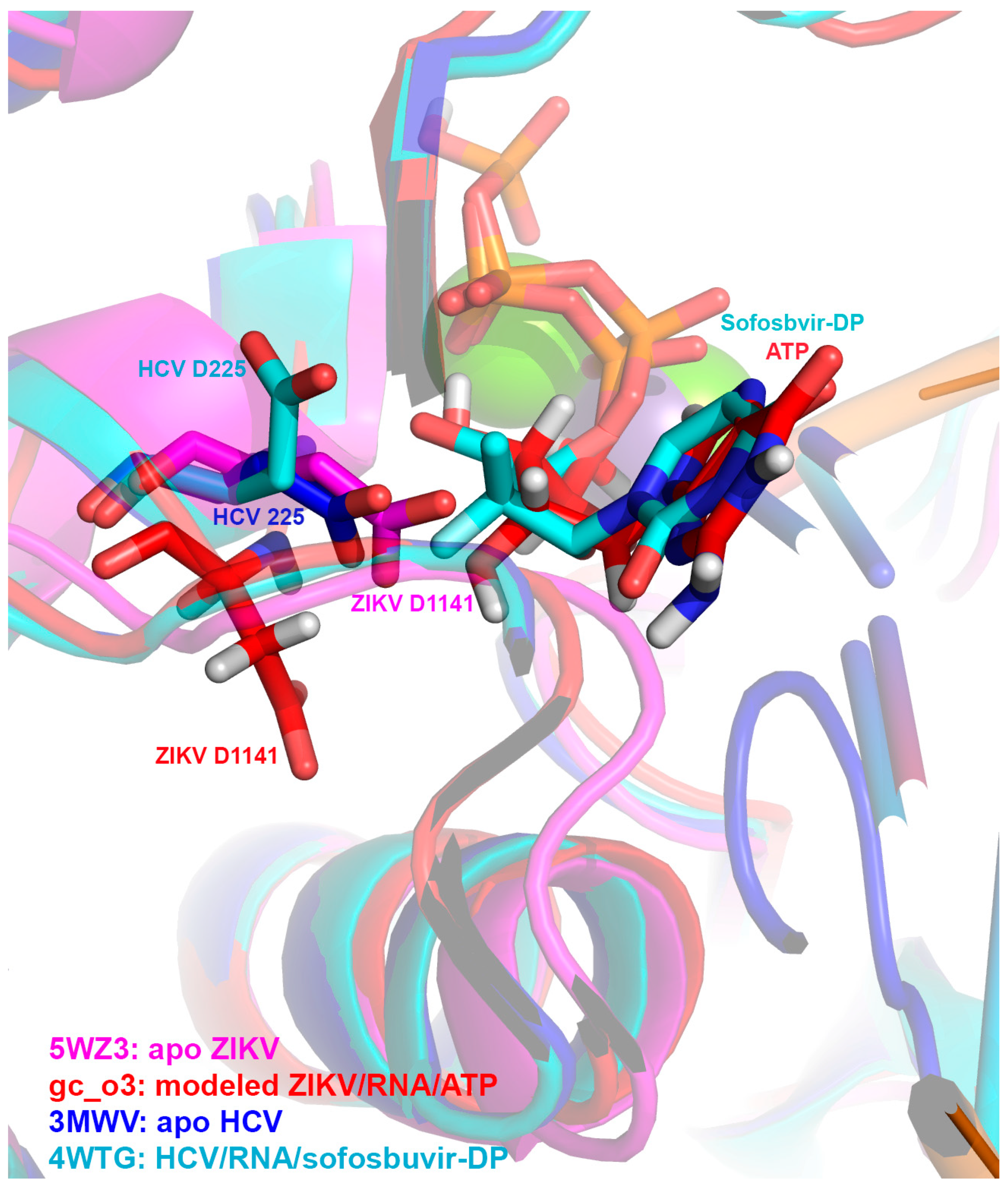
2.9. Structural Alignment
3. Results
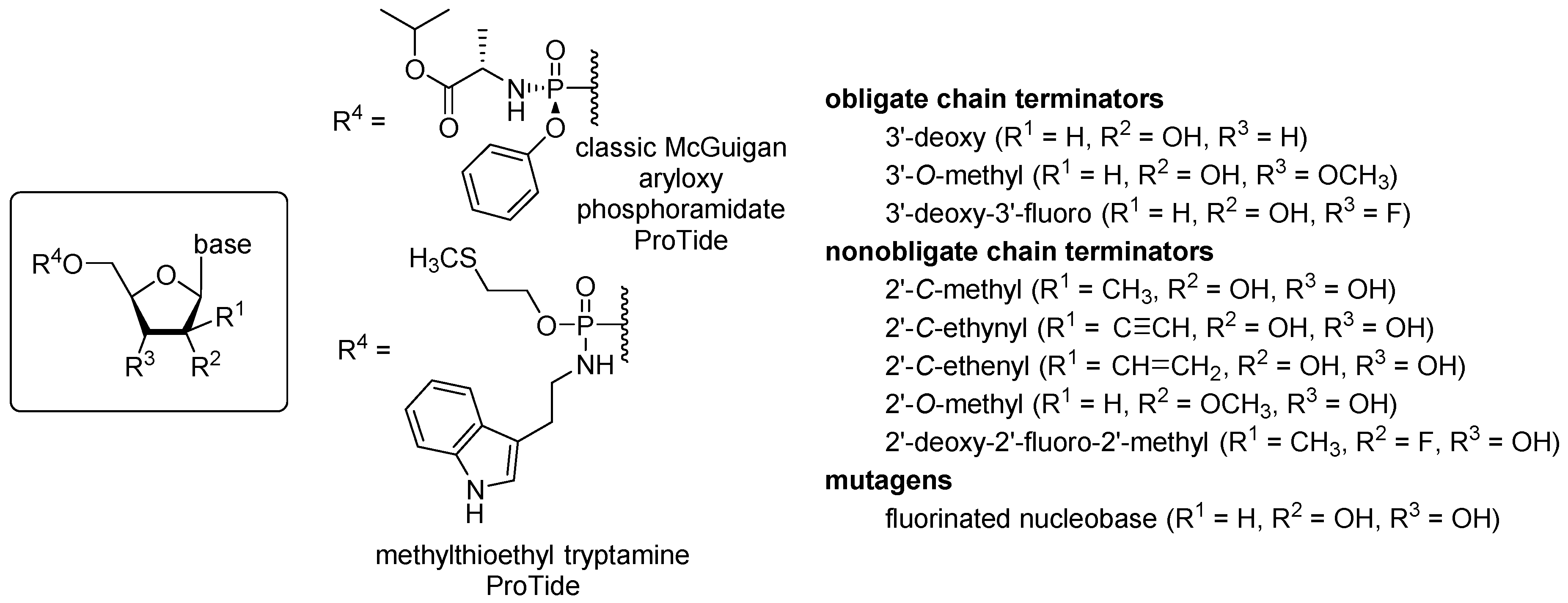
3.1. ProTide Library Design
3.2. Activity against ZIKV and Toxicity of Nucleoside Analogue ProTides in Human Neural Stem Cells
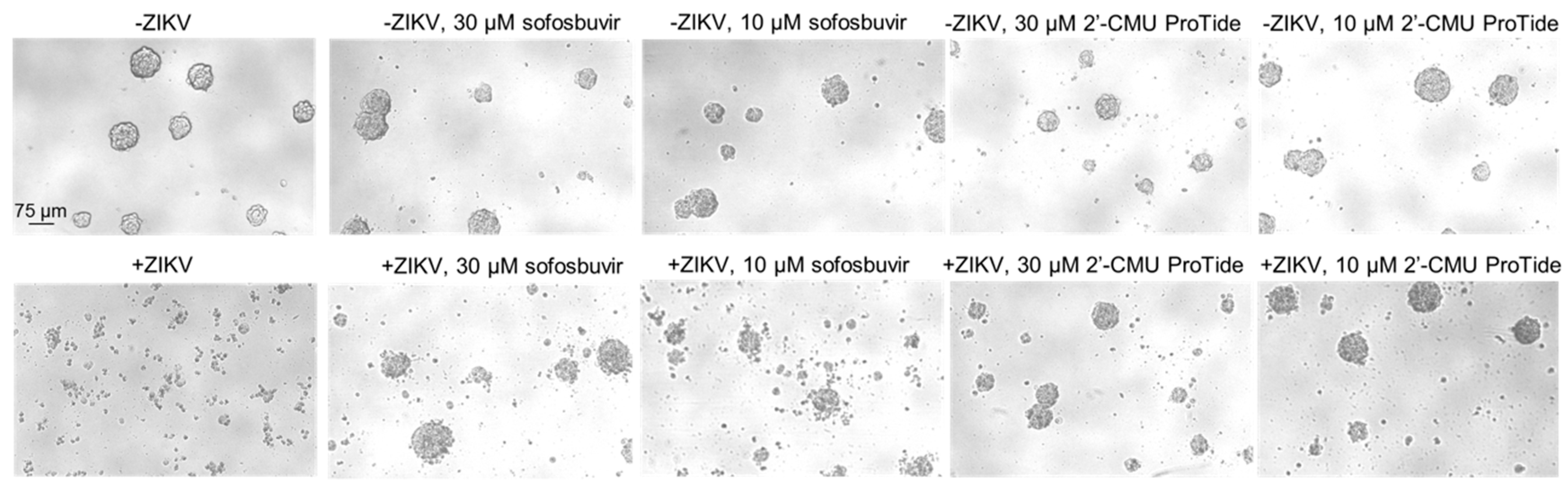
3.3. Protection from ZIKV-Induced Cytopathic Effect by 2′-C-methyluridine Aryloxyl Phosphoramidate ProTide and Sofosbuvir in a ZIKV-Sensitive Glioblastoma Stem Cell Line
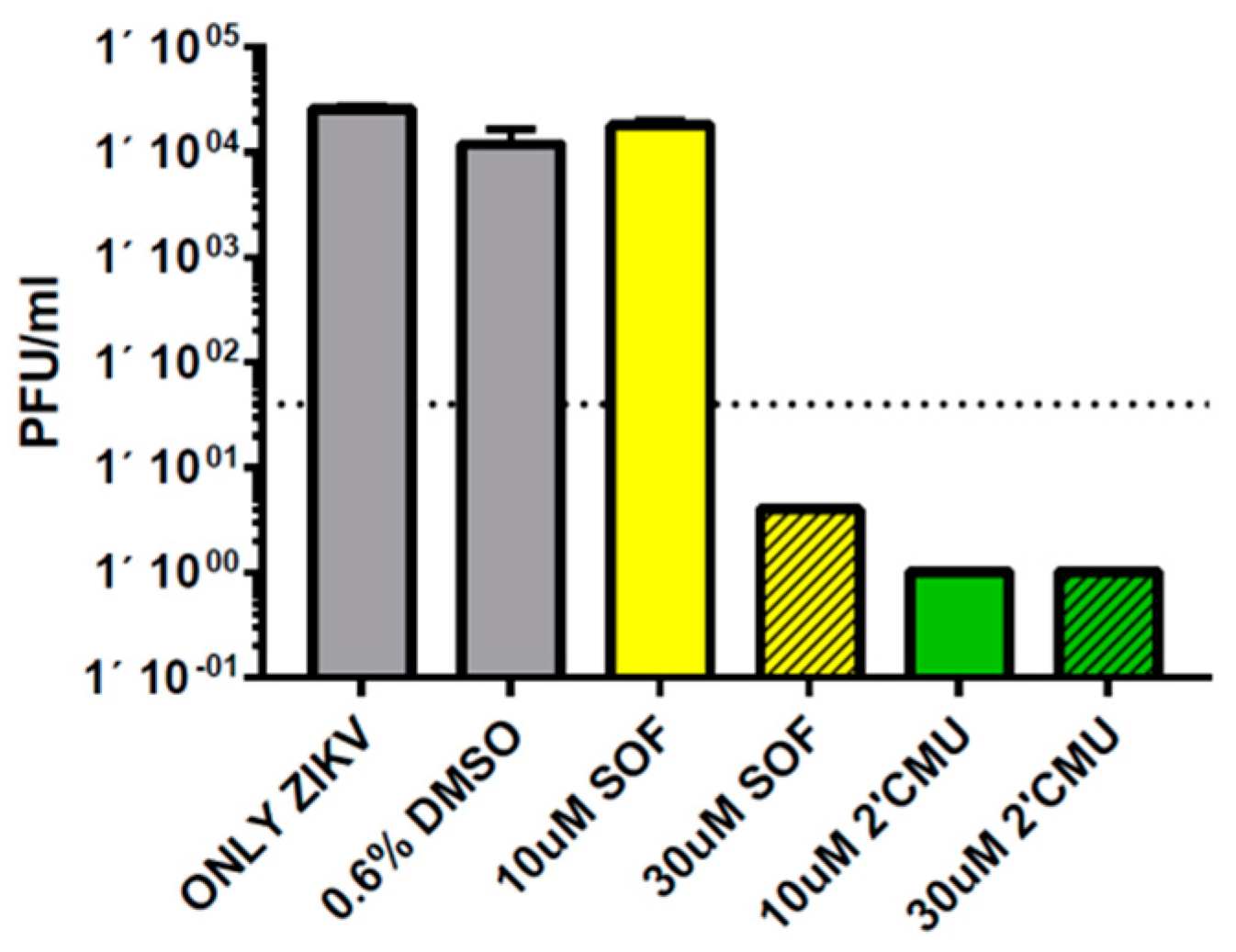
3.4. Repression of ZIKV Titers by 2′-C-methyluridine Aryloxyl Phosphoramidate ProTide and Sofosbuvir in Neural Stem Cells
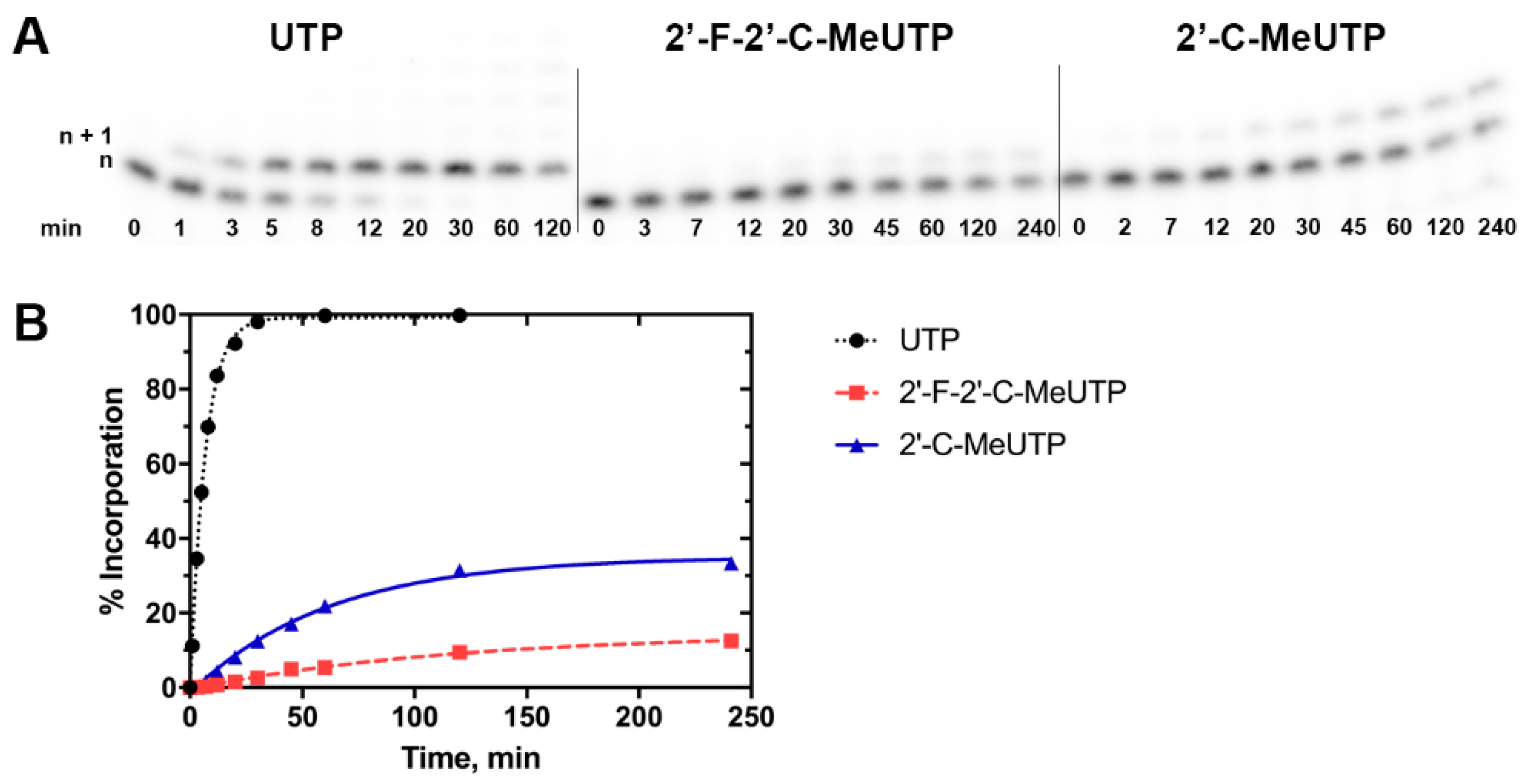
3.5. Enzymatic Incorporation of the Active Triphosphate Metabolites of 2′-C-methyluridine and Sofosbuvir Over Time Reveals Higher Levels of Incorporation for the Former Compound
3.6. Molecular Superpositioning of the ZIKV NS5 Active Site: Consequences for 2′-C-Derivatitized Nucleoside Analogues
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Araújo, T.V.B.; de Alencar Ximenes, R.A.; de Barros Miranda-Filho, D.; Souza, W.V.; Montarroyos, U.R.; de Melo, A.P.L.; Valongueiro, S.; Braga, C.; Brandão Filho, S.P.; Cordeiro, M.T.; et al. Association between microcephaly, Zika virus infection, and other risk factors in Brazil: Final report of a case-control study. Lancet Infect. Dis. 2018, 18, 328–336. [Google Scholar]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef]
- Ferrero, D.; Ferrer-Orta, C.; Verdaguer, N. Viral RNA-Dependent RNA Polymerases: A Structural Overview. Subcell. Biochem. 2018, 88, 39–71. [Google Scholar] [PubMed]
- Zarrouk, K.; Piret, J.; Boivin, G. Herpesvirus DNA polymerases: Structures, functions and inhibitors. Virus Res. 2017, 234, 177–192. [Google Scholar] [CrossRef]
- Menéndez-Arias, L.; Sebastián-Martín, A.; Álvarez, M. Viral reverse transcriptases. Virus Res. 2017, 234, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Mayberry, J.; Lee, W.M. The revolution in treatment of hepatitis C. Med. Clin. N. Am. 2019, 103, 43–55. [Google Scholar] [CrossRef]
- Sluis-Cremer, N.; Wainberg, M.A.; Schinazi, R.F. Resistance to reverse transcriptase inhibitors used in the treatment and prevention of HIV-1 infection. Future Microbiol. 2015, 10, 1773–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuigan, C.; Pathirana, R.N.; Mahmood, N.; Devine, K.G.; Hay, A.J. Aryl phosphate derivatives of AZT retain activity against HIV1 in cell lines which are resistant to the action of AZT. Antivir. Res. 1992, 17, 311–321. [Google Scholar] [CrossRef]
- Cahard, D.; McGuigan, C.; Balzarini, J. Aryloxy phosphoramidate triesters as pro-tides. Mini. Rev. Med. Chem. 2004, 4, 371–381. [Google Scholar] [CrossRef]
- Mehellou, Y.; Balzarini, J.; McGuigan, C. Aryloxy phosphoramidate triesters: A technology for delivering monophosphorylated nucleosides and sugars into cells. Chem. Med. Chem. 2009, 4, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Ovadia, R.; Khalil, A.; Li, H.; De Schutter, C.; Mengshetti, S.; Zhou, S.; Bassit, L.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis and anti-HCV activity of β-D -2′-deoxy-2′-α-chloro-2′-β-fluoro and β-d-2′-deoxy-2′-α-bromo-2′-β-fluoro nucleosides and their phosphoramidate prodrugs. Bioorg. Med. Chem. 2019, 27, 664–676. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Harris, S.A.; Daluge, S.M.; Gudmundsson, K.S.; McLean, E.W.; Burnette, T.C.; Marr, H.; Hazen, R.; Condreay, L.D.; Johnson, L.; et al. Application of phosphoramidate pronucleotide technology to abacavir leads to a significant enhancement of antiviral potency. J. Med. Chem. 2005, 48, 3504–3515. [Google Scholar] [CrossRef] [PubMed]
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Steuer, H.M.M.; Bao, D.; Chang, W.; Espiritu, C.; Bansal, S.; Lam, A.M.; et al. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285, 34337–34347. [Google Scholar] [CrossRef]
- Bullard-Feibelman, K.M.; Govero, J.; Zhu, Z.; Salazar, V.; Veselinovic, M.; Diamond, M.S.; Geiss, B.J. The FDA-approved drug sofosbuvir inhibits Zika virus infection. Antiviral Res. 2017, 137, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.C.; Zaverucha-do-Valle, C.; Reis, P.A.; Barbosa-Lima, G.; Vieira, Y.R.; Mattos, M.; de Paiva Silva, P.; Sacramento, C.; de Castro Faria Neto, H.C.; Campanati, L.; et al. Sofosbuvir protects Zika virus-infected mice from mortality, preventing short- and long-term sequelae. Sci. Rep. 2017, 7, 9409. [Google Scholar] [CrossRef] [Green Version]
- Mesci, P.; Macia, A.; Moore, S.M.; Shiryaev, S.A.; Pinto, A.; Huang, C.-T.; Tejwani, L.; Fernandes, I.R.; Suarez, N.A.; Kolar, M.J.; et al. Blocking Zika virus vertical transmission. Sci. Rep. 2018, 8, 1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumtaz, N.; Jimmerson, L.C.; Bushman, L.R.; Kiser, J.J.; Aron, G.; Reusken, C.B.E.M.; Koopmans, M.P.G.; van Kampen, J.J.A. Cell-line dependent antiviral activity of sofosbuvir against Zika virus. Antiviral Res. 2017, 146, 161–163. [Google Scholar] [CrossRef]
- Eyer, L.; Nencka, R.; Huvarová, I.; Palus, M.; Joao Alves, M.; Gould, E.A.; De Clercq, E.; Růžek, D. Nucleoside inhibitors of zika virus. J. Infect. Dis. 2016, 214, 707–711. [Google Scholar] [CrossRef]
- Lu, G.; Bluemling, G.R.; Collop, P.; Hager, M.; Kuiper, D.; Gurale, B.P.; Painter, G.R.; De La Rosa, A.; Kolykhalov, A.A. Analysis of Ribonucleotide 5′-Triphosphate Analogs as Potential Inhibitors of Zika Virus RNA-Dependent RNA Polymerase by Using Nonradioactive Polymerase Assays. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Hercík, K.; Kozak, J.; Šála, M.; Dejmek, M.; Hřebabecký, H.; Zborníková, E.; Smola, M.; Ruzek, D.; Nencka, R.; Boura, E. Adenosine triphosphate analogs can efficiently inhibit the Zika virus RNA-dependent RNA polymerase. Antiviral Res. 2017, 137, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.-Q.; Zhang, N.-N.; Li, C.-F.; Tian, M.; Hao, J.-N.; Xie, X.-P.; Shi, P.-Y.; Qin, C.-F. Adenosine analog NITD008 is a potent inhibitor of zika virus. Open Forum Infect. Dis. 2016, 3, ofw175. [Google Scholar] [CrossRef]
- Zmurko, J.; Marques, R.E.; Schols, D.; Verbeken, E.; Kaptein, S.J.F.; Neyts, J. The Viral Polymerase Inhibitor 7-Deaza-2′-C-Methyladenosine Is a Potent Inhibitor of In Vitro Zika Virus Replication and Delays Disease Progression in a Robust Mouse Infection Model. PLoS Negl. Trop. Dis. 2016, 10, e0004695. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, N.; Soma, R.; Hidano, S.; Watanabe, K.; Umekita, H.; Fukuda, C.; Noguchi, K.; Gendo, Y.; Ozaki, T.; Sonoda, A.; et al. Ribavirin inhibits Zika virus (ZIKV) replication in vitro and suppresses viremia in ZIKV-infected STAT1-deficient mice. Antiviral Res. 2017, 146, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, J.A.; Yang, Z.; Coste, M.; Li, J.; Beck, S.; Liu, Y.; Clark, A.E.; Zhu, Z.; Luna, L.A.; Sohl, C.D.; et al. Development and validation of a phenotypic high-content imaging assay for assessing the antiviral activity of small-molecule inhibitors targeting the Zika virus. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Ross, B.S.; Reddy, P.G.; Zhang, H.-R.; Rachakonda, S.; Sofia, M.J. Synthesis of diastereomerically pure nucleotide phosphoramidates. J. Org. Chem. 2011, 76, 8311–8319. [Google Scholar] [CrossRef]
- Okon, A.; Han, J.; Dawadi, S.; Demosthenous, C.; Aldrich, C.C.; Gupta, M.; Wagner, C.R. Anchimerically Activated ProTides as Inhibitors of Cap-Dependent Translation and Inducers of Chemosensitization in Mantle Cell Lymphoma. J. Med. Chem. 2017, 60, 8131–8144. [Google Scholar] [CrossRef] [PubMed]
- Anandan, S.K.; Aulakh, V.S.; Fenaux, M.; Lin, X.; Mao, L.; Saunders, O.; Sweeney, Z.K.; Yokokawa, F.; Zhong, W. 2′-Ethynyl Nucleoside Derivatives for Treatment of Viral Infections 2017. Available online: https://app.dimensions.ai/details/patent/US-9814739-B2 (accessed on 1 March 2019).
- Itoh, T.; Melik-Ohanjanian, R.G.; Ishikawa, I.; Kawahara, N.; Mizuno, Y.; Honma, Y.; Hozumi, M.; Ogura, H. Improved procedures for the syntheses of pyrido-and pyrrolo(2,3-d) pyrimidines, and ribosides thereof. Chem. Pharm. Bull. 1989, 37, 3184–3190. [Google Scholar] [CrossRef]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.J.B.; Ghosh, S.K.; Bevilacqua, P.C.; Cameron, C.E. Single-nucleotide resolution of RNA strands in the presence of their RNA complements. BioTechniques 1999, 27, 450–456. [Google Scholar]
- Potisopon, S.; Ferron, F.; Fattorini, V.; Selisko, B.; Canard, B. Substrate selectivity of Dengue and Zika virus NS5 polymerase towards 2′-modified nucleotide analogues. Antiviral Res. 2017, 140, 25–36. [Google Scholar] [CrossRef]
- Jin, Z.; Leveque, V.; Ma, H.; Johnson, K.A.; Klumpp, K. Assembly, purification, and pre-steady-state kinetic analysis of active RNA-dependent RNA polymerase elongation complex. J. Biol. Chem. 2012, 287, 10674–10683. [Google Scholar] [CrossRef] [PubMed]
- Kamkaew, M.; Chimnaronk, S. Characterization of soluble RNA-dependent RNA polymerase from dengue virus serotype 2: The polyhistidine tag compromises the polymerase activity. Protein Expr. Purif. 2015, 112, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Appleby, T.C.; Perry, J.K.; Murakami, E.; Barauskas, O.; Feng, J.; Cho, A.; Fox, D.; Wetmore, D.R.; McGrath, M.E.; Ray, A.S.; et al. Viral replication. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 2015, 347, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger PyMOL: The PyMOL Molecular Graphics System, Version 1.8, Schrödinger, LLC. Available online: http://www.sciepub.com/reference/159710 (accessed on 1 March 2019).
- Šebera, J.; Dubankova, A.; Sychrovský, V.; Ruzek, D.; Boura, E.; Nencka, R. The structural model of Zika virus RNA-dependent RNA polymerase in complex with RNA for rational design of novel nucleotide inhibitors. Sci. Rep. 2018, 8, 11132. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.K.; Cyr, M.; Longenecker, K.; Tripathi, R.; Sun, C.; Kempf, D.J. Crystal structure of full-length Zika virus NS5 protein reveals a conformation similar to Japanese encephalitis virus NS5. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 116–122. [Google Scholar] [CrossRef]
- Duan, W.; Song, H.; Wang, H.; Chai, Y.; Su, C.; Qi, J.; Shi, Y.; Gao, G.F. The crystal structure of Zika virus NS5 reveals conserved drug targets. EMBO J. 2017, 36, 919–933. [Google Scholar] [CrossRef] [Green Version]
- LaPlante, S.R.; Gillard, J.R.; Jakalian, A.; Aubry, N.; Coulombe, R.; Brochu, C.; Tsantrizos, Y.S.; Poirier, M.; Kukolj, G.; Beaulieu, P.L. Importance of ligand bioactive conformation in the discovery of potent indole-diamide inhibitors of the hepatitis C virus NS5B. J. Am. Chem. Soc. 2010, 132, 15204–15212. [Google Scholar] [CrossRef]
- De Clercq, E.; Neyts, J. Antiviral agents acting as DNA or RNA chain terminators. Handb. Exp. Pharmacol. 2009, 189, 53–84. [Google Scholar]
- Bonnac, L.F.; Mansky, L.M.; Patterson, S.E. Structure-activity relationships and design of viral mutagens and application to lethal mutagenesis. J. Med. Chem. 2013, 56, 9403–9414. [Google Scholar] [CrossRef]
- Sofia, M.J. Nucleotide prodrugs for the treatment of HCV infection. Adv. Pharmacol. 2013, 67, 39–73. [Google Scholar]
- Mehellou, Y.; Rattan, H.S.; Balzarini, J. The protide prodrug technology: From the concept to the clinic. J. Med. Chem. 2018, 61, 2211–2226. [Google Scholar] [CrossRef] [PubMed]
- Eldrup, A.B.; Allerson, C.R.; Bennett, C.F.; Bera, S.; Bhat, B.; Bhat, N.; Bosserman, M.R.; Brooks, J.; Burlein, C.; Carroll, S.S.; et al. Structure-activity relationship of purine ribonucleosides for inhibition of hepatitis C virus RNA-dependent RNA polymerase. J. Med. Chem. 2004, 47, 2283–2295. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lim, S.P.; Chen, Y.-L.; Hunziker, J.; Rao, R.; Gu, F.; Seh, C.C.; Ghafar, N.A.; Xu, H.; Chan, K.; et al. Structure-activity relationship of uridine-based nucleoside phosphoramidate prodrugs for inhibition of dengue virus RNA-dependent RNA polymerase. Bioorg. Med. Chem. Lett. 2018, 28, 2324–2327. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Kinkade, A.; Behera, I.; Chaudhuri, S.; Tucker, K.; Dyatkina, N.; Rajwanshi, V.K.; Wang, G.; Jekle, A.; Smith, D.B.; et al. Structure-activity relationship analysis of mitochondrial toxicity caused by antiviral ribonucleoside analogs. Antiviral Res. 2017, 143, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kaid, C.; Goulart, E.; Caires-Júnior, L.C.; Araujo, B.H.S.; Soares-Schanoski, A.; Bueno, H.M.S.; Telles-Silva, K.A.; Astray, R.M.; Assoni, A.F.; Júnior, A.F.R.; et al. Zika virus selectively kills aggressive human embryonal CNS tumor cells in vitro and in vivo. Cancer Res. 2018, 78, 3363–3374. [Google Scholar] [CrossRef]
- Mazar, J.; Li, Y.; Rosado, A.; Phelan, P.; Kedarinath, K.; Parks, G.D.; Alexander, K.A.; Westmoreland, T.J. Zika virus as an oncolytic treatment of human neuroblastoma cells requires CD24. PLoS ONE 2018, 13, e0200358. [Google Scholar] [CrossRef]
- Chen, Q.; Wu, J.; Ye, Q.; Ma, F.; Zhu, Q.; Wu, Y.; Shan, C.; Xie, X.; Li, D.; Zhan, X.; et al. Treatment of Human Glioblastoma with a Live Attenuated Zika Virus Vaccine Candidate. MBio 2018, 9. [Google Scholar] [CrossRef]
- Xu, H.-T.; Hassounah, S.A.; Colby-Germinario, S.P.; Oliveira, M.; Fogarty, C.; Quan, Y.; Han, Y.; Golubkov, O.; Ibanescu, I.; Brenner, B.; et al. Purification of Zika virus RNA-dependent RNA polymerase and its use to identify small-molecule Zika inhibitors. J. Antimicrob. Chemother. 2017, 72, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Ehteshami, M.; Zhou, L.; Amiralaei, S.; Shelton, J.R.; Cho, J.H.; Zhang, H.; Li, H.; Lu, X.; Ozturk, T.; Stanton, R.; et al. Nucleotide Substrate Specificity of Anti-Hepatitis C Virus Nucleoside Analogs for Human Mitochondrial RNA Polymerase. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleoside Analogue ProTide | EC50 PRVABC59 ZIKV, µM | EC50 H/PAN ZIKV, µM | CC50, µM | SI 1 PRVABC59 ZIKV | SI 1 H/PAN ZIKV |
|---|---|---|---|---|---|
| 2′-C-Methylcytidine aryloxyl phosphoramidate | 48 ± 1 | >50 | >50 | >1 | >1 |
| 3′-Deoxyadenosine aryloxyl phosphoramidate | >50 | >50 | >50 | >1 | >1 |
| 5′-Fluorocytidine aryloxyl phosphoramidate | >50 | >50 | 10 ± 1 | >0.2 | >0.2 |
| 3′-O-Methyluridine aryloxyl phosphoramidate | >50 | >50 | >50 | >1 | >1 |
| 2′-O-Methylcytidine aryloxyl phosphoramidate | >50 | >50 | >50 | >1 | >1 |
| 2′-C-methyluridinearyloxyl phosphoramidate | 1 ± 1 | 2 ± 1 | 47 ± 1 | >47 | >23 |
| 2′-C-Methyladenosine aryloxyl phosphoramidate | 10 ± 1 | 18 ± 1 | 42 ± 1 | >4 | >2 |
| 3′-Deoxycytidine aryloxyl phosphoramidate | >50 | >50 | >50 | >1 | >1 |
| 3′-Deoxy-3′-Fluoroguanosine aryloxyl phosphoramidate | >50 | >50 | >50 | >1 | >1 |
| 2′-C-Ethenyluridine aryloxyl phosphoramidate | >50 | >50 | 10 ± 1 | >0.1 | >0.1 |
| 2′-C-Ethynyluridine aryloxyl phosphoramidate | 0.3 ± 1 | 0.8 ± 1 | >50 | >166 | >62 |
| 2′-C-methyluridine 2-(methylthio)ethyl phosphoramidate | >50 | >50 | >50 | >1 | >1 |
| Sofosbuvir | 35 ± 1 | 46 ± 1 | >50 | >1.4 | >1.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernatchez, J.A.; Coste, M.; Beck, S.; Wells, G.A.; Luna, L.A.; Clark, A.E.; Zhu, Z.; Hecht, D.; Rich, J.N.; Sohl, C.D.; et al. Activity of Selected Nucleoside Analogue ProTides against Zika Virus in Human Neural Stem Cells. Viruses 2019, 11, 365. https://0-doi-org.brum.beds.ac.uk/10.3390/v11040365
Bernatchez JA, Coste M, Beck S, Wells GA, Luna LA, Clark AE, Zhu Z, Hecht D, Rich JN, Sohl CD, et al. Activity of Selected Nucleoside Analogue ProTides against Zika Virus in Human Neural Stem Cells. Viruses. 2019; 11(4):365. https://0-doi-org.brum.beds.ac.uk/10.3390/v11040365
Chicago/Turabian StyleBernatchez, Jean A., Michael Coste, Sungjun Beck, Grace A. Wells, Lucas A. Luna, Alex E. Clark, Zhe Zhu, David Hecht, Jeremy N. Rich, Christal D. Sohl, and et al. 2019. "Activity of Selected Nucleoside Analogue ProTides against Zika Virus in Human Neural Stem Cells" Viruses 11, no. 4: 365. https://0-doi-org.brum.beds.ac.uk/10.3390/v11040365