Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, α- and β-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display


Abstract
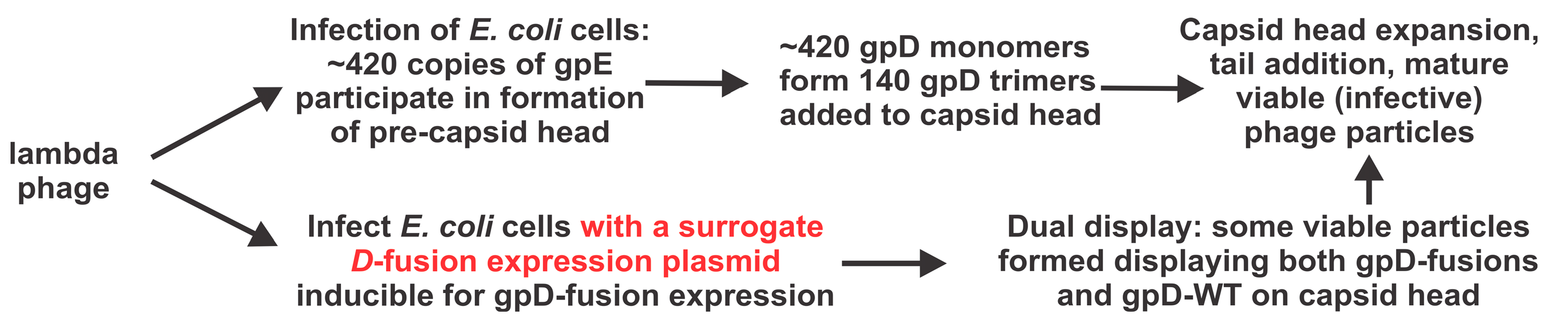
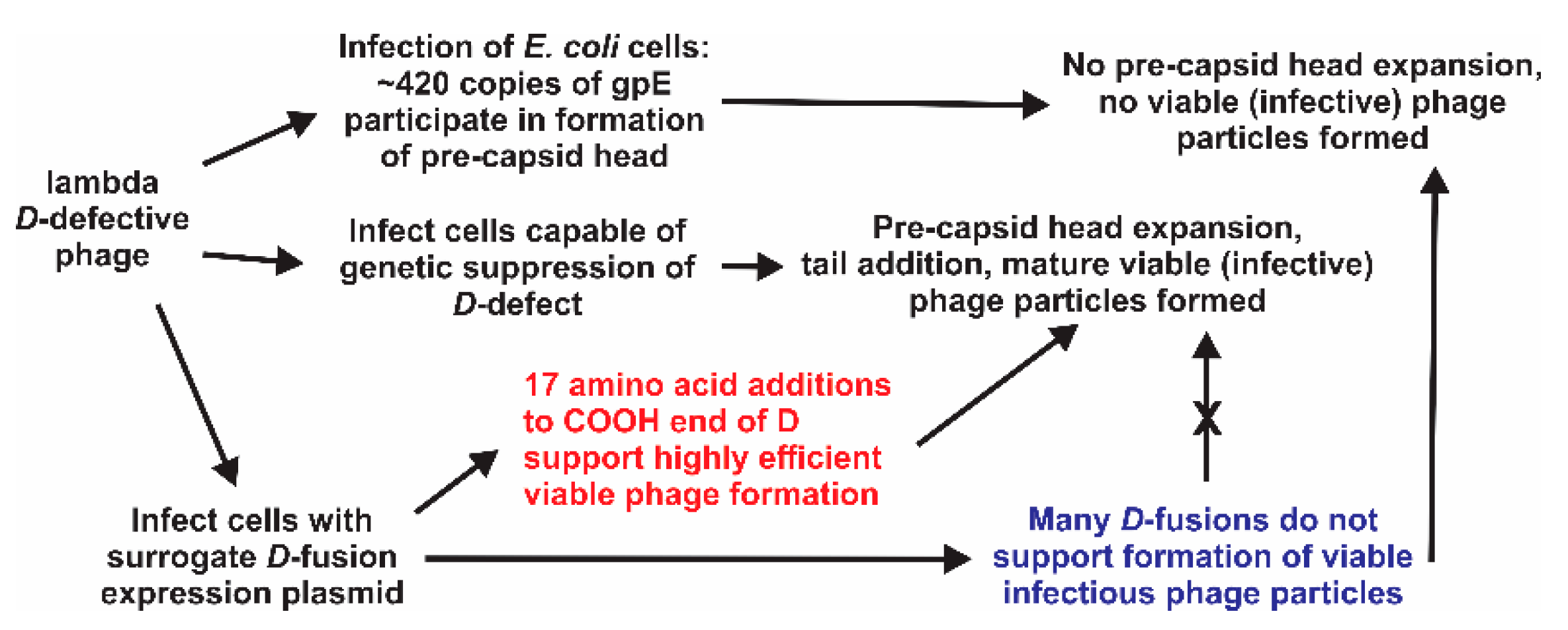
:1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Growth Medium and General Dilution “Buffer”
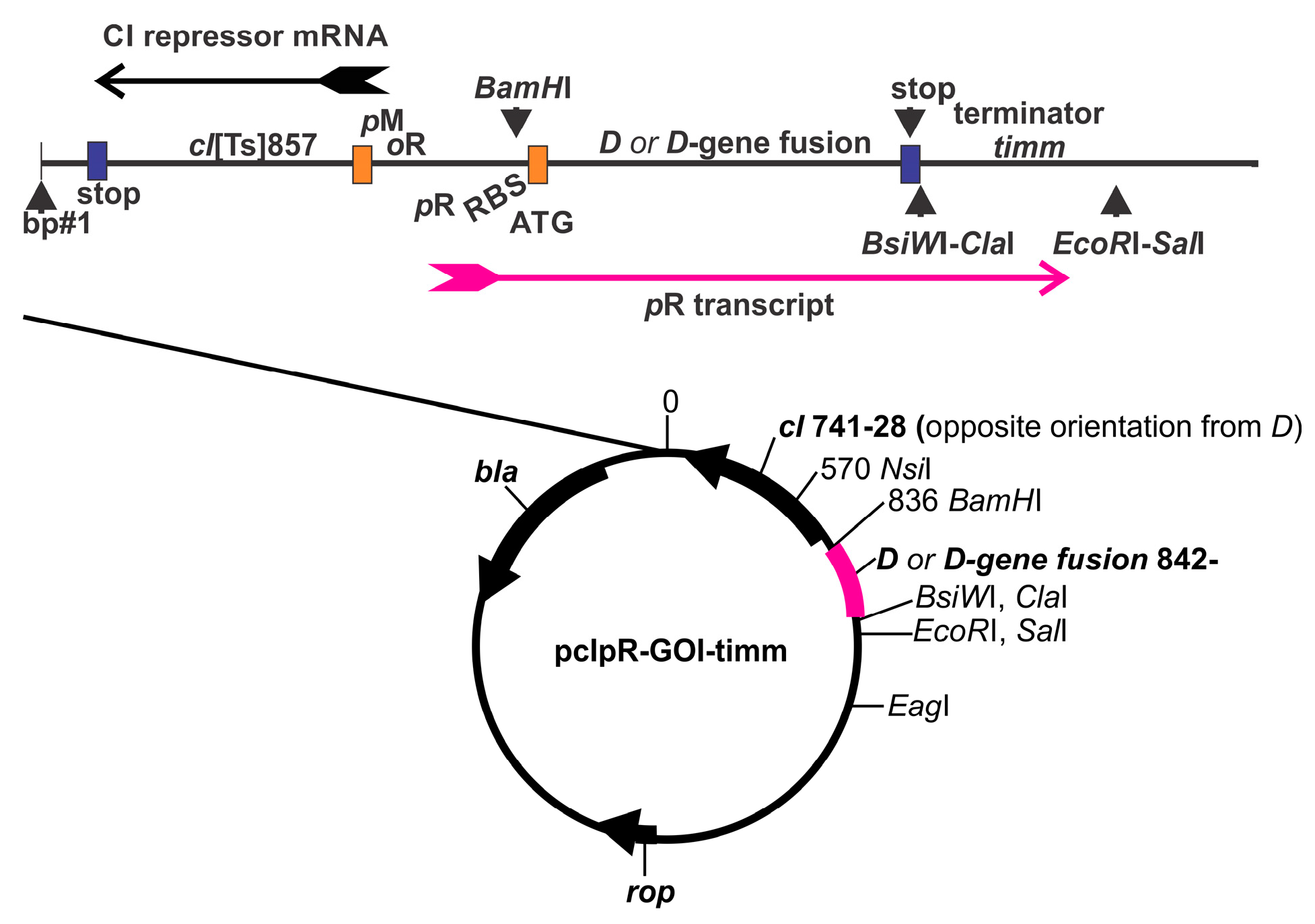
2.3. Plasmid Construction; D-Fusion Plasmids and Encoded Amino Acids
2.4. Single-Burst LDP-Vaccine and SEV Production
2.5. Infection-Complementation Using Transformed Cells
2.6. Complementation Methodology
2.7. Complementation Versus Reversion, Versus Marker rescue
2.7.1. D-Amber Reversion
2.7.2. D-Amber Marker Rescue
2.7.3. Complementation Temperature
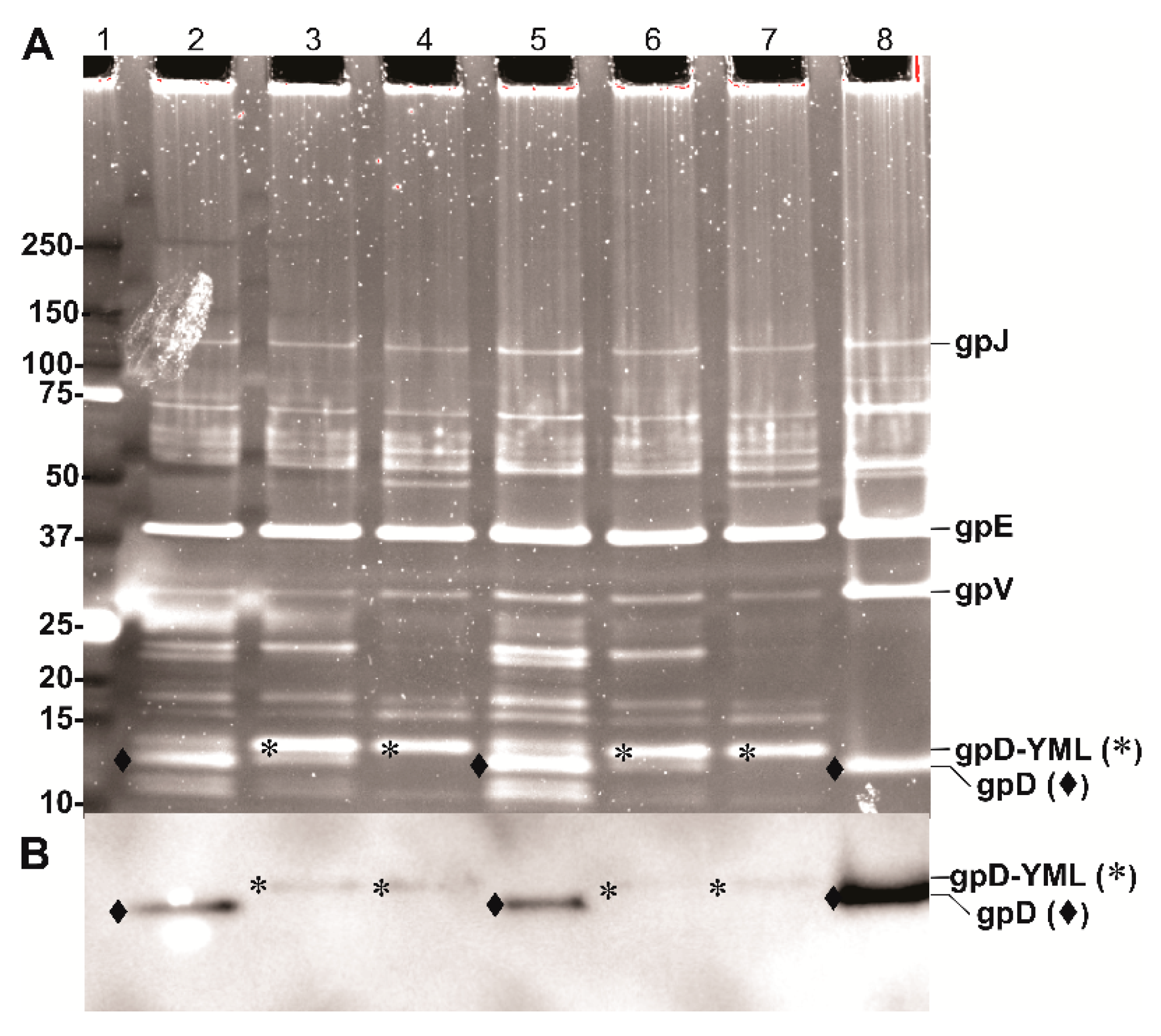
2.8. Protein Gels and Western Blots
3. Results and Discussion
3.1. Temperature Dependent Gene Expression from Plasmid pcIpR-GOI-Timm
3.2. Evaluating D-Fusion Cathelicidins for Cellular Toxicity
3.3. Temperature-Dependent Complementation for Phage Growth in D-Defective Phage mutant by Plasmids Expressing gpD or gpD-Fusions
3.4. Evaluating the Influence of D-fusion Size and Toxicity on LDP Production
4. Further Considerations and Summary
Supplementary Materials
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Casjens, S.R.; Hendrix, R.W. Locations and amounts of major structural proteins in bacteriophage lambda. J. Mol. Biol. 1974, 88, 535–545. [Google Scholar] [CrossRef]
- Imber, R.; Tsugita, A.; Wurtz, M.; Hohn, T. Outer surface protein of bacteriophage lambda. J. Mol. Biol. 1980, 139, 277–295. [Google Scholar] [CrossRef]
- Georgopoulos, C.; Tilly, K.; Casjens, S. Lambdoid phage head assembly. In Lambda ii; Hendrix, R.W., Roberts, J.R., Stahl, F.S., Weisberg, R.A., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1983; pp. 279–304. [Google Scholar]
- Sternberg, N.; Hoess, R.H. Display of peptides and proteins on the surface of bacteriophage lambda. Proc. Natl. Acad. Sci. USA 1995, 92, 1609–1613. [Google Scholar] [CrossRef]
- Sternberg, N.; Weisberg, R. Packaging of coliphage lambda DNA. Ii. The role of the gene d protein. J. Mol. Biol. 1977, 117, 733–759. [Google Scholar] [CrossRef]
- Ansuini, H.; Cicchini, C.; Nicosia, A.; Tripodi, M.; Cortese, R.; Luzzago, A. Biotin-tagged cdna expression libraries displayed on lambda phage: A new tool for the selection of natural protein ligands. Nucleic Acids Res. 2002, 30, e78. [Google Scholar] [CrossRef]
- Cicchini, C.; Ansuini, H.; Amicone, L.; Alonzi, T.; Nicosia, A.; Cortese, R.; Tripodi, M.; Luzzago, A. Searching for DNA-protein interactions by lambda phage display. J. Mol. Biol. 2002, 322, 697–706. [Google Scholar] [CrossRef]
- Cortese, R.; Monaci, P.; Luzzago, A.; Santini, C.; Bartoli, F.; Cortese, I.; Fortugno, P.; Galfre, G.; Nicosia, A.; Felici, F. Selection of biologically active peptides by phage display of random peptide libraries. Curr. Opin. Biotechnol. 1996, 7, 616–621. [Google Scholar] [CrossRef]
- Kong, B.; Ma, W.J. Display of aggregation-prone ligand binding domain of human ppar gamma on surface of bacteriophage lambda. Acta Pharmacol. Sin. 2006, 27, 91–99. [Google Scholar] [CrossRef]
- Maruyama, I.N.; Maruyama, H.I.; Brenner, S. Lambda foo: A lambda phage vector for the expression of foreign proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 8273–8277. [Google Scholar] [CrossRef]
- Mikawa, Y.G.; Maruyama, I.N.; Brenner, S. Surface display of proteins on bacteriophage lambda heads. J. Mol. Biol. 1996, 262, 21–30. [Google Scholar] [CrossRef]
- Niwa, M.; Maruyama, H.; Fujimoto, T.; Dohi, K.; Maruyama, I.N. Affinity selection of cdna libraries by lambda phage surface display. Gene 2000, 256, 229–236. [Google Scholar] [CrossRef]
- Santi, E.; Capone, S.; Mennuni, C.; Lahm, A.; Tramontano, A.; Luzzago, A.; Nicosia, A. Bacteriophage lambda display of complex cdna libraries: A new approach to functional genomics. J. Mol. Biol 2000, 296, 497–508. [Google Scholar] [CrossRef]
- Santini, C.; Brennan, D.; Mennuni, C.; Hoess, R.H.; Nicosia, A.; Cortese, R.; Luzzago, A. Efficient display of an hcv cdna expression library as c-terminal fusion to the capsid protein d of bacteriophage lambda. J. Mol. Biol. 1998, 282, 125–135. [Google Scholar] [CrossRef]
- Vilchez, S.; Jacoby, J.; Ellar, D.J. Display of biologically functional insecticidal toxin on the surface of lambda phage. Appl. Environ. Microbiol. 2004, 70, 6587–6594. [Google Scholar] [CrossRef]
- Zucconi, A.; Dente, L.; Santonico, E.; Castagnoli, L.; Cesareni, G. Selection of ligands by panning of domain libraries displayed on phage lambda reveals new potential partners of synaptojanin 1. J. Mol. Biol. 2001, 307, 1329–1339. [Google Scholar] [CrossRef]
- Gupta, A.; Onda, M.; Pastan, I.; Adhya, S.; Chaudhary, V.K. High-density functional display of proteins on bacteriophage lambda. J. Mol. Biol. 2003, 334, 241–254. [Google Scholar] [CrossRef]
- Zanghi, C.N.; Lankes, H.A.; Bradel-Tretheway, B.; Wegman, J.; Dewhurst, S. A simple method for displaying recalcitrant proteins on the surface of bacteriophage lambda. Nucleic Acids Res. 2005, 33, e160. [Google Scholar] [CrossRef]
- Zanghi, C.N.; Sapinoro, R.; Bradel-Tretheway, B.; Dewhurst, S. A tractable method for simultaneous modifications to the head and tail of bacteriophage lambda and its application to enhancing phage-mediated gene delivery. Nucleic Acids Res. 2007, 35, e59. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Liu, Z. Phage display an its application in vaccine design. Ann. Microbiol. 2010, 60, 13–19. [Google Scholar] [CrossRef]
- Ren, Z.J.; Lewis, G.K.; Wingfield, P.T.; Locke, E.G.; Steven, A.C.; Black, L.W. Phage display of intact domains at high copy number: A system based on soc, the small outer capsid protein of bacteriophage t4. Protein Sci. 1996, 5, 1833–1843. [Google Scholar] [CrossRef]
- Ren, Z.J.; Tian, C.J.; Zhu, Q.S.; Zhao, M.Y.; Xin, A.G.; Nie, W.X.; Ling, S.R.; Zhu, M.W.; Wu, J.Y.; Lan, H.Y.; et al. Orally delivered foot-and-mouth disease virus capsid protomer vaccine displayed on t4 bacteriophage surface: 100% protection from potency challenge in mice. Vaccine 2008, 26, 1471–1481. [Google Scholar] [CrossRef]
- Sathaliyawala, T.; Rao, M.; Maclean, D.M.; Birx, D.L.; Alving, C.R.; Rao, V.B. Assembly of human immunodeficiency virus (hiv) antigens on bacteriophage t4: A novel in vitro approach to construct multicomponent hiv vaccines. J. Virol. 2006, 80, 7688–7698. [Google Scholar] [CrossRef]
- Shivachandra, S.B.; Rao, M.; Janosi, L.; Sathaliyawala, T.; Matyas, G.R.; Alving, C.R.; Leppla, S.H.; Rao, V.B. In vitro binding of anthrax protective antigen on bacteriophage t4 capsid surface through hoc-capsid interactions: A strategy for efficient display of large full-length proteins. Virology 2006, 345, 190–198. [Google Scholar] [CrossRef]
- Wu, J.; Tu, C.; Yu, X.; Zhang, M.; Zhang, N.; Zhao, M.; Nie, W.; Ren, Z. Bacteriophage t4 nanoparticle capsid surface soc and hoc bipartite display with enhanced classical swine fever virus immunogenicity: A powerful immunological approach. J. Virol. Methods 2007, 139, 50–60. [Google Scholar] [CrossRef]
- Hayes, S.; Gamage, L.N.; Hayes, C. Dual expression system for assembling phage lambda display particle (ldp) vaccine to porcine circovirus 2 (pcv2). Vaccine 2010, 28, 6789–6799. [Google Scholar] [CrossRef]
- Gamage, L.N.; Ellis, J.; Hayes, S. Immunogenicity of bacteriophage lambda particles displaying porcine circovirus 2 (pcv2) capsid protein epitopes. Vaccine 2009, 27, 6595–6604. [Google Scholar] [CrossRef]
- Gonzalez-Cano, P.; Gamage, L.N.A.; Marciniuk, K.; Hayes, C.; Napper, S.; Hayes, S.; Griebel, P.J. Lambda display phage as a mucosal vaccine delivery vehicle for peptide antigens. Vaccine 2017, 35, 7256–7263. [Google Scholar] [CrossRef]
- Hayes, S.; Erker, C.; Horbay, M.A.; Marciniuk, K.; Wang, W.; Hayes, C. Phage lambda p protein: Trans-activation, inhibition phenotypes and their suppression. Viruses 2013, 5, 619–653. [Google Scholar] [CrossRef]
- Hayes, S.; Rajamanickam, K.; Hayes, C. Complementation studies of bacteriophage lambda o amber mutants by allelic forms of o expressed from plasmid, and o-p interaction phenotypes. Antibiotics 2018, 7, 31. [Google Scholar] [CrossRef]
- Rajamanickam, K.; Hayes, S. The bacteriophage lambda cii phenotypes for complementation, cellular toxicity and replication inhibition are suppressed in cii-oop constructs expressing the small rna oop. Viruses 2018, 10, 115. [Google Scholar] [CrossRef]
- Braff, M.H.; Hawkins, M.A.; Di Nardo, A.; Lopez-Garcia, B.; Howell, M.D.; Wong, C.; Lin, K.; Streib, J.E.; Dorschner, R.; Leung, D.Y.; et al. Structure-function relationships among human cathelicidin peptides: Dissociation of antimicrobial properties from host immunostimulatory activities. J. Immunol. 2005, 174, 4271–4278. [Google Scholar] [CrossRef]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A comprehensive summary of ll-37, the factotum human cathelicidin peptide. Cell Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef]
- Yang, D.; Chertov, O.; Oppenheim, J.J. Participation of mammalian defensins and cathelicidins in anti-microbial immunity: Receptors and activities of human defensins and cathelicidin (ll-37). J. Leukoc. Biol. 2001, 69, 691–697. [Google Scholar]
- Zanetti, M. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 2004, 75, 39–48. [Google Scholar] [CrossRef]
- Sang, Y.; Blecha, F. Porcine host defense peptides: Expanding repertoire and functions. Dev. Comp. Immunol. 2009, 33, 334–343. [Google Scholar] [CrossRef]
- Selsted, M.E.; Harwig, S.S.; Ganz, T.; Schilling, J.W.; Lehrer, R.I. Primary structures of three human neutrophil defensins. J. Clin. Investig. 1985, 76, 1436–1439. [Google Scholar] [CrossRef]
- Schneider, J.J.; Unholzer, A.; Schaller, M.; Schafer-Korting, M.; Korting, H.C. Human defensins. J. Mol. Med. 2005, 83, 587–595. [Google Scholar] [CrossRef]
- Tollner, T.L.; Bevins, C.L.; Cherr, G.N. Multifunctional glycoprotein defb126—A curious story of defensin-clad spermatozoa. Nat. Rev. Urol. 2012, 9, 365–375. [Google Scholar] [CrossRef]
- Tollner, T.L.; Venners, S.A.; Hollox, E.J.; Yudin, A.I.; Liu, X.; Tang, G.; Xing, H.; Kays, R.J.; Lau, T.; Overstreet, J.W.; et al. A common mutation in the defensin defb126 causes impaired sperm function and subfertility. Sci. Transl. Med. 2011, 3, 92ra65. [Google Scholar] [CrossRef]
- Bessette, P.H.; Aslund, F.; Beckwith, J.; Georgiou, G. Efficient folding of proteins with multiple disulfide bonds in the escherichia coli cytoplasm. Proc. Natl. Acad. Sci. USA 1999, 96, 13703–13708. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Barton, A.; Daher, K.A.; Harwig, S.S.; Ganz, T.; Selsted, M.E. Interaction of human defensins with escherichia coli. Mechanism of bactericidal activity. J. Clin. Investig. 1989, 84, 553–561. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Wu, Z.; Nuding, S.; Groscurth, S.; Marcinowski, M.; Beisner, J.; Buchner, J.; Schaller, M.; Stange, E.F.; Wehkamp, J. Reduction of disulphide bonds unmasks potent antimicrobial activity of human beta-defensin 1. Nature 2011, 469, 419–423. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Duda, R.L. Bacteriophage lambda papa: Not the mother of all lambda phages. Science 1992, 258, 1145–1148. [Google Scholar] [CrossRef]
- Lemon, D.J.; Kay, M.K.; Titus, J.K.; Ford, A.A.; Chen, W.; Hamlin, N.J.; Hwang, Y.Y. Construction of a genetically modified T7 select phage system to express the antimicrobial peptide 1018. J. Microbiol. 2019, 57, 532–538. [Google Scholar] [CrossRef]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef]
- Knappe, D.; Henklein, P.; Hoffmann, R.; Hilpert, K. Easy strategy to protect antimicrobial peptides from fast degradation in serum. Antimicrob. Agents Chemother. 2010, 54, 4003–4005. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D-Fusion Name (Plasmid Number) | |
|---|---|
| COOH-terminal additions: a | NH2-terminal additions: a |
| His-TAGZ-Dcoe-TEV-LL37 (p619) b | LL37-TEV-Dcoe-His (p620) |
| Dcoe-LL37 (p627) c | LL37-Dcoe (p617) c |
| Dcoe-PR39 (p625) c | PR39-Dcoe (p623) c |
| Dcoe-DEFB126ΔC (p618) d | DEFB126ΔC-Dcoe (p622) d |
| Dcoe-HBD3 (p616) | HBD3-Dcoe (p624) |
| Dcoe-HD5 (p628) | HD5-Dcoe (p621) |
| Dcoe-HNP1 (p615) [see text] | HNP1-Dcoe (p626) [see text] |
| Dcoe-YML (p674) | YML-Dcoe (p676) |
| D-YML (p675) | His-Dcoe (p614) |
| Dcoe-5EV2 (p521,p629) e | His-Dcoe- (p614*) f |
| Plasmid In E. coli, Encoded D or D-Fusion Protein | Non-Induces (0 Time) | Non-Induced, 140 min Cell Growth at 30 °C | Induced, 140 min Cell Growth after Shifting Culture to 42 °C |
|---|---|---|---|
| p613, gpDcoe | 0.10 | 0.44 | 0.79 |
| p627, gpD-PR39 | 0.14 | 0.51 | 0.70 |
| p625, gpD-LL37 | 0.15 | 0.71 | 0.70 |
| Strain 594[pcIpR-D-fusion-timm] Plasmid Construct a | Relative Culture Viability at 42 °C (D-fusion Toxicity) b | EOP c of λimm434 Dam123 at 42 °C on Assay Strain d |
|---|---|---|
| p613: Dcoe | 0.65 | 0.56 |
| p613*: Dcoe- (67 bp deletion within D) e | 0.79 | <0.0001 |
| p614: His-Dcoe (11AA N-terminal) | 0.09 | <0.0001 f |
| p614*: His- Dcoe-:*(Pro48Glu mutation in D) | 0.87 | <0.0001 |
| p615: Dcoe-HNP1 (35AA C-term.) | 0.83 | <0.0001 f |
| p626: HNP1- Dcoe (36AA N-term.) | 1.00 | <0.0001 f |
| p616: Dcoe-HBD3 (50AA C-term.) | <0.0001 | <0.0005 |
| p624: HBD3- Dcoe (53AA N-term.) | 0.78 | <0.0001 f |
| p627: Dcoe-LL37 (45AA C-term.) | 0.0002 | g |
| p617: LL37-Dcoe (46AA N-term.) | 0.93 | <0.0001 f |
| p618: Dcoe-DEFB126-Δ (84AA C-term.) | 0.0001 | g |
| p622: DEFB126-ΔC- Dcoe (85AA N-term.) | 0.85 | <0.0001 f |
| p619: His-TAGZ-Dcoe-TEV-LL37 (11AA N-, 55AA C-term.) | 0.0003 | <0.000 1f |
| p620: LL37-TEV- Dcoe-His (50AA N-, 12AA C-term.) | 0.87 | <0.0001 f |
| p628: Dcoe-HD5 (40AA C-term.) | 0.0034 | <0.0001 f |
| p621: HD5- Dcoe (41AA N-term.) | 0.77 | <0.0001 f |
| p625: Dcoe-PR39 (48AA C-term.) | <0.0001 | g |
| p623: PR39-Dcoe (49AA N-term.) | 0.97 | <0.0001 f |
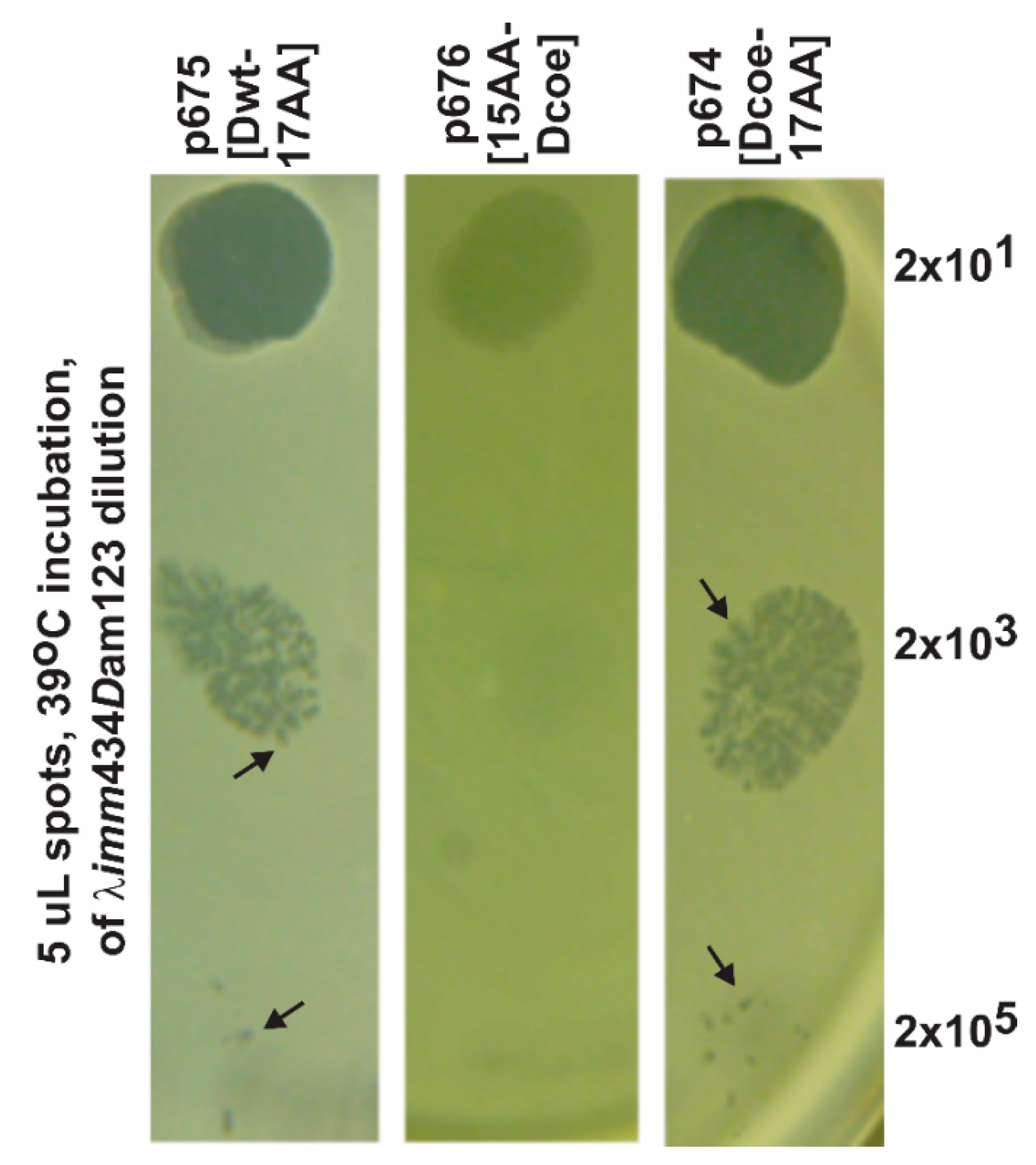
| p674: Dcoe-YML (17AA C-term.) | 0.05 | ≥0.90 h |
| p675: D(wild type)-YML (17AA C-term.) | 0.88 | 0.93 |
| p676: YML-Dcoe (15AA N-term.) | 1.00 | 0.06 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayes, S. Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, α- and β-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display. Viruses 2019, 11, 869. https://0-doi-org.brum.beds.ac.uk/10.3390/v11090869
Hayes S. Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, α- and β-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display. Viruses. 2019; 11(9):869. https://0-doi-org.brum.beds.ac.uk/10.3390/v11090869
Chicago/Turabian StyleHayes, Sidney. 2019. "Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, α- and β-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display" Viruses 11, no. 9: 869. https://0-doi-org.brum.beds.ac.uk/10.3390/v11090869