Virus-Like Particles and Nanoparticles for Vaccine Development against HCMV


1
Faculty of Biomedical Sciences, Institute for Research in Biomedicine, Università della Svizzera Italiana, 6500 Bellinzona, Switzerland
2
Institute of Microbiology, ETH Zürich, 8093 Zürich, Switzerland
3
European Virus Bioinformatics Center, 07743 Jena, Germany
*
Author to whom correspondence should be addressed.
Viruses 2020, 12(1), 35; https://0-doi-org.brum.beds.ac.uk/10.3390/v12010035
Submission received: 22 November 2019
/
Revised: 21 December 2019
/
Accepted: 25 December 2019
/
Published: 28 December 2019
(This article belongs to the Special Issue Virus-Like Particle Vaccines)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Human cytomegalovirus (HCMV) infects more than 70% of the human population worldwide. HCMV is responsible for high morbidity and mortality in immunocompromised patients and remains the leading viral cause of congenital birth defects. Despite considerable efforts in vaccine and therapeutic development, HCMV infection still represents an unmet clinical need and a life-threatening disease in immunocompromised individuals and newborns. Immune repertoire interrogation of HCMV seropositive patients allowed the identification of several potential antigens for vaccine design. However, recent HCMV vaccine clinical trials did not lead to a satisfactory outcome in term of efficacy. Therefore, combining antigens with orthogonal technologies to further increase the induction of neutralizing antibodies could improve the likelihood of a vaccine to reach protective efficacy in humans. Indeed, presentation of multiple copies of an antigen in a repetitive array is known to drive a more robust humoral immune response than its soluble counterpart. Virus-like particles (VLPs) and nanoparticles (NPs) are powerful platforms for multivalent antigen presentation. Several self-assembling proteins have been successfully used as scaffolds to present complex glycoprotein antigens on their surface. In this review, we describe some key aspects of the immune response to HCMV and discuss the scaffolds that were successfully used to increase vaccine efficacy against viruses with unmet medical need.
1. Introduction
Human cytomegalovirus (HCMV) is a ubiquitously distributed member of the Herpesviridae family belonging to the Betaherpesvirinae sub-family; it infects 70% of the human adult population worldwide [1,2] and primary infection is usually asymptomatic in immunocompetent individuals. Nevertheless, the virus establishes a lifelong latency of the host after a primary infection and viral reactivation is common, also it does not necessarily imply clinical symptoms [1,3]. However, infection or viral reactivation in immunodeficient individuals such as AIDS patients, solid organ (SOT) or hematopoietic stem cells (HSCT) transplant patients causes morbidity and mortality [4,5]. Furthermore, HCMV is the most common viral cause of congenital birth defects affecting 0.7% of the newborns with permanent sequelae such as sensorineural hearing loss, growth restriction, and cognitive disabilities [6]. Current antiviral therapies and transfusion with hyper-immune globulins to control viremia are not efficient [7]. Therefore, given the severity and importance of these diseases, as well as the associated socioeconomic cost, the need for an HCMV vaccine has been assigned in the highest priority category by the Institute of Medicine and it represents the second highest priority target after HIV by the Centers for Disease Control [8,9]. Over the last decades, considerable efforts were deployed to develop a vaccine capable of preventing HCMV infection, congenital transmission and viral spreading after SOT or HSCT from seropositive donors to seronegative recipients [10]. The potential vaccine included live virus, attenuated or vectored viral vaccines expressing HCMV immunogens as well as purified recombinant protein vaccines that have been evaluated in clinical trials [11,12]. The abundant virion envelope glycoprotein B (gB) was shown to elicit vigorous T cell and antibody responses and represents the basis of most vaccines developed so far [13]. In a recent phase II clinical trial the recombinant gB vaccine formulated in MF59 adjuvant (gB/MF59), an oil-in-water emulsion, generated antibody titers comparable to natural infection. However, the vaccine demonstrated only modest efficacy in preventing primary HCMV infections in seronegative women and in the reduction of viremia in transplant recipients [14,15]. Surprisingly, a recent study demonstrated that seronegative patients vaccinated with the gB/MF59 vaccine developed a faster humoral response against gB after solid organ transplantation from seropositive donors [16]. Still, it is of the utmost importance to evaluate possible strategies for HCMV next generation vaccines. In this review, we will summarize the current findings on the adaptive immune response to HCMV and provide an update on the new methodologies available to boost the immune response against infectious diseases using virus-like particles (VLPs) and nanoparticles (NPs).
2. Immune Response against HCMV
The establishment of a long-lasting immunity in response to a primary HCMV infection is required to control subsequent HCMV reactivation and prevent uncontrolled viral replication or serious HCMV diseases [17,18]. HCMV infection activates both humoral and cellular immune responses. It is commonly believed that cellular immunity controls most of HCMV replication. Nonetheless, HCMV-specific antibodies have been associated with the prevention and protection from reinfection as well as the congenital transmission of HCMV [6,19,20]. Despite this, it is clear that the humoral response on its own is not able to clear the virus and prevent viral reactivation [21,22]. Nevertheless, the two arms of the adaptive immune response appear to be necessary for protection and resolution of HCMV infection [23].
2.1. Cellular Immune Response against HCMV
The primary cellular response to HCMV infection arises from natural killer (NK) cells, which trigger the release of inflammatory cytokines and cause lysis or apoptosis of infected cells [24]. Secondly, T cells restrict HCMV viral replication and viral spreading, although they are not able to definitively clear the virus [25,26,27]. HCMV-specific T cells represent 10% of both the CD4+ and CD8+ memory compartments in the peripheral blood of CMV-seropositive individuals [28]. CD8+ T cells release perforin and granzyme B to promote lysis of infected cells that present CMV peptides on MHC-I [29,30], while CD4+ specific T cells present a typical Th1 cytokine secretion signature (IFN-γ and TNF-α) [31]. Immune repertoire interrogation using 13,687 spanning peptides to cover 213 predicted CMV-proteins [32] revealed that human T cells recognize at least 151 HCMV proteins. Among these, CD4+ T cells recognize 125 proteins, while CD8+ T cells recognize 107 proteins, and 81 proteins are recognized by both cell types. Although the HCMV-specific T cell response is very broad, the vast majority of the results points toward antigens that are highly conserved among different HCMV strains, such as the 65 kDa phosphoprotein (pp65) and the immediate early 1 (IE1) proteins, which are recognized by more than 50% of seropositive individuals [28]. CD8+ T cells were proven to have a protective role in both HSCT and SOT recipients [33,34,35] and a function in preventing congenital HCMV infection [36]. However, one of the paradoxes of the cellular immunity against HCMV comes from the observation that HCMV does induce a very large T cell response, meanwhile the virus also possesses sophisticated and numerous immune evasion mechanisms [37]. Among those, the capacity of the virus to enter a latency state has been the subject of intense investigation. Indeed, while primary infection of immunocompetent individuals by HCMV is rarely the source of disease, the opposite is true for individuals with an immune system that is still immature (fetus) or compromised (HIV/AIDS and transplantations). Consequently, several studies sought to identify the antigens that are relevant for the latency phase. A vaccine able to boost the immune system against the latter would help clearing the latent reservoir of the virus. It is known that HCMV establishes latency in the myeloid cell lineage and viral genome has been detected in peripheral blood monocytes and in CD34+ progenitors in the bone marrow [38,39,40]. The viral gene expression program in these cells is restricted and extremely different from the one established during lytic infections. HCMV latent transcriptome is an area of extensive research, and at present, the exact roles of many latency-associated genes remain unclear. Nevertheless, several studies identified a number of viral genes that are specifically associated with latency [41,42]. Notably, RNAs from the major IE region (UL122–123 CLTs) [43], Latency Unique Natural Antigen (LUNA) [44], UL138 [41], UL111a (encoding a viral IL-10 homologue (LAcmvIL-10)) [45], UL144 [46] and US28 [47] were identified [22]. Moreover, viral reactivation is also associated with the expression of the major immediate early proteins (IE72 and IE86) [48]. T-cell repertoire interrogation identified four viral proteins expressed during latency (LUNA, UL138, US28, and LAcmvIL-10). Interestingly, these proteins can be recognized by CD4+ T cells and T-cell responses specific for all four proteins are detectable in healthy HCMV-positive donors [49]. Accordingly, future vaccine candidates should boost cellular immunity against some of these proteins representing key targets for viral clearance.
2.2. Humoral Immune Response against HCMV
Upon HCMV infection, a large quantity of the antibodies generated target the most abundant viral proteins. These include proteins from the tegument (i.e., pp65), the immediate early proteins (i.e., IE1), and envelope glycoproteins such as gB, gM/gN, and the gH/gL complexes (trimer and pentamer). Antibodies targeting antigens that are not exposed on the surface of the virion are unlikely to generate an efficient protection against the virus [10]. Nonetheless, antibodies targeting the envelope glycoproteins are able to neutralize viral infection in vitro and correlate with a decrease of viral transmission in primary infected pregnant women [50]. It is known that antibodies targeting the pentamer are the most potent neutralizers at least in vitro [51,52], while antibodies targeting gHgL and gB are a thousand times less potent [51]. This result is still not fully understood, since the gHgL and gB proteins form the core machinery for viral membrane fusion [53] and monoclonal antibodies (mAbs) targeting the fusion machinery of other herpesviruses, such as Epstein–Barr virus, are potent neutralizers [54]. Immune repertoire interrogation of HCMV seropositive donors allowed the identification of multiple mAbs targeting the pULs subunits. We recently demonstrated that these mAbs neutralize the virus by blocking the molecular recognition of the pentamer to its cellular host receptor Neuropilin 2 [55]. In contrast, mAbs targeting gH are supposed to prevent the activation of the fusion machinery [56]. These mAbs were assigned to only two antigenic regions of gH, indeed 3G16 and MSL-109 mAbs bind toward the C-terminus of gH, while the 13H11 mAb binds close to the gL interaction site [57].
Recently, two studies reported the results of the recombinant gB vaccine formulated in MF59 adjuvant, which demonstrated partial efficacy in reducing viraemia after SOT and preventing primary infection in women and adolescents [14,15]. Sera analysis from gB-vaccinated individuals allowed the characterization of at least five gB antigenic domains (AD-1 to AD-5). The AD-1 is a domain of 80 amino acids between amino acid 560 and 640 whereas AD-2 consists of two discontinuous binding sites: Site I, located between amino acids 68 and 77, is highly conserved among strains and is considered as a target of neutralizing responses, while Site II, located between amino acids 50 and 54, is less conserved. AD-3 is a linear epitope corresponding to amino acids 798–805 in the intraluminal region of gB. AD-4, like AD-2, is a discontinuous domain corresponding to amino acids 121–132 and 344–438. AD-5 matches to amino acids between 133 and 343 [58,59,60]. Interestingly, Baraniak et. al. demonstrated that vaccination only boosted AD-2 responses in the 50% of CMV+ individuals with a preexisting response and did not induce a new AD-2 response in those who lacked AD-2 antibodies following natural infection [15,61]. In parallel, Nelson et al. observed an increase of anti-gB IgG titer induced by the same vaccine in a cohort of CMV-post-partum women [14]. However, the authors also subsequently observed that 76% of the vaccine-induced IgG response recognized AD-3. Considering that AD-3 is located in the intraluminal part of the virus, antibodies against this region will be most probably non-protective [12]. The hypothesis that AD-3 is diverting the immune response away from antigenic sites that more likely induce protective antibody responses (AD-2), was recently proposed [62]. The next HCMV vaccines will have to contain multiple antigens that should induce a strong neutralizing antibody response protecting from primary infections and an efficient T-cell response to prevent viral reactivation and to clear the latent virus.
3. HCMV Antigens as Vaccine Candidates
HCMV is the largest virus infecting humans, with a genome containing a linear double-stranded DNA of about 230 kb, from which at least 170 open reading frames (ORFs) have been identified [63,64]. The virus contains over 30 structural proteins and encodes at least 20 membrane glycoproteins expressed on the virion envelope, which are involved in immune evasion, cell attachment, and viral entry. HCMV exhibits an extremely broad cellular tropism being able to infect different cell types ranging from epithelial, endothelial, fibroblasts, myeloid hematopoietic precursors, monocytes/macrophages to smooth muscle cells, stromal cells, neurons, astrocytes, hepatocytes and glial cells [65]. Identification of the host cellular receptors has been a subject of debate during the last decade. It is now accepted that the two main host cell receptors for HCMV involve the platelet-derived growth factor receptor α (PDGFRα) [66,67,68] and Neuropilin 2 (Nrp2) [55]. In addition, other cellular host factors were described as facilitators of viral entry such as the olfactory receptor family member OR14I1 [69], the adipocyte plasma membrane-associated protein (APMAP) [70], CD46 [71], and CD147 [72]. However, the biological relevance of this multitude of receptors remains to be understood. In contrast, the viral ligands responsible for the broad cellular tropism were clearly identified and correspond to two viral envelope complexes: the gHgLgO (trimer) and the gHgLpUL128pUL130pUL131A (pentamer) (Figure 1) [73]. The trimer is a heterocomplex, in which the heterodimer consisting of gH (UL75) and gL (UL115) is disulfide-linked to glycoprotein O (gO), a heavily N-glycosylated polypeptide encoded by UL74 [74,75,76]. The trimer complex binds PDGFRα and is required for entry in all cell types [77], representing an attractive target for vaccine design. However, the gO sequence heterogeneity and the large glycan shield present on this subunit are impairing the generation of potent neutralizing mAbs. Along with others, we speculate that these issues will probably slow down possible efforts to use the trimer as a vaccine candidate [78]. The pentamer complex is composed of the gHgL heterodimer bound to three additional glycoproteins encoded by pUL128, pUL130, and pUL131A. The pentamer complex is required for viral entry in epithelial, endothelial, and myeloid cells and binds Nrp2 [55,65,79]. It represents the main target of HCMV neutralizing antibodies, eliciting a protective response targeting the pULs subunits, which is several orders of magnitude more potent than any other response against HCMV glycoprotein [51,52,80]. However, anti-pULs mAbs are not able to block infection in all cell types, in contrast to mAbs directed against gH and gB subunits [81]. Indeed, membrane fusion and cell entry in all cell types is dictated by the core fusion machinery composed of gHgL and the glycoprotein B (gB) which are thus considered as primary targets for vaccine development [2]. gB is a class III viral fusion protein that forms homotrimers (Figure 1) and was initially reported to bind cell–surface proteins such as PDGFRα and EGFR. However, it is more likely that gB functions as a viral fusogen that is triggered when viral and host membranes are in close proximity [82]. In addition to the HCMV membrane glycoproteins, other viral antigens have been considered as potential vaccine targets. The phosphoprotein 65 (pp65) encoded by pUL83 and the immediate-early protein 1 (IE1) encoded by pUL123, both eliciting a robust T cell response, have undergone evaluation as subunit or vectored vaccines in human [83,84]. In conclusion, the aforementioned vaccine candidates failed to confer an appropriate protection from infection, viral reinfection or reactivation. We believe that the development of a protective vaccine should include antigens from the viral envelope to trigger the humoral response (mainly pentamer and newly designed gB) and antigens from the tegument or proteins that are specifically associated with latency (i.e., US28) to prompt the cellular response able to control viral reactivation.
4. Virus-Like Particles and Nanoparticles for Antigen Display
Virus-like particles (VLPs) and nanoparticles (NPs) are protein structures that resemble wild type viruses but do not have a viral genome nor infectious ability, creating in principle safer vaccine candidates. Both VLPs and NPs are composed of self-assembling proteins displaying the epitope of interest at a high density on their surface. Antigen presentation as a repetitive array is known to drive stronger humoral immune responses when compared to the single soluble antigen [85]. This effect is thought to derive from some key properties of the VLPs/NPs presentation such as the increased antigen density that creates avidity leading to a stronger B cell activation through antigen-driven cross-linking of B cell receptors (BCRs) [86]. Moreover, VLPs/NPs are also supposed to enhance antigen uptake by antigen presenting cells (APCs) which subsequently support the adaptive arms of the immune system [87]. Finally, VLPs/NPs have also been proven, in some cases, to generate an epitope-focusing effect in which antigenic sites promoting the generation of potent neutralizing mAbs were preferentially targeted [88,89]. Over the last decades, several platforms for VLPs/NPs design have been generated, including the usage of viral core proteins, covalent links of individual folded proteins through site-specific ligations and de novo protein nanostructure formation via non-covalent intramolecular and intermolecular interactions. VLPs/NPs are currently recognized as one of the most promising and extensively studied molecular carriers for new vaccine development. Here we will present the current VLP HCMV vaccines in development and provide some examples that can be relevant for novel vaccine design.
4.1. Developed HCMV VLP Vaccine
Enveloped virus-like particles (eVLPs) are VLP structures wrapped with a lipid envelope obtained by transfection of the Moloney murine leukemia virus (MLV) gag protein in human or hamster cell lines (i.e., HEK or CHO cells). The MLV gag expression induces burgeoning of particles from membranes of transfected cells. Variation Biotechnologies Incorporated (VBI) laboratories generated an eVLP vaccine expressing an HCMV gB full-length or a chimeric gB protein, where the extracellular domain (ECD) of gB is membrane-anchored using the transmembrane and cytoplasmic domains of the vesicular stomatitis virus (VSV) G protein [90]. The chimeric gB-VSV-G protein is thought to maintain gB in a prefusion conformation. Both vaccines were used to immunize mice and induce a neutralizing antibody response 10-fold higher compared to their soluble recombinant protein counterpart [90]. The vaccine was entered in phase I clinical studies enrolling HCMV-seronegative individuals for evaluation of safety and immunogenicity in early 2016 [91]. An additional eVLP vaccine candidate, targeting both the gB and pp65 antigens, has also been manufactured by VBI. However, we believe that a combination of gB, pentamer, and pp65 or US28 would be more appealing. Finally, another eVLP HCMV vaccine candidate was developed by Redvax GmbH, a Swiss biopharmaceutical company. Unlike VBI, that used mammalian cells, they used the rePAX baculovirus co-expression technology minimizing the purification issues due to the relative large size of VLPs for the efficient generation of a VLP vaccine candidate containing HCMV pp65 and gB [92]. Redvax GmbH was later acquired by Pfizer and the vaccine was able to elicit high-titer neutralizing antibodies and good T cell responses after immunization. Nonetheless, no protection was demonstrated from viremia upon challenge.
4.2. Hepatitis B Core Antigen Nanoparticle
The core protein of the hepatitis B virus (HBc) self-assembles into highly immunogenic virus-like structures with icosahedral symmetry. The hepatitis B core nanoparticle (HBc) is assembled from dimers of 183 or 185 amino acids and occurs in two size classes with a T = 3 or T = 4 symmetry. Depending on the symmetry, HBc is formed by 90 or 120 dimeric subunits with a respective diameter of 24 or 31 nm [93]. The antigen of interest needs to be inserted between alpha helices 3 and 4 of the major immune-dominant region (MIR) domain (amino acids 78–82) (Figure 2A) of the HBc protein. This insertion allows the antigen to be exposed on spike structures at the surface of the assembled nanoparticle. HBc protomers first dimerize and then multimerize (Figure 2B,C). Several developments led to the success of this platform, such as splitting of the HBc protein between the α3 and α4 helices or fusing two monomers together using a flexible glycine serine linker that allowed the introduction of larger protein domains [94,95]. However, one limitation of this technology is the need for both the N- and C-termini of the inserted antigen to fit with the geometry of the core acceptor site, a requirement that is not always successfully met by natively folded proteins.
In summary, the HBc is a powerful platform to generate nanoparticles and it is particularly useful in the case of small antigens. Indeed, it is unlikely that complex envelope glycoproteins such as gB or the gHgL complexes could be correctly displayed on HBc particles. Nevertheless, HBc could be very efficient to generate strong T cell responses [96]. For example, the viral-encoded G protein-coupled receptor US28 was recently shown to aid in the establishment and the maintenance of viral latency. US28 modulates host–cell proteins to suppress viral processes associated with lytic replication and thereby represent an important target promoting latent infection [97]. HBc particles displaying US28 peptides recognized by T cells could provide an important advancement to boost the existing immune responses necessary to control reactivation of latently infected individuals [98].
4.3. Ferritin-Based Nanoparticle
Ferritin is an intracellular globular protein that stores iron and releases it in a controlled fashion [99]. The protein is expressed in many tissues, mainly as a cytosolic protein, but it can be also found at low concentration in the serum where it functions as an iron carrier [100]. Ferritin is a four-helix bundle protein consisting of two couples of anti-parallel α-helices connected by a long loop and a short C-terminal α-helix (Figure 3A). The monomer can dimerize and further self-assemble to form a nanocage of 24 protein subunits, which has an internal and external diameter of 8 and 12 nm, respectively [101]. Ferritin nanoparticle (ferritin-NP) presents the advantage of being resistant to thermal and chemical stress [102]. Moreover, antigen display is achieved through genetic fusion at the N-terminus of each ferritin protomer. Ferritin-NP self-arranges as an octahedron composed of eight trimeric units (Figure 3B), each with a 3-fold symmetry axis that allows the correct presentation of trimeric antigens at the nanoparticle surface (Figure 3C).
A recent development and one of the major advantages of this carrier is the possibility to generate multivalent displays of different antigenic proteins. For example, ferritin-NP was first used to present HIV-1 and influenza antigens on the same particle [103]. Furthermore, Kanekiyo and coworkers at the Vaccine Research Center in the United States. further developed and exploited the ferritin-NP system using a ferritin hybrid protein [89] to generate a universal vaccine against influenza. The team co-displayed the receptor binding domain (RBD) of the last 90 years H1N1 influenza viruses [104] and showed that immunization with this mosaic RBD-NP elicited a broad antibody response covering all the known H1N1 strains. Multiple displays on ferritin-NP have opened up new possibilities for novel vaccines against different pathogens or against different antigens of a given pathogen.
In the case of HCMV, we can speculate that a mosaic ferritin-NP displaying both pentamer and gB ECDs should generate a potent humoral immune reaction. The antibodies generated targeting the gHgL and gB subunits will block infection of connective tissues and stroma (containing fibroblasts). On the other hand, antibodies directed against the pUL subunits will block infection of epithelial tissues.
4.4. Qβ Nanoparticle
The bacteriophage Qβ is a member of the Leviviridae family, forming 30 nm icosahedral nanoparticles of 180 copies from the 14 kDa coat protein [105]. The coat protein monomer (Figure 4A) forms Qβ dimers (Figure 4B) that further assemble into pentamers and hexamers by disulfide and hydrogen bond interactions to form a Qβ nanoparticle under physiological conditions (Figure 4C). This nanoparticle is highly stable and is used to display ligands and proteins on its exterior surface [106,107]. Co-expression of Qβ proteins displaying different antigens of interest or engineered Qβ proteins displaying functionalized groups for subsequent direct antigen conjugation can be easily produced in bacterial cells. Of note, the genetic fusion occurs at the C-terminus of the protein, a property that might impair the correct folding of some antigens of interest. Genes encoding the viral Qβ protein have been also successfully expressed in yeast cells but the possibility to generate a Qβ nanoparticle in prokaryotic hosts, maintaining therefore a low production cost, will be of great interest for the generation of a personalized T cell vaccine targeting the proteins associated to latency. Regarding mammalian cell expression, there is currently no evidence that the system is also compatible with it, which is a prerequisite for the production of most of the HCMV glycoprotein antigens.
4.5. De Novo Design-Based Nanoparticle
Structure-based design of nanoparticle immunogens has been limited by the restricted number of scaffolds available and the fact that their physico-chemical properties are fixed (i.e., ferritin, encapsulins). Moreover, most of the self-assembling scaffolds spontaneously do so upon expression in the transfected host cell, a property that might lead to poor production yield. These constraints have pushed the exploration of new structural and functional geometries (i.e., icosahedron, dodecahedron) in the nanoparticle immunogen design field. Recent computational methods for designing novel self-assembling proteins with atomic-level accuracy offer the possibility to design self-assembling proteins with customized structures, offering new opportunities for structure-based vaccine design [108,109,110]. For instance, the HIV gp41 ectodomain trimer was displayed onto the I3–01 particle [108,111]. The gp41 antigen exposed on NPs, stimulated mAb-expressing B cells more effectively than the soluble trimers [111]. Two additional publications highlighted the potential of this de novo self-assembling nanoparticle for antigen display. The I5350 nanoparticle (Figure 5) was used to generate an enhanced vaccine against the respiratory syncytial virus (RSV). Self-assembling protein nanoparticles displaying 20 copies of the stabilized version of the RSV fusion glycoprotein trimer (DS-Cav1) induced a neutralizing antibody response ∼10-fold higher than soluble trimeric DS-Cav1 [112]. In the second case, native-like HIV-1 envelope trimer antigens were displayed in a multivalent fashion on the I5350 nanoparticle scaffold and immunization studies revealed a more effective priming compared to the soluble SOSIP trimers [113]. SOSIP corresponds to a stabilized trimer with mutations that cross-link the cleaved gp120 and gp41. In contrast, the natural HIV trimer is comprised of three copies of a non-covalently linked gp120/gp41 heterodimer arising from cleavage of the viral gp160 precursor protein. It can be speculated that de novo design-based nanoparticles will have an increasing impact on next generation vaccines since they are customizable at will, supported by a multitude of expression platforms and able to generate multivalent display as well as encapsulation of small proteins or compounds [114,115].
Ideally, I5350 particles should present on their surface the pentamer and gB to generate a humoral response against the gH, gL, pULs, and gB molecules. Moreover, as I5350 nanoparticles are hollow, the in vitro assembly step with I53–50B.4PT1 could be performed in the presence of T-cell stimulating peptides (i.e., pp65, IE1 and US28) that would be encapsulated inside the particles. The resulting vaccine candidates should in this way stimulate both arms of the immune system, offering a maximal protection.
4.6. SpyCatcher and SpyTag for Nanoparticle Formation
The SpyTag/SpyCatcher system (referred as Spy-System) is based on the internal isopeptide bond of the CnaB2 domain of FbaB protein from Streptococcus pyogenes. An internal isopeptide bond forms spontaneously in this domain between the reactive amine of lysine K31 and the side chain carboxyl of aspartate D117. This reaction is catalyzed by the spatially adjacent glutamate E77. The resulting isopeptide bond confers high stability to the CnaB2 domain. The CnaB2 domain is cleaved into two subcomponents, the large and incomplete immunoglobulin-like domain, named SpyCatcher, composed of 138 amino acids, and the short beta strand, named SpyTag, composed of 13 amino acids [116]. While the SpyCatcher contains the reactive lysine (K31) and catalytic glutamate (E77), the SpyTag includes the reactive aspartate (D117). The two components can still recognize each other with high affinity and the isopeptide is formed within minutes. Despite the fact that this system is not able to generate nanoparticles on its own, the two components of the Spy-System can be genetically fused to a number of viral or designed self-assembling proteins leading to the generation of a stable nanoparticle displaying a large variety of antigens. Moreover, the Spy-System offers a great flexibility since components can be inserted at the N- or C-termini of the target proteins. Its simplicity is of particular interest to quickly screen if a nanoparticle formulation will be of benefit for vaccine design, before generating genetic fusion on the nanoparticle of interest. The first nanoparticle assembly with Spy-System was performed by genetically fusing the SpyCatcher to the N-terminus of the viral coat protein (CP3) of the RNA bacteriophage AP205 [117]. AP205 VLPs have been then further investigated as vaccine candidates against a number of targets, including malaria and cancer [117,118,119].
5. Conclusions and Future Directions
We described a large variety of systems to generate both VLPs and NPs. Self-assembling scaffolds have been used to present complex glycoprotein antigens and for vaccination studies against influenza [104], HIV [113,120,121,122,123], Epstein–Barr virus [89], and RSV [112]. In all cases, antigen immunogenicity was increased by multivalent presentation and in some cases, an epitope focusing effect could be observed. The rise of computational design to generate novel self-assembling proteins has now paved the way for new possibilities to display antigens without limitation in term of quantity or oligomeric state of the antigen. HCMV is still the principal viral cause of congenital malformations. About one quarter of infants infected by HCMV in utero will develop severe sequelae including microcephaly, sensorineural hearing and vision loss, or cognitive delay [1,6]. The recent identification of key HCMV antigenic targets for both humoral and cellular immune responses and the capacity to display and encapsulate proteins or nucleic acids (RNAs) on VLPs/NPs is likely to generate improved vaccine candidates able to redirect the immune response against specific targets.
Author Contributions
M.P. and L.P. conceptualized and wrote the review. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Novartis Foundation for Medical–Biological Research, Novartis grant (application #19B116 to LP).
Acknowledgments
The authors acknowledge Mathilde Foglierini and Jessica Marcandalli for critical reading and helpful comments on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Boppana, S.B. Vaccination against the human cytomegalovirus. Vaccine 2018, 37, 7437–7442. [Google Scholar] [CrossRef] [PubMed]
- Britt, W. Manifestations of human cytomegalovirus infection: Proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 2008, 325, 417–470. [Google Scholar] [CrossRef] [PubMed]
- Gerna, G.; Zavattoni, M.; Baldanti, F.; Furione, M.; Chezzi, L.; Revello, M.G.; Percivalle, E. Circulating cytomegalic endothelial cells are associated with high human cytomegalovirus (HCMV) load in AIDS patients with late-stage disseminated HCMV disease. J. Med. Virol. 1998, 55, 64–74. [Google Scholar] [CrossRef]
- Lilleri, D.; Gerna, G. Strategies to control human cytomegalovirus infection in adult hematopoietic stem cell transplant recipients. Immunotherapy 2016, 8, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Boppana, S.B.; Britt, W.J. Antiviral antibody responses and intrauterine transmission after primary maternal cytomegalovirus infection. J. Infect. Dis. 1995, 171, 1115–1121. [Google Scholar] [CrossRef]
- Spinillo, A.; Gerna, G. Hyperimmune globulin to prevent congenital CMV infection. N. Engl. J. Med. 2014, 370, 2544–2545. [Google Scholar] [CrossRef]
- James, S.H.; Kimberlin, D.W. Advances in the prevention and treatment of congenital cytomegalovirus infection. Curr. Opin. Pediatrics 2016, 28, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Marsico, C.; Kimberlin, D.W. Congenital Cytomegalovirus infection: Advances and challenges in diagnosis, prevention and treatment. Ital. J. Pediatrics 2017, 43, 38. [Google Scholar] [CrossRef]
- Gomes, A.C.; Griffiths, P.D.; Reeves, M.B. The Humoral Immune Response Against the gB Vaccine: Lessons Learnt from Protection in Solid Organ Transplantation. Vaccines 2019, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Schleiss, M.R. Recombinant cytomegalovirus glycoprotein B vaccine: Rethinking the immunological basis of protection. Proc. Natl. Acad. Sci. USA 2018, 115, 6110–6112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.S.; Herold, B.C.; Permar, S.R. A new era in cytomegalovirus vaccinology: Considerations for rational design of next-generation vaccines to prevent congenital cytomegalovirus infection. Npj Vaccines 2018, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foglierini, M.; Marcandalli, J.; Perez, L. HCMV Envelope Glycoprotein Diversity Demystified. Front. Microbiol. 2019, 10, 1005. [Google Scholar] [CrossRef]
- Nelson, C.S.; Huffman, T.; Jenks, J.A.; Cisneros de la Rosa, E.; Xie, G.; Vandergrift, N.; Pass, R.F.; Pollara, J.; Permar, S.R. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc. Natl. Acad. Sci. USA 2018, 115, 6267–6272. [Google Scholar] [CrossRef] [Green Version]
- Baraniak, I.; Kropff, B.; Ambrose, L.; McIntosh, M.; McLean, G.R.; Pichon, S.; Atkinson, C.; Milne, R.S.B.; Mach, M.; Griffiths, P.D.; et al. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2018, 115, 6273–6278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baraniak, I.; Gomes, A.C.; Sodi, I.; Langstone, T.; Rothwell, E.; Atkinson, C.; Pichon, S.; Piras-Douce, F.; Griffiths, P.D.; Reeves, M.B. Seronegative patients vaccinated with cytomegalovirus gB-MF59 vaccine have evidence of neutralising antibody responses against gB early post-transplantation. EBioMedicine 2019, 50, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerna, G.; Lilleri, D.; Fornara, C.; Bruno, F.; Gabanti, E.; Cane, I.; Furione, M.; Revello, M.G. Differential kinetics of human cytomegalovirus load and antibody responses in primary infection of the immunocompetent and immunocompromised host. J. Gen. Virol. 2015, 96, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.J.; Quinn, M.; Snyder, C.M. CMV-Specific CD8 T Cell Differentiation and Localization: Implications for Adoptive Therapies. Front. Immunol. 2016, 7, 352. [Google Scholar] [CrossRef] [Green Version]
- Bialas, K.M.; Westreich, D.; Cisneros de la Rosa, E.; Nelson, C.S.; Kauvar, L.M.; Fu, T.M.; Permar, S.R. Maternal Antibody Responses and Nonprimary Congenital Cytomegalovirus Infection of HIV-1-Exposed Infants. J. Infect. Dis. 2016, 214, 1916–1923. [Google Scholar] [CrossRef] [Green Version]
- Itell, H.L.; Nelson, C.S.; Martinez, D.R.; Permar, S.R. Maternal immune correlates of protection against placental transmission of cytomegalovirus. Placenta 2017, 60 (Suppl. 1), S73–S79. [Google Scholar] [CrossRef]
- Stern, L.; Withers, B.; Avdic, S.; Gottlieb, D.; Abendroth, A.; Blyth, E.; Slobedman, B. Human Cytomegalovirus Latency and Reactivation in Allogeneic Hematopoietic Stem Cell Transplant Recipients. Front. Microbiol. 2019, 10, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins-McMillen, D.; Buehler, J.; Peppenelli, M.; Goodrum, F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 2018, 10, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, B.A.; Wills, M.R.; Sinclair, J.H. Advances in the treatment of cytomegalovirus. Br. Med. Bull. 2019, 131, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.W.; Tomasec, P.; Stanton, R.J.; Armstrong, M.; Prod’homme, V.; Aicheler, R.; McSharry, B.P.; Rickards, C.R.; Cochrane, D.; Llewellyn-Lacey, S.; et al. Modulation of natural killer cells by human cytomegalovirus. J. Clin. Virol. 2008, 41, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Rist, M.; Cooper, L.; Elkington, R.; Walker, S.; Fazou, C.; Tellam, J.; Crough, T.; Khanna, R. Ex vivo expansion of human cytomegalovirus-specific cytotoxic T cells by recombinant polyepitope: Implications for HCMV immunotherapy. Eur. J. Immunol. 2005, 35, 996–1007. [Google Scholar] [CrossRef]
- Rohrlich, P.S.; Cardinaud, S.; Lule, J.; Montero-Julian, F.A.; Prodhomme, V.; Firat, H.; Davignon, J.L.; Perret, E.; Monseaux, S.; Necker, A.; et al. Use of a lentiviral vector encoding a HCMV-chimeric IE1-pp65 protein for epitope identification in HLA-Transgenic mice and for ex vivo stimulation and expansion of CD8(+) cytotoxic T cells from human peripheral blood cells. Hum. Immunol. 2004, 65, 514–522. [Google Scholar] [CrossRef]
- Boppana, S.B.; Britt, W.J. Recognition of human cytomegalovirus gene products by HCMV-specific cytotoxic T cells. Virology 1996, 222, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Hertoghs, K.M.; Moerland, P.D.; van Stijn, A.; Remmerswaal, E.B.; Yong, S.L.; van de Berg, P.J.; van Ham, S.M.; Baas, F.; ten Berge, I.J.; van Lier, R.A. Molecular profiling of cytomegalovirus-induced human CD8+ T cell differentiation. J. Clin. Invest. 2010, 120, 4077–4090. [Google Scholar] [CrossRef]
- Gillespie, G.M.; Wills, M.R.; Appay, V.; O’Callaghan, C.; Murphy, M.; Smith, N.; Sissons, P.; Rowland-Jones, S.; Bell, J.I.; Moss, P.A. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J. Virol. 2000, 74, 8140–8150. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, E.M.; Remmerswaal, E.B.; Vossen, M.T.; Rowshani, A.T.; Wertheim-van Dillen, P.M.; van Lier, R.A.; ten Berge, I.J. Emergence of a CD4+CD28- granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J. Immunol. 2004, 173, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Kern, F.; Faulhaber, N.; Frommel, C.; Khatamzas, E.; Prosch, S.; Schonemann, C.; Kretzschmar, I.; Volkmer-Engert, R.; Volk, H.D.; Reinke, P. Analysis of CD8 T cell reactivity to cytomegalovirus using protein-spanning pools of overlapping pentadecapeptides. Eur. J. Immunol. 2000, 30, 1676–1682. [Google Scholar] [CrossRef]
- Lilleri, D.; Zelini, P.; Fornara, C.; Zavaglio, F.; Rampino, T.; Perez, L.; Gabanti, E.; Gerna, G. Human cytomegalovirus (HCMV)-specific T cell but not neutralizing or IgG binding antibody responses to glycoprotein complexes gB, gHgLgO, and pUL128L correlate with protection against high HCMV viral load reactivation in solid-organ transplant recipients. J. Med. Virol. 2018, 90, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Reusser, P.; Gambertoglio, J.G.; Lilleby, K.; Meyers, J.D. Phase I-II trial of foscarnet for prevention of cytomegalovirus infection in autologous and allogeneic marrow transplant recipients. J. Infect. Dis. 1992, 166, 473–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, P.D.; Reusser, P.; Goodrich, J.M.; Riddell, S.R. Development of a treatment regimen for human cytomegalovirus (CMV) infection in bone marrow transplantation recipients by adoptive transfer of donor-derived CMV-specific T cell clones expanded in vitro. Ann. N. Y. Acad. Sci. 1991, 636, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Fornara, C.; Cassaniti, I.; Zavattoni, M.; Furione, M.; Adzasehoun, K.M.G.; De Silvestri, A.; Comolli, G.; Baldanti, F. Human Cytomegalovirus-Specific Memory CD4+ T-Cell Response and Its Correlation With Virus Transmission to the Fetus in Pregnant Women With Primary Infection. Clin. Infect. Dis. 2017, 65, 1659–1665. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J. Antigens and immunoevasins: Opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2002, 2, 831–844. [Google Scholar] [CrossRef]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 1991, 72 Pt 9, 2059–2064. [Google Scholar] [CrossRef]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 1996, 77 Pt 12, 3099–3102. [Google Scholar] [CrossRef]
- Hargett, D.; Shenk, T.E. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl Acad. Sci. USA 2010, 107, 20039–20044. [Google Scholar] [CrossRef] [Green Version]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.B.; Sinclair, J.H. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kaneshima, H.; Mocarski, E.S. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl Acad. Sci. USA 1994, 91, 11879–11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bego, M.; Maciejewski, J.; Khaiboullina, S.; Pari, G.; St Jeor, S. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J. Virol. 2005, 79, 11022–11034. [Google Scholar] [CrossRef] [Green Version]
- Avdic, S.; Cao, J.Z.; Cheung, A.K.; Abendroth, A.; Slobedman, B. Viral interleukin-10 expressed by human cytomegalovirus during the latent phase of infection modulates latently infected myeloid cell differentiation. J. Virol. 2011, 85, 7465–7471. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.; Walther, A.; Raven, K.; Benedict, C.A.; Mason, G.M.; Sinclair, J. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J. Virol. 2013, 87, 4261–4271. [Google Scholar] [CrossRef] [Green Version]
- Beisser, P.S.; Laurent, L.; Virelizier, J.L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef] [Green Version]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.; et al. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef] [Green Version]
- Mason, G.M.; Jackson, S.; Okecha, G.; Poole, E.; Sissons, J.G.; Sinclair, J.; Wills, M.R. Human cytomegalovirus latency-associated proteins elicit immune-suppressive IL-10 producing CD4(+) T cells. PLoS Pathog. 2013, 9, e1003635. [Google Scholar] [CrossRef]
- Lilleri, D.; Kabanova, A.; Revello, M.G.; Percivalle, E.; Sarasini, A.; Genini, E.; Sallusto, F.; Lanzavecchia, A.; Corti, D.; Gerna, G. Fetal human cytomegalovirus transmission correlates with delayed maternal antibodies to gH/gL/pUL128-130-131 complex during primary infection. PLoS ONE 2013, 8, e59863. [Google Scholar] [CrossRef] [Green Version]
- Kabanova, A.; Perez, L.; Lilleri, D.; Marcandalli, J.; Agatic, G.; Becattini, S.; Preite, S.; Fuschillo, D.; Percivalle, E.; Sallusto, F.; et al. Antibody-driven design of a human cytomegalovirus gHgLpUL128L subunit vaccine that selectively elicits potent neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2014, 111, 17965–17970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macagno, A.; Bernasconi, N.L.; Vanzetta, F.; Dander, E.; Sarasini, A.; Revello, M.G.; Gerna, G.; Sallusto, F.; Lanzavecchia, A. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 2010, 84, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanarsdall, A.L.; Howard, P.W.; Wisner, T.W.; Johnson, D.C. Human Cytomegalovirus gH/gL Forms a Stable Complex with the Fusion Protein gB in Virions. PLoS Pathog. 2016, 12, e1005564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, W.; Joyce, M.G.; Nguyen, H.; Banh, D.V.; Aguilar, F.; Tariq, Z.; Yap, M.L.; Tsujimura, Y.; Gillespie, R.A.; Tsybovsky, Y.; et al. Immunization with Components of the Viral Fusion Apparatus Elicits Antibodies That Neutralize Epstein-Barr Virus in B Cells and Epithelial Cells. Immunity 2019. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e1119. [Google Scholar] [CrossRef] [Green Version]
- Malito, E.; Chandramouli, S.; Carfi, A. From recognition to execution-the HCMV Pentamer from receptor binding to fusion triggering. Curr. Opin. Virol. 2018, 31, 43–51. [Google Scholar] [CrossRef]
- Ciferri, C.; Chandramouli, S.; Leitner, A.; Donnarumma, D.; Cianfrocco, M.A.; Gerrein, R.; Friedrich, K.; Aggarwal, Y.; Palladino, G.; Aebersold, R.; et al. Antigenic Characterization of the HCMV gH/gL/gO and Pentamer Cell Entry Complexes Reveals Binding Sites for Potently Neutralizing Human Antibodies. PLoS Pathog. 2015, 11, e1005230. [Google Scholar] [CrossRef]
- Wiegers, A.K.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Identification of a neutralizing epitope within antigenic domain 5 of glycoprotein B of human cytomegalovirus. J. Virol. 2015, 89, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Spindler, N.; Diestel, U.; Stump, J.D.; Wiegers, A.K.; Winkler, T.H.; Sticht, H.; Mach, M.; Muller, Y.A. Structural basis for the recognition of human cytomegalovirus glycoprotein B by a neutralizing human antibody. PLoS Pathog. 2014, 10, e1004377. [Google Scholar] [CrossRef]
- Potzsch, S.; Spindler, N.; Wiegers, A.K.; Fisch, T.; Rucker, P.; Sticht, H.; Grieb, N.; Baroti, T.; Weisel, F.; Stamminger, T.; et al. B cell repertoire analysis identifies new antigenic domains on glycoprotein B of human cytomegalovirus which are target of neutralizing antibodies. PLoS Pathog. 2011, 7, e1002172. [Google Scholar] [CrossRef] [Green Version]
- Baraniak, I.; Kropff, B.; McLean, G.R.; Pichon, S.; Piras-Douce, F.; Milne, R.S.B.; Smith, C.; Mach, M.; Griffiths, P.D.; Reeves, M.B. Epitope-Specific Humoral Responses to Human Cytomegalovirus Glycoprotein-B Vaccine With MF59: Anti-AD2 Levels Correlate With Protection From Viremia. J. Infect. Dis. 2018, 217, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, I.; Kern, F.; Holenya, P.; Griffiths, P.; Reeves, M. Original Antigenic Sin Shapes the Immunological Repertoire Evoked by Human Cytomegalovirus Glycoprotein B/MF59 Vaccine in Seropositive Recipients. J. Infect. Dis. 2019, 220, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pasa-Tolic, L.; Wang, D.; Camp, D.G., 2nd; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.; Hein, M.Y.; Huang, S.X.; Ma, M.; Shen, B.; Qian, S.B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Oberstein, A.; Wang, W.; Shenk, T. Role of PDGF receptor-alpha during human cytomegalovirus entry into fibroblasts. Proc. Natl. Acad. Sci. USA 2018, 115, E9889–E9898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-alpha as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-alpha receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef]
- E, X.; Meraner, P.; Lu, P.; Perreira, J.M.; Aker, A.M.; McDougall, W.M.; Zhuge, R.; Chan, G.C.; Gerstein, R.M.; Caposio, P.; et al. OR14I1 is a receptor for the human cytomegalovirus pentameric complex and defines viral epithelial cell tropism. Proc. Natl. Acad. Sci. USA 2019, 116, 7043–7052. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Gui, X.; Freed, D.C.; Ku, Z.; Li, L.; Chen, Y.; Xiong, W.; Fan, X.; Su, H.; He, X.; et al. Identification of adipocyte plasma membrane-associated protein as a novel modulator of human cytomegalovirus infection. PLoS Pathog. 2019, 15, e1007914. [Google Scholar] [CrossRef] [Green Version]
- Stein, K.R.; Gardner, T.J.; Hernandez, R.E.; Kraus, T.A.; Duty, J.A.; Ubarretxena-Belandia, I.; Moran, T.M.; Tortorella, D. CD46 facilitates entry and dissemination of human cytomegalovirus. Nat. Commun. 2019, 10, 2699. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Pritchard, S.R.; Wisner, T.W.; Liu, J.; Jardetzky, T.S.; Johnson, D.C. CD147 Promotes Entry of Pentamer-Expressing Human Cytomegalovirus into Epithelial and Endothelial Cells. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.T.; Compton, T. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J. Virol. 1998, 72, 8191–8197. [Google Scholar]
- Wille, P.T.; Knoche, A.J.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J. Virol. 2010, 84, 2585–2596. [Google Scholar] [CrossRef] [Green Version]
- Ciferri, C.; Chandramouli, S.; Donnarumma, D.; Nikitin, P.A.; Cianfrocco, M.A.; Gerrein, R.; Feire, A.L.; Barnett, S.W.; Lilja, A.E.; Rappuoli, R.; et al. Structural and biochemical studies of HCMV gH/gL/gO and Pentamer reveal mutually exclusive cell entry complexes. Proc. Natl. Acad. Sci. USA 2015, 112, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Lanchy, J.M.; Ryckman, B.J. Human Cytomegalovirus gH/gL/gO Promotes the Fusion Step of Entry into All Cell Types, whereas gH/gL/UL128-131 Broadens Virus Tropism through a Distinct Mechanism. J. Virol. 2015, 89, 8999–9009. [Google Scholar] [CrossRef] [Green Version]
- Bagdonaite, I.; Norden, R.; Joshi, H.J.; King, S.L.; Vakhrushev, S.Y.; Olofsson, S.; Wandall, H.H. Global Mapping of O-Glycosylation of Varicella Zoster Virus, Human Cytomegalovirus, and Epstein-Barr Virus. J. Biol. Chem. 2016, 291, 12014–12028. [Google Scholar] [CrossRef] [Green Version]
- Nogalski, M.T.; Chan, G.C.; Stevenson, E.V.; Collins-McMillen, D.K.; Yurochko, A.D. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog. 2013, 9, e1003463. [Google Scholar] [CrossRef]
- Fouts, A.E.; Chan, P.; Stephan, J.P.; Vandlen, R.; Feierbach, B. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J. Virol. 2012, 86, 7444–7447. [Google Scholar] [CrossRef] [Green Version]
- Zydek, M.; Petitt, M.; Fang-Hoover, J.; Adler, B.; Kauvar, L.M.; Pereira, L.; Tabata, T. HCMV infection of human trophoblast progenitor cells of the placenta is neutralized by a human monoclonal antibody to glycoprotein B and not by antibodies to the pentamer complex. Viruses 2014, 6, 1346–1364. [Google Scholar] [CrossRef] [PubMed]
- Wille, P.T.; Wisner, T.W.; Ryckman, B.; Johnson, D.C. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. MBio 2013, 4, e00332-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selinsky, C.; Luke, C.; Wloch, M.; Geall, A.; Hermanson, G.; Kaslow, D.; Evans, T. A DNA-based vaccine for the prevention of human cytomegalovirus-associated diseases. Hum. Vaccin. 2005, 1, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Y.; Kaur, A.; Zhou, S.S.; Barry, P.A. Characterization and immunological analysis of the rhesus cytomegalovirus homologue (Rh112) of the human cytomegalovirus UL83 lower matrix phosphoprotein (pp65). J. Gen. Virol. 2006, 87, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Reviews. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Lopez-Sagaseta, J.; Malito, E.; Rappuoli, R.; Bottomley, M.J. Self-assembling protein nanoparticles in the design of vaccines. Comput Struct Biotechnol J. 2016, 14, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Duan, H.; Chen, X.; Boyington, J.C.; Cheng, C.; Zhang, Y.; Jafari, A.J.; Stephens, T.; Tsybovsky, Y.; Kalyuzhniy, O.; Zhao, P.; et al. Glycan Masking Focuses Immune Responses to the HIV-1 CD4-Binding Site and Enhances Elicitation of VRC01-Class Precursor Antibodies. Immunity 2018, 49, 301–311.e305. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, M.; Bu, W.; Joyce, M.G.; Meng, G.; Whittle, J.R.; Baxa, U.; Yamamoto, T.; Narpala, S.; Todd, J.P.; Rao, S.S.; et al. Rational Design of an Epstein-Barr Virus Vaccine Targeting the Receptor-Binding Site. Cell 2015, 162, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Kirchmeier, M.; Fluckiger, A.C.; Soare, C.; Bozic, J.; Ontsouka, B.; Ahmed, T.; Diress, A.; Pereira, L.; Schodel, F.; Plotkin, S.; et al. Enveloped virus-like particle expression of human cytomegalovirus glycoprotein B antigen induces antibodies with potent and broad neutralizing activity. Clin. Vaccine Immunol. Cvi. 2014, 21, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Snapper, C.M. Development of novel vaccines against human cytomegalovirus. Hum. Vaccines Immunother. 2019, 15, 2673–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente, T.; Burri, S.; Wellnitz, S.; Walsh, K.; Rothe, S.; Liderfelt, J. Fully aseptic single-use cross flow filtration system for clarification and concentration of cytomegalovirus-like particles. Eng. Life Sci. 2014, 14, 318–326. [Google Scholar] [CrossRef]
- Crowther, R.A.; Kiselev, N.A.; Bottcher, B.; Berriman, J.A.; Borisova, G.P.; Ose, V.; Pumpens, P. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 1994, 77, 943–950. [Google Scholar] [CrossRef]
- Roseman, A.M.; Berriman, J.A.; Wynne, S.A.; Butler, P.J.; Crowther, R.A. A structural model for maturation of the hepatitis B virus core. Proc. Natl. Acad. Sci. USA 2005, 102, 15821–15826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.; Skamel, C.; Vorreiter, J.; Nassal, M. Internal core protein cleavage leaves the hepatitis B virus capsid intact and enhances its capacity for surface display of heterologous whole chain proteins. J. Biol. Chem. 2008, 283, 33508–33515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, K.; Meijerink, E.; Speiser, D.E.; Tissot, A.C.; Cielens, I.; Renhof, R.; Dishlers, A.; Pumpens, P.; Bachmann, M.F. Efficient homologous prime-boost strategies for T cell vaccination based on virus-like particles. Eur. J. Immunol. 2005, 35, 816–821. [Google Scholar] [CrossRef]
- Krishna, B.A.; Humby, M.S.; Miller, W.E.; O’Connor, C.M. Human cytomegalovirus G protein-coupled receptor US28 promotes latency by attenuating c-fos. Proc. Natl. Acad. Sci. USA 2019, 116, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Krishna, B.A.; Miller, W.E.; O’Connor, C.M. US28: HCMV’s Swiss Army Knife. Viruses 2018, 10, 445. [Google Scholar] [CrossRef] [Green Version]
- Watt, G.D.; Jacobs, D.; Frankel, R.B. Redox reactivity of bacterial and mammalian ferritin: Is reductant entry into the ferritin interior a necessary step for iron release? Proc. Natl. Acad. Sci. USA 1988, 85, 7457–7461. [Google Scholar] [CrossRef] [Green Version]
- Theil, E.C. Ferritin: Structure, gene regulation, and cellular function in animals, plants, and microorganisms. Annu. Rev. Biochem. 1987, 56, 289–315. [Google Scholar] [CrossRef]
- Cho, K.J.; Shin, H.J.; Lee, J.H.; Kim, K.J.; Park, S.S.; Lee, Y.; Lee, C.; Park, S.S.; Kim, K.H. The crystal structure of ferritin from Helicobacter pylori reveals unusual conformational changes for iron uptake. J. Mol. Biol. 2009, 390, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Pulsipher, K.W.; Villegas, J.A.; Roose, B.W.; Hicks, T.L.; Yoon, J.; Saven, J.G.; Dmochowski, I.J. Thermophilic Ferritin 24mer Assembly and Nanoparticle Encapsulation Modulated by Interdimer Electrostatic Repulsion. Biochemistry 2017, 56, 3596–3606. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, I.S.; Joyce, M.G.; Chen, R.E.; Leung, K.; McKee, K.; Druz, A.; Van Galen, J.G.; Kanekiyo, M.; Tsybovsky, Y.; Yang, E.S.; et al. Two-Component Ferritin Nanoparticles for Multimerization of Diverse Trimeric Antigens. Acs. Infect. Dis. 2018, 4, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, M.; Joyce, M.G.; Gillespie, R.A.; Gallagher, J.R.; Andrews, S.F.; Yassine, H.M.; Wheatley, A.K.; Fisher, B.E.; Ambrozak, D.R.; Creanga, A.; et al. Mosaic nanoparticle display of diverse influenza virus hemagglutinins elicits broad B cell responses. Nat. Immunol. 2019, 20, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Golmohammadi, R.; Fridborg, K.; Bundule, M.; Valegard, K.; Liljas, L. The crystal structure of bacteriophage Q beta at 3.5 A resolution. Structure 1996, 4, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Cornuz, J.; Zwahlen, S.; Jungi, W.F.; Osterwalder, J.; Klingler, K.; van Melle, G.; Bangala, Y.; Guessous, I.; Muller, P.; Willers, J.; et al. A vaccine against nicotine for smoking cessation: A randomized controlled trial. PLoS ONE 2008, 3, e2547. [Google Scholar] [CrossRef]
- Vasiljeva, I.; Kozlovska, T.; Cielens, I.; Strelnikova, A.; Kazaks, A.; Ose, V.; Pumpens, P. Mosaic Qbeta coats as a new presentation model. Febs. Lett. 1998, 431, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Hsia, Y.; Bale, J.B.; Gonen, S.; Shi, D.; Sheffler, W.; Fong, K.K.; Nattermann, U.; Xu, C.; Huang, P.S.; Ravichandran, R.; et al. Corrigendum: Design of a hyperstable 60-subunit protein icosahedron. Nature 2016, 540, 150. [Google Scholar] [CrossRef]
- King, N.P.; Bale, J.B.; Sheffler, W.; McNamara, D.E.; Gonen, S.; Gonen, T.; Yeates, T.O.; Baker, D. Accurate design of co-assembling multi-component protein nanomaterials. Nature 2014, 510, 103–108. [Google Scholar] [CrossRef]
- Bale, J.B.; Gonen, S.; Liu, Y.; Sheffler, W.; Ellis, D.; Thomas, C.; Cascio, D.; Yeates, T.O.; Gonen, T.; King, N.P.; et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 2016, 353, 389–394. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Kumar, S.; Allen, J.D.; Huang, D.; Lin, X.; Mann, C.J.; Saye-Francisco, K.L.; Copps, J.; Sarkar, A.; Blizard, G.S.; et al. HIV-1 vaccine design through minimizing envelope metastability. Sci. Adv. 2018, 4, eaau6769. [Google Scholar] [CrossRef] [Green Version]
- Marcandalli, J.; Fiala, B.; Ols, S.; Perotti, M.; de van der Schueren, W.; Snijder, J.; Hodge, E.; Benhaim, M.; Ravichandran, R.; Carter, L.; et al. Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell 2019, 176, 1420–1431.e1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwer, P.J.M.; Antanasijevic, A.; Berndsen, Z.; Yasmeen, A.; Fiala, B.; Bijl, T.P.L.; Bontjer, I.; Bale, J.B.; Sheffler, W.; Allen, J.D.; et al. Enhancing and shaping the immunogenicity of native-like HIV-1 envelope trimers with a two-component protein nanoparticle. Nat. Commun. 2019, 10, 4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyken, S.E.; Benhaim, M.A.; Busch, F.; Jia, M.; Bick, M.J.; Choi, H.; Klima, J.C.; Chen, Z.; Walkey, C.; Mileant, A.; et al. De novo design of tunable, pH-driven conformational changes. Science 2019, 364, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Ogohara, C.; Yi, S.; Hsia, Y.; Nattermann, U.; Belnap, D.M.; King, N.P.; Sundquist, W.I. Designed proteins induce the formation of nanocage-containing extracellular vesicles. Nature 2016, 540, 292–295. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [Green Version]
- Brune, K.D.; Leneghan, D.B.; Brian, I.J.; Ishizuka, A.S.; Bachmann, M.F.; Draper, S.J.; Biswas, S.; Howarth, M. Plug-and-Display: Decoration of Virus-Like Particles via isopeptide bonds for modular immunization. Sci. Rep. 2016, 6, 19234. [Google Scholar] [CrossRef] [Green Version]
- Palladini, A.; Thrane, S.; Janitzek, C.M.; Pihl, J.; Clemmensen, S.B.; de Jongh, W.A.; Clausen, T.M.; Nicoletti, G.; Landuzzi, L.; Penichet, M.L.; et al. Virus-like particle display of HER2 induces potent anti-cancer responses. Oncoimmunology 2018, 7, e1408749. [Google Scholar] [CrossRef] [Green Version]
- Thrane, S.; Janitzek, C.M.; Matondo, S.; Resende, M.; Gustavsson, T.; de Jongh, W.A.; Clemmensen, S.; Roeffen, W.; van de Vegte-Bolmer, M.; van Gemert, G.J.; et al. Bacterial superglue enables easy development of efficient virus-like particle based vaccines. J. Nanobiotechnology 2016, 14, 30. [Google Scholar] [CrossRef] [Green Version]
- Abbott, R.K.; Lee, J.H.; Menis, S.; Skog, P.; Rossi, M.; Ota, T.; Kulp, D.W.; Bhullar, D.; Kalyuzhniy, O.; Havenar-Daughton, C.; et al. Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested with Germline-Targeting HIV Vaccine Immunogens. Immunity 2018, 48, 133–146.e6. [Google Scholar] [CrossRef] [Green Version]
- He, L.; de Val, N.; Morris, C.D.; Vora, N.; Thinnes, T.C.; Kong, L.; Azadnia, P.; Sok, D.; Zhou, B.; Burton, D.R.; et al. Presenting native-like trimeric HIV-1 antigens with self-assembling nanoparticles. Nat. Commun. 2016, 7, 12041. [Google Scholar] [CrossRef] [PubMed]
- McGuire, A.T.; Gray, M.D.; Dosenovic, P.; Gitlin, A.D.; Freund, N.T.; Petersen, J.; Correnti, C.; Johnsen, W.; Kegel, R.; Stuart, A.B.; et al. Specifically modified Env immunogens activate B-cell precursors of broadly neutralizing HIV-1 antibodies in transgenic mice. Nat. Commun. 2016, 7, 10618. [Google Scholar] [CrossRef] [PubMed]
- Sliepen, K.; Ozorowski, G.; Burger, J.A.; van Montfort, T.; Stunnenberg, M.; LaBranche, C.; Montefiori, D.C.; Moore, J.P.; Ward, A.B.; Sanders, R.W. Presenting native-like HIV-1 envelope trimers on ferritin nanoparticles improves their immunogenicity. Retrovirology 2015, 12, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
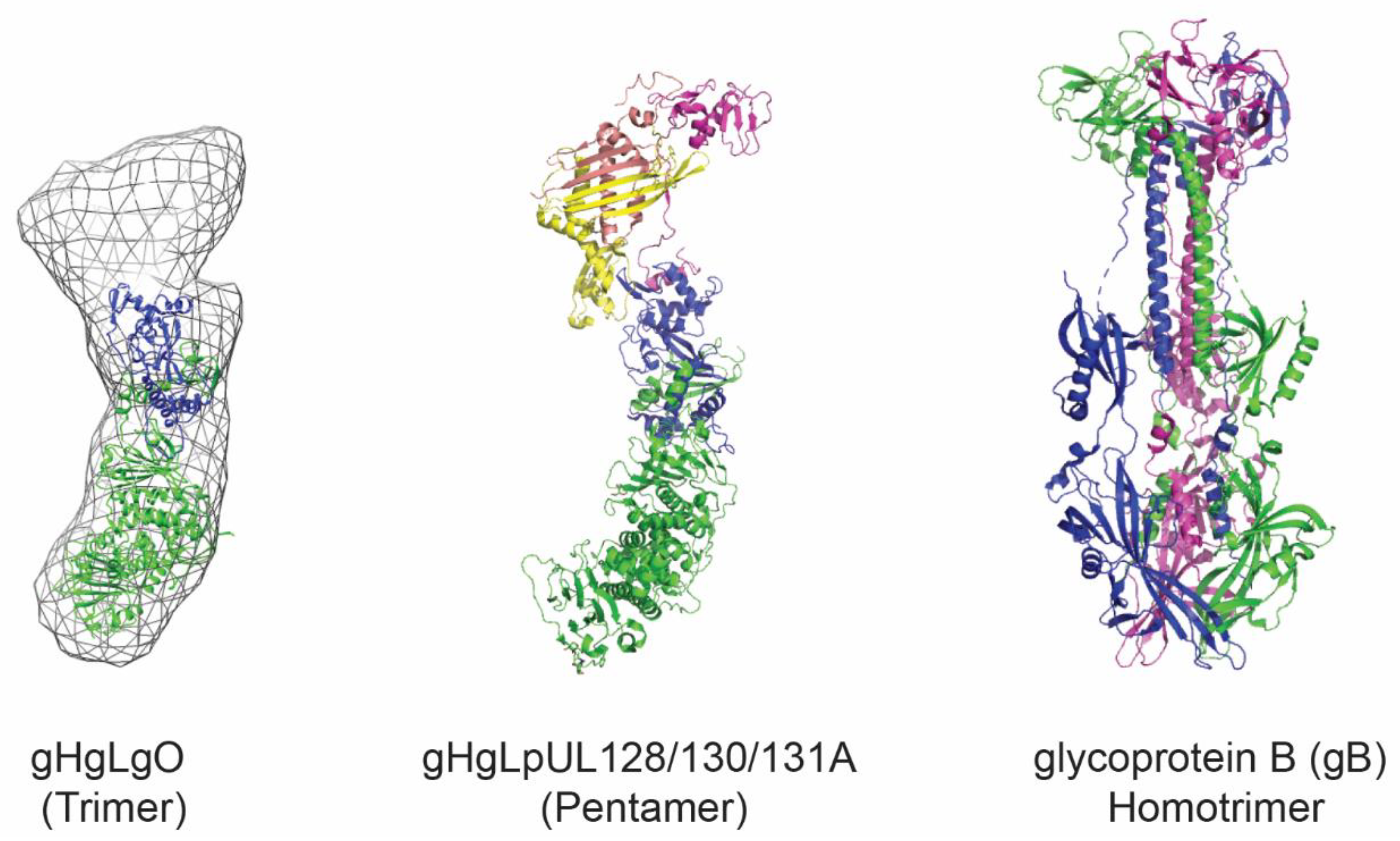
Figure 1.
Structural representation of HCMV glycoproteins. Shown is the trimer, composed of the gHgL heterodimer and gO. Representation prepared with PDB: 5voc and EMD-3391. The pentamer is composed of the gHgL heterodimer and the three additional subunits pUL128, pUL130, and pUL131A. Representation prepared with PDB: 5voc. The gB homotrimer in its postfusion conformation is shown based on PDB: 5cxf. The figure was prepared using PyMOL software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).
Figure 1.
Structural representation of HCMV glycoproteins. Shown is the trimer, composed of the gHgL heterodimer and gO. Representation prepared with PDB: 5voc and EMD-3391. The pentamer is composed of the gHgL heterodimer and the three additional subunits pUL128, pUL130, and pUL131A. Representation prepared with PDB: 5voc. The gB homotrimer in its postfusion conformation is shown based on PDB: 5cxf. The figure was prepared using PyMOL software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).

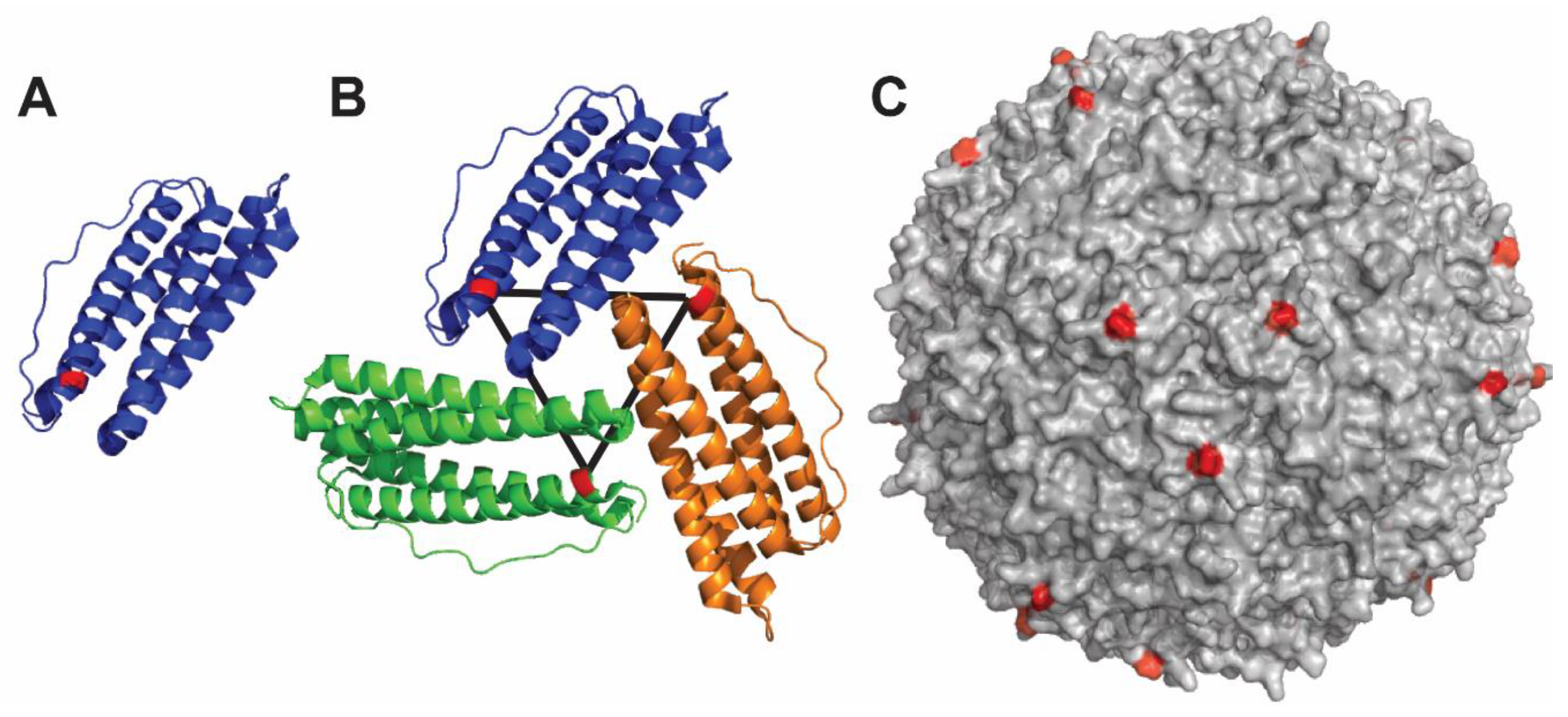
Figure 2.
Hepatitis B core antigen and relative nanoparticle. (A) Polypeptide fold and structure of HBcAg monomer. The antigen of interest needs to be inserted in the MIR loop connecting alpha helices 3 and 4. (B) Two HBcAg monomers spontaneously interact to form a dimer, which acts as an intermediate in the nanoparticle formation. (C) Assembled nanoparticle containing either 90 or 120 dimers. The figure was prepared with PDB: 6htx using Pymol software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).
Figure 2.
Hepatitis B core antigen and relative nanoparticle. (A) Polypeptide fold and structure of HBcAg monomer. The antigen of interest needs to be inserted in the MIR loop connecting alpha helices 3 and 4. (B) Two HBcAg monomers spontaneously interact to form a dimer, which acts as an intermediate in the nanoparticle formation. (C) Assembled nanoparticle containing either 90 or 120 dimers. The figure was prepared with PDB: 6htx using Pymol software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).

Figure 3.
Structure of ferritin nanoparticle. (A) The antigen of interest is fused to the N-terminus of the ferritin polypeptide (shown in red). (B) Ferritin assembles to form symmetry units with threefold axes. (C) Fully assembled nanoparticle with surface-exposed N-termini colored in red. The figure was prepared using PDB: 3bve using PyMOL software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).
Figure 3.
Structure of ferritin nanoparticle. (A) The antigen of interest is fused to the N-terminus of the ferritin polypeptide (shown in red). (B) Ferritin assembles to form symmetry units with threefold axes. (C) Fully assembled nanoparticle with surface-exposed N-termini colored in red. The figure was prepared using PDB: 3bve using PyMOL software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).

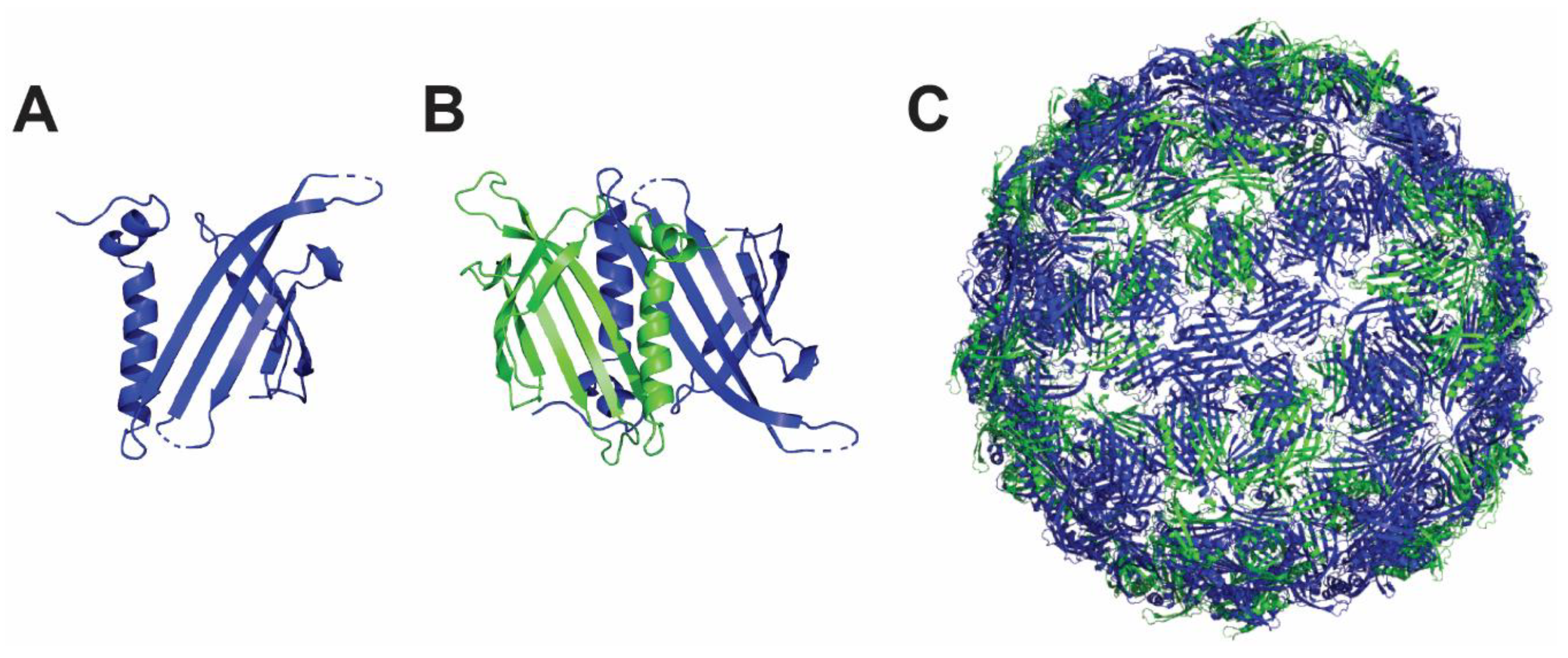
Figure 4.
Structure and assembly of Qβ nanoparticle. Qβ protein is depicted as a monomer (A) and noncovalent dimer (B). (C) The fully self-assembled Qβ nanoparticle consists of 20 hexamers and 12 pentamers. The figure was prepared using 1qbe PDB structure and PyMOL software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).
Figure 4.
Structure and assembly of Qβ nanoparticle. Qβ protein is depicted as a monomer (A) and noncovalent dimer (B). (C) The fully self-assembled Qβ nanoparticle consists of 20 hexamers and 12 pentamers. The figure was prepared using 1qbe PDB structure and PyMOL software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).

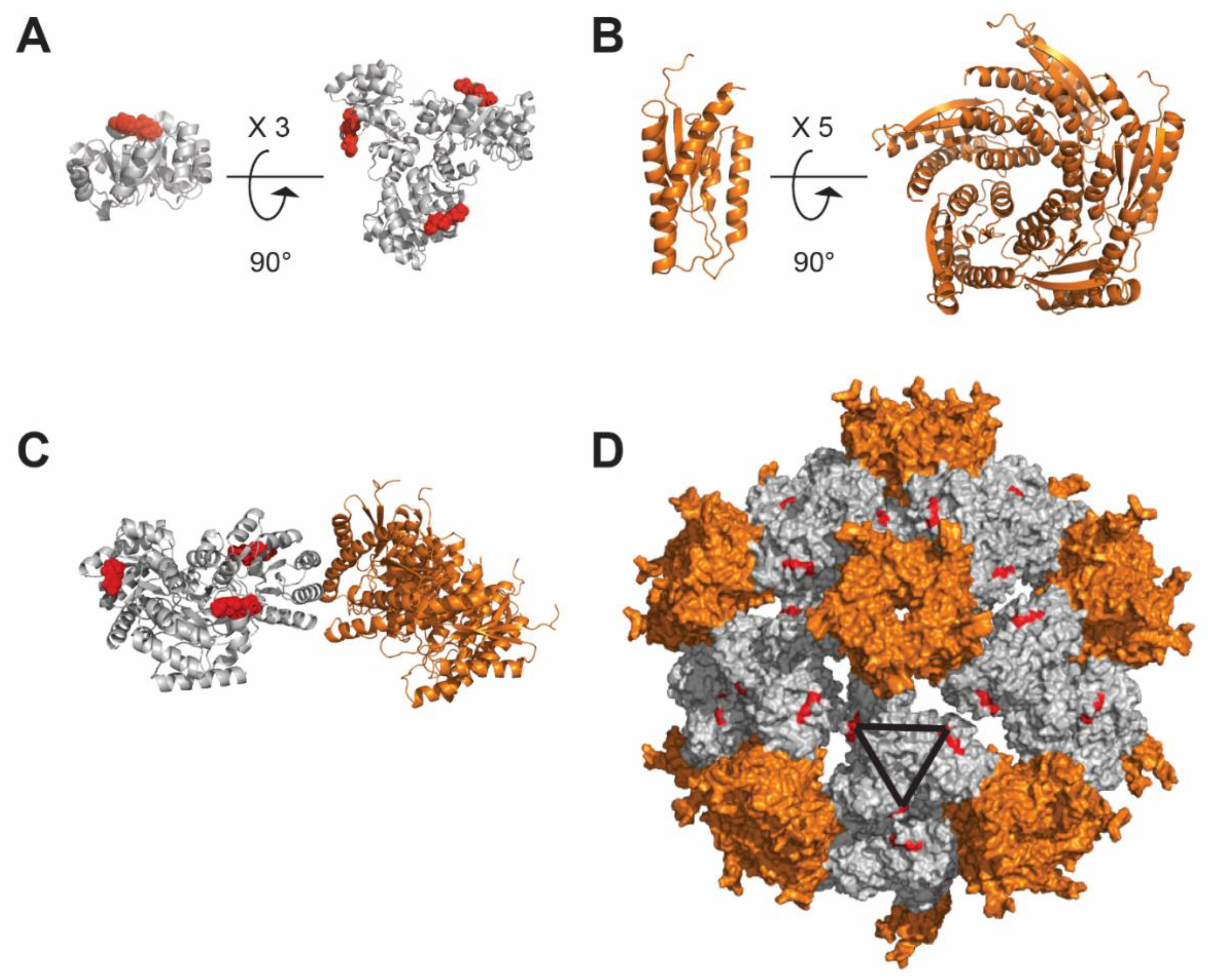
Figure 5.
Self-assembly of the I5350 nanoparticle. (A) Polypeptide I53–50A.1NT1; shown in red is the N-terminus projecting outward, where antigens can be displayed. I53–50A.1NT1 naturally trimerizes forming a 3-fold symmetry axis. (B) The I53–50B.4PT1 protein (orange) assembles into a pentamer. (C) I53–50A.1NT1 trimer and I53–50B.4PT1 pentamer self-assemble as a nanoparticle. (D) Assembled nanoparticle formed by 20 trimers and 12 pentamers, the N-termini of I53–50A.1NT1 is shown in red, and the black triangle represents the 3-fold symmetry axis of each pair of trimers. The figure was prepared using PyMOL software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).
Figure 5.
Self-assembly of the I5350 nanoparticle. (A) Polypeptide I53–50A.1NT1; shown in red is the N-terminus projecting outward, where antigens can be displayed. I53–50A.1NT1 naturally trimerizes forming a 3-fold symmetry axis. (B) The I53–50B.4PT1 protein (orange) assembles into a pentamer. (C) I53–50A.1NT1 trimer and I53–50B.4PT1 pentamer self-assemble as a nanoparticle. (D) Assembled nanoparticle formed by 20 trimers and 12 pentamers, the N-termini of I53–50A.1NT1 is shown in red, and the black triangle represents the 3-fold symmetry axis of each pair of trimers. The figure was prepared using PyMOL software (The PyMOL Molecular Graphics System, Version 4.5 Schrödinger, LLC).

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Perotti, M.; Perez, L. Virus-Like Particles and Nanoparticles for Vaccine Development against HCMV. Viruses 2020, 12, 35. https://0-doi-org.brum.beds.ac.uk/10.3390/v12010035
AMA Style
Perotti M, Perez L. Virus-Like Particles and Nanoparticles for Vaccine Development against HCMV. Viruses. 2020; 12(1):35. https://0-doi-org.brum.beds.ac.uk/10.3390/v12010035
Chicago/Turabian StylePerotti, Michela, and Laurent Perez. 2020. "Virus-Like Particles and Nanoparticles for Vaccine Development against HCMV" Viruses 12, no. 1: 35. https://0-doi-org.brum.beds.ac.uk/10.3390/v12010035
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.