Identification of Modulators of HIV-1 Proviral Transcription from a Library of FDA-Approved Pharmaceuticals

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Isolation and Culture of Peripheral Blood Mononuclear Cells (PBMCs)
2.3. Transfections and Luciferase Assay for Initial Drug Screen
2.4. Cell Viability Assay
2.5. FDA-Approved Drugs and Other Chemicals
2.6. Protein Extracts and Immunoblotting
2.7. RNA Extraction, RT-PCR and qRT-PCR
2.8. Jurkat E4 GFP Analysis by Plate Reader and Flow Cytometry
2.9. ChIP Assay
3. Results
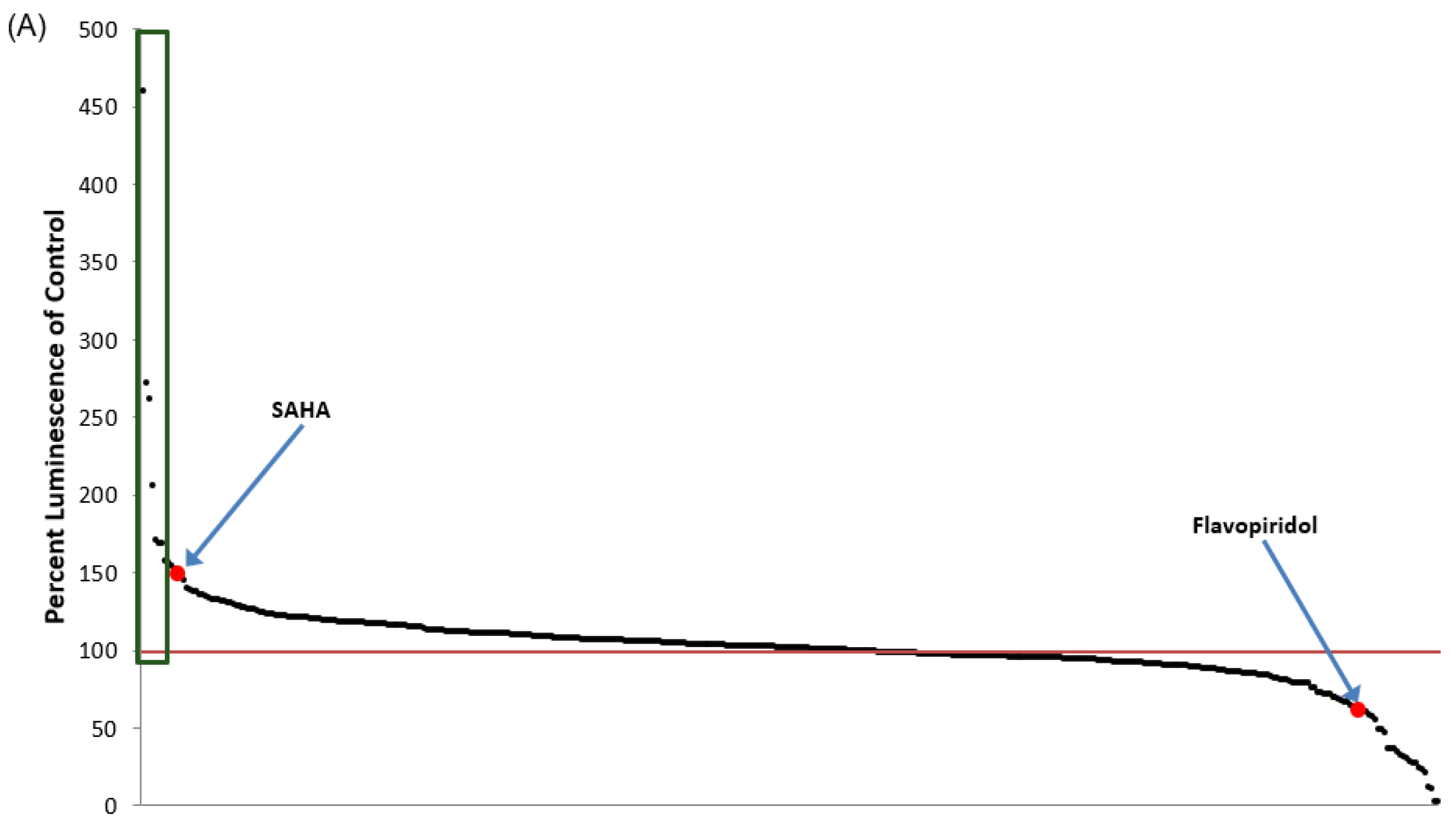
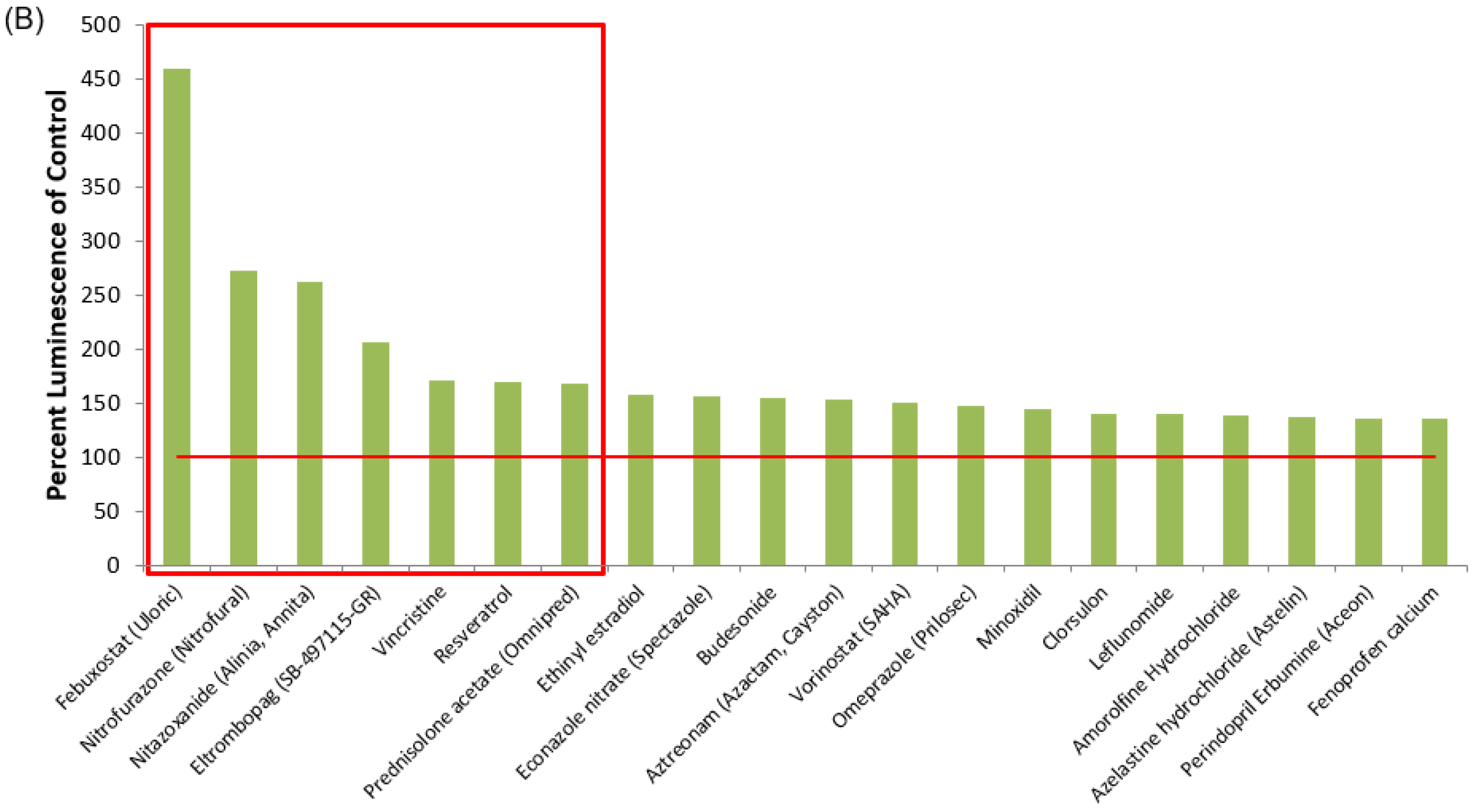
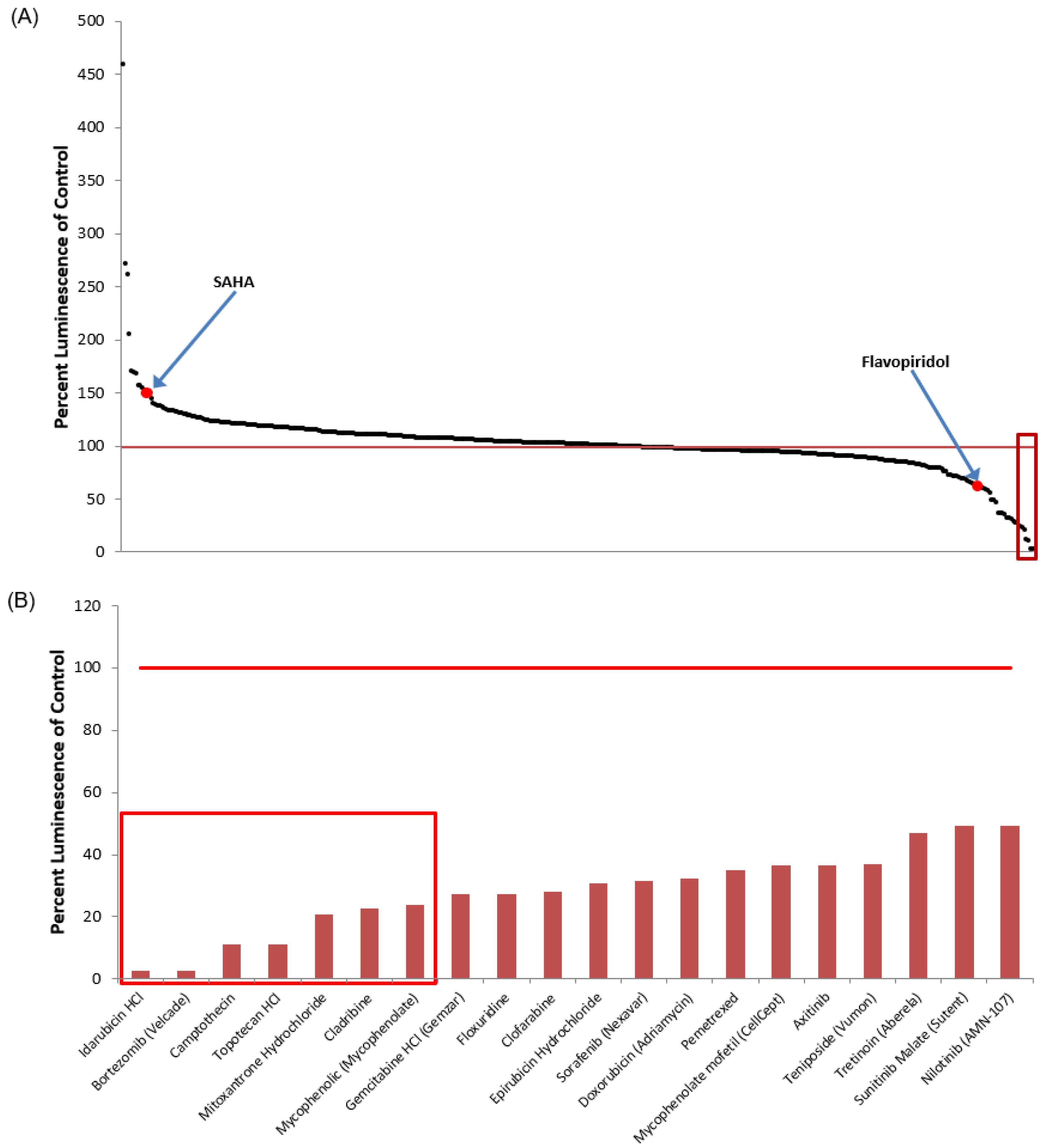
3.1. Identification of Lead HIV-1 Transcriptional Activators from TZM-bl Screen of FDA-Approved Drugs
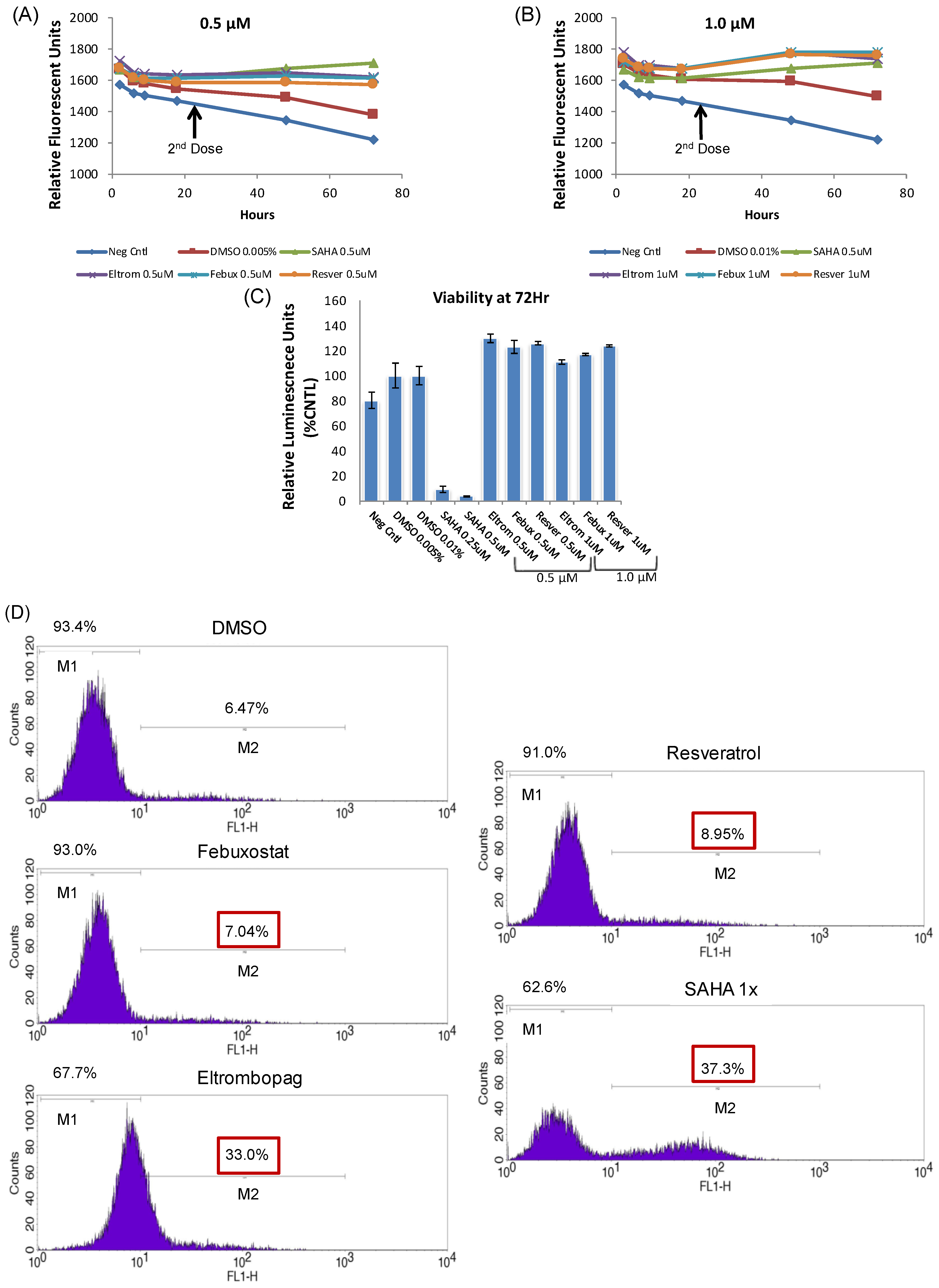
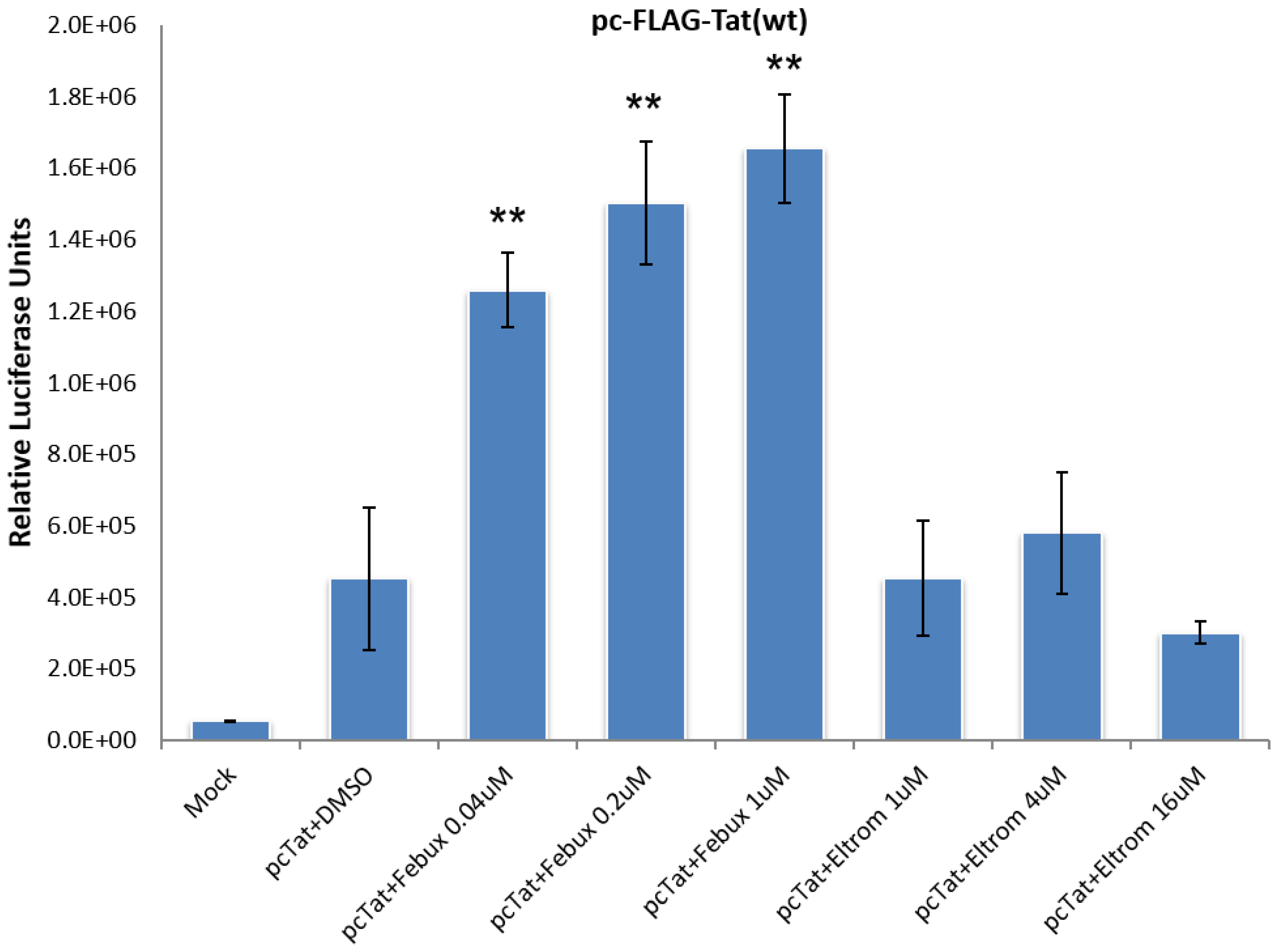
3.2. Lead HIV-1 Activator pc-Luciferase and Toxicity Screens
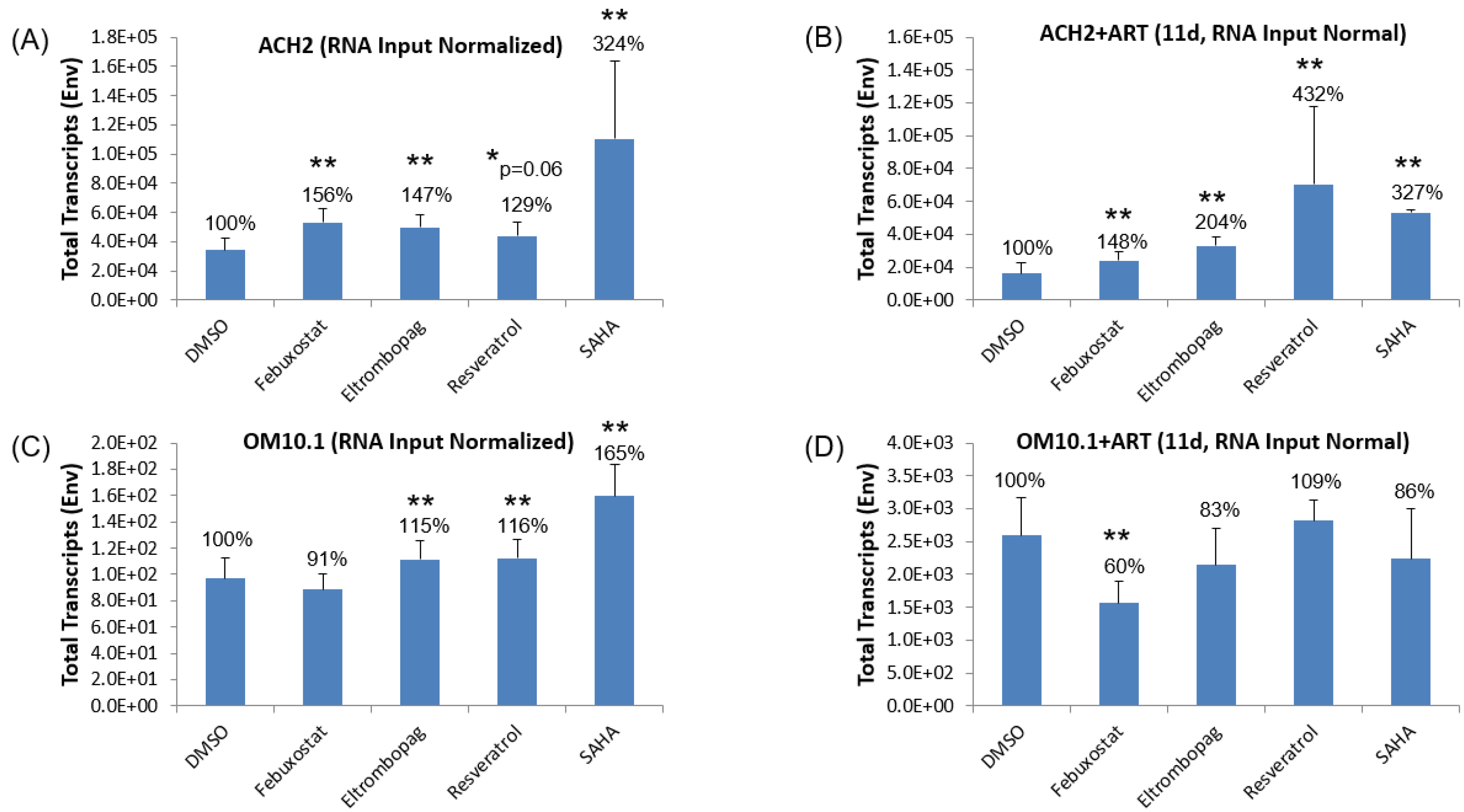
3.3. Testing Lead Transcriptional Activators in Latently HIV-1-Infected Cell Lines
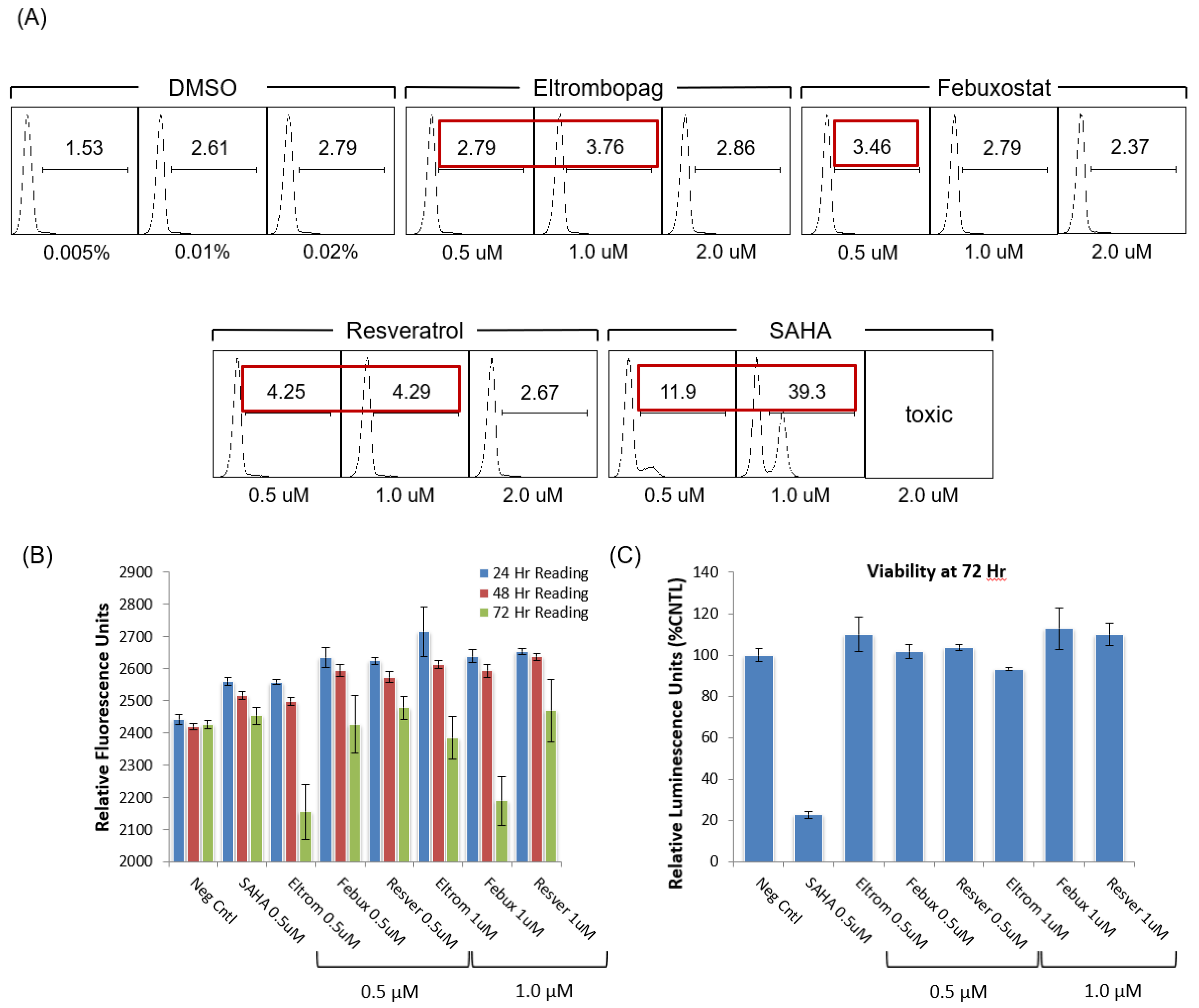
3.4. Testing Jurkat E4 Latent T Cell Model with Single and Multi-Dose Regimens of Lead HIV-1 Transcriptional Activators
3.5. Identification of Lead HIV-1 Transcriptional Inhibitors from TZM-bl Screen of FDA-Approved Drugs
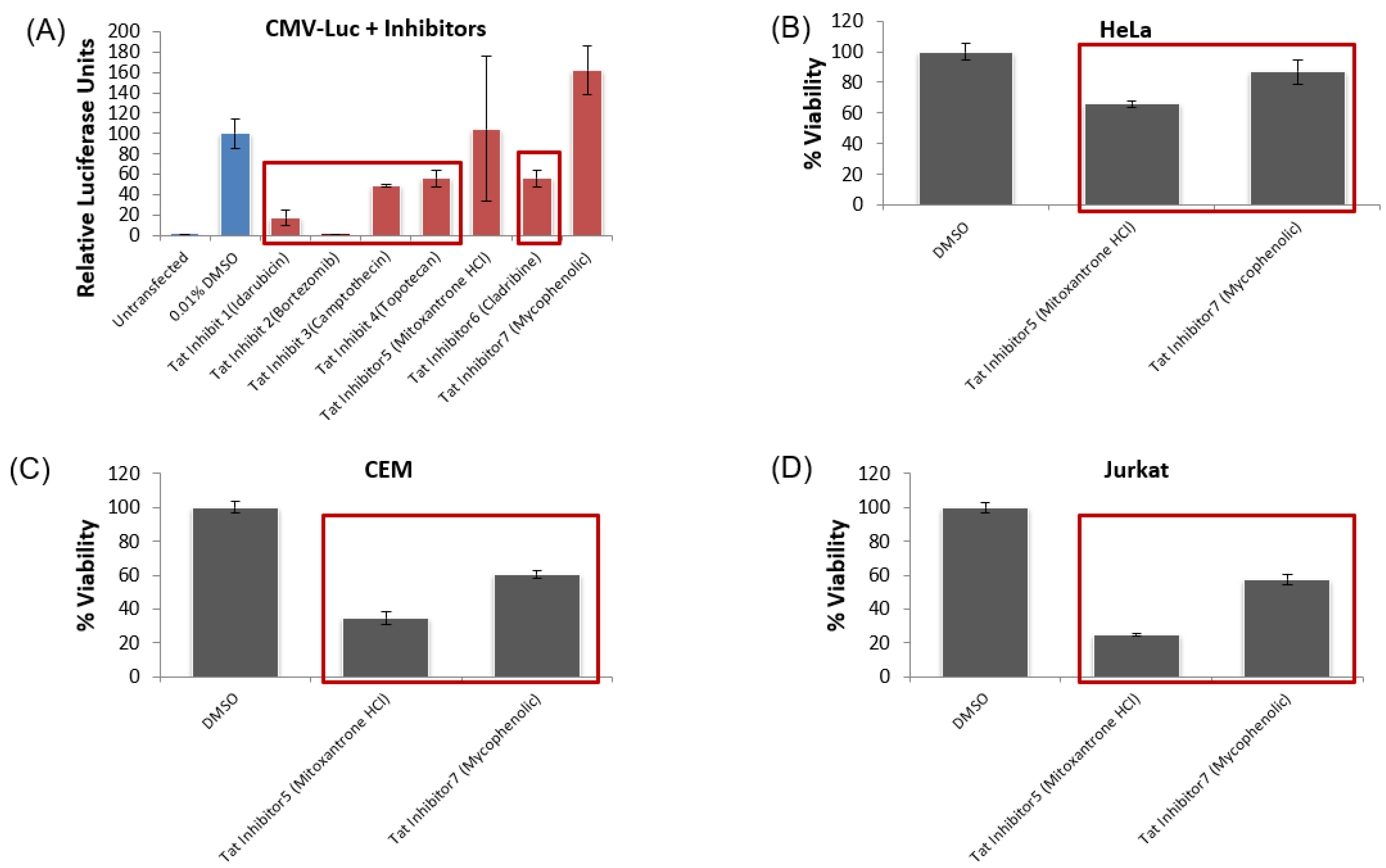
3.6. Lead HIV-1 Inhibitor pc-Luciferase and Toxicity Screens
3.7. Testing Lead Transcriptional Inhibitors in Chronically HIV-1-Infected Cell Lines
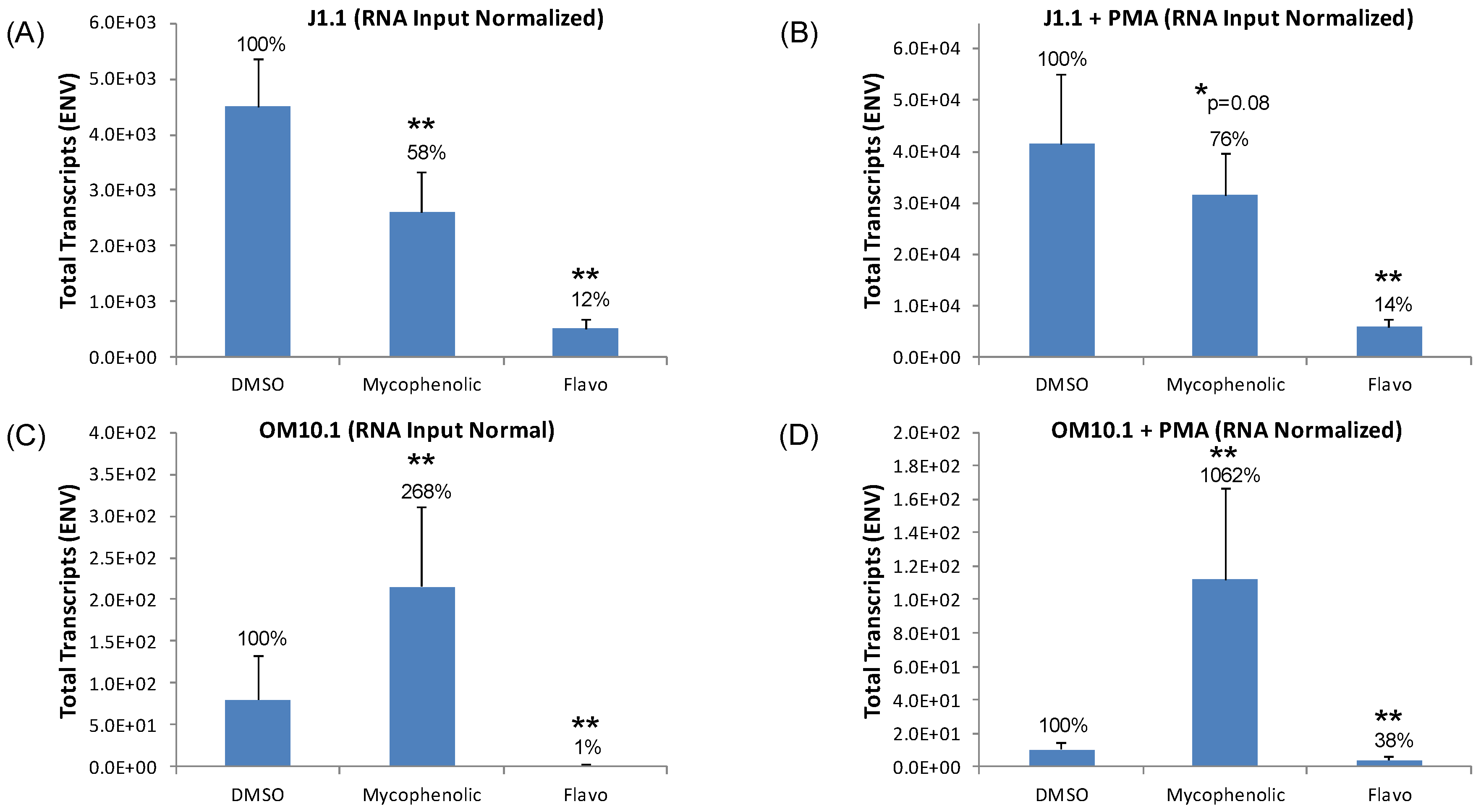
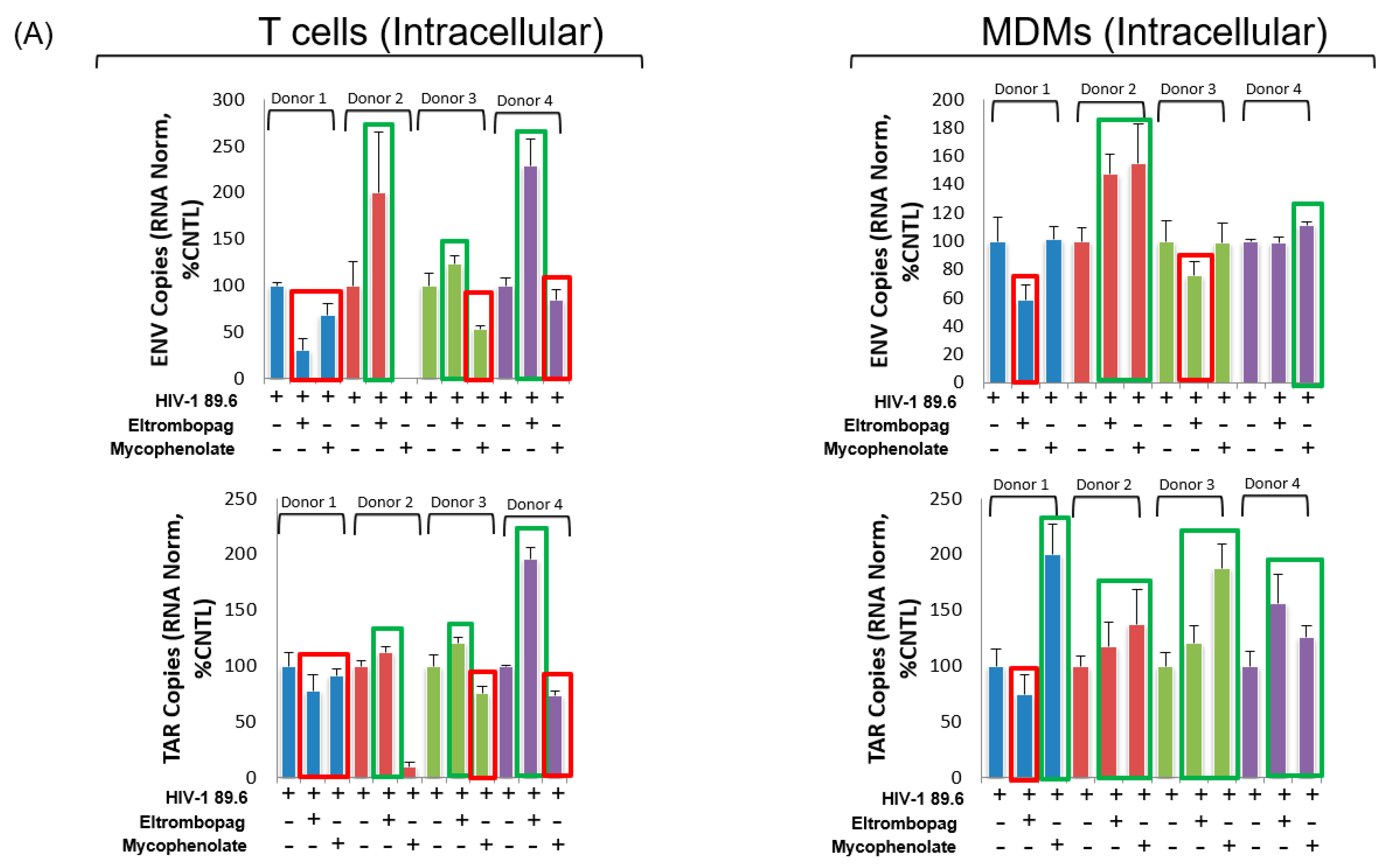
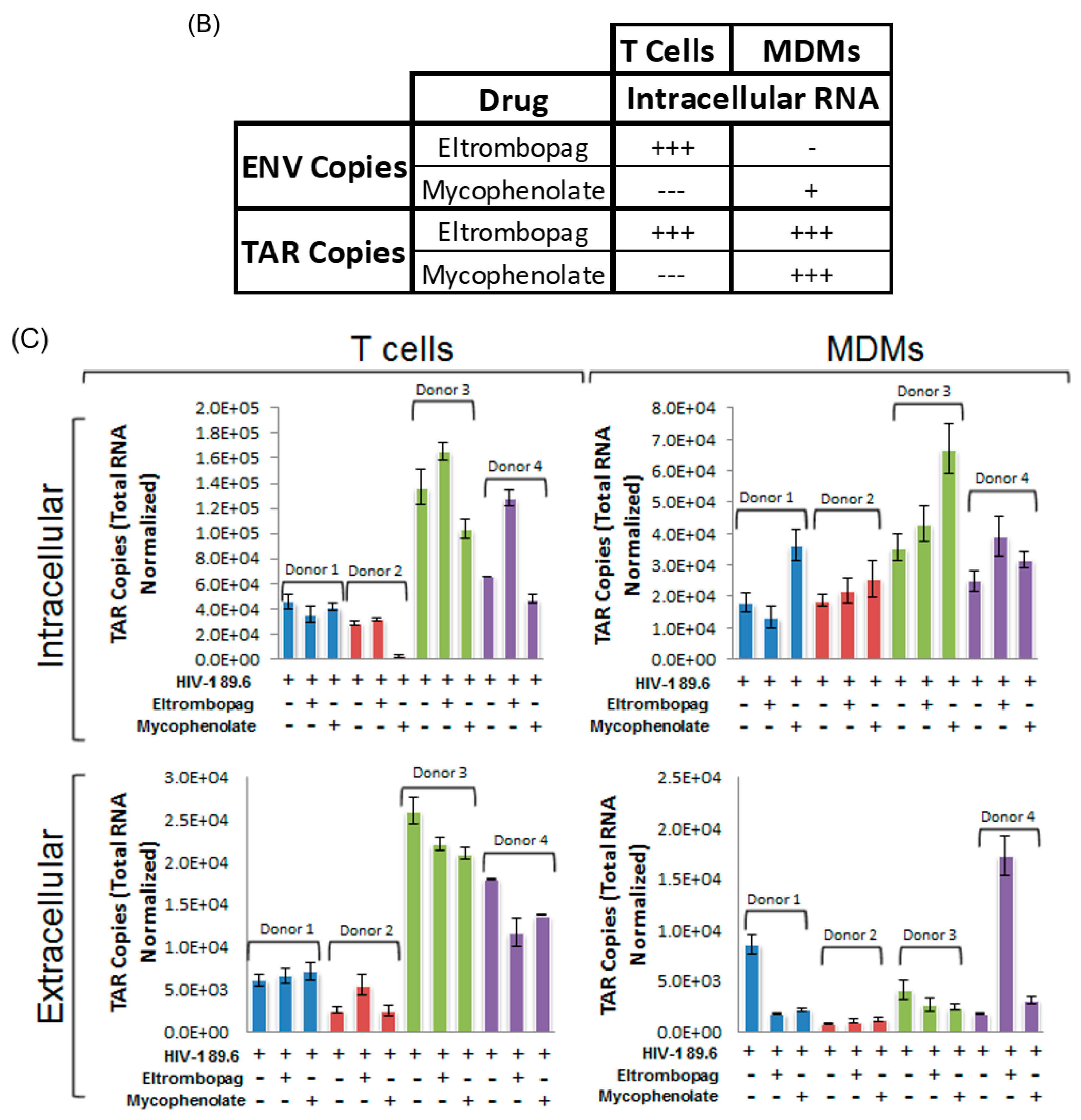
3.8. Eltrombopag and Mycophenolate Treatment of Infected T Cells and Macrophages
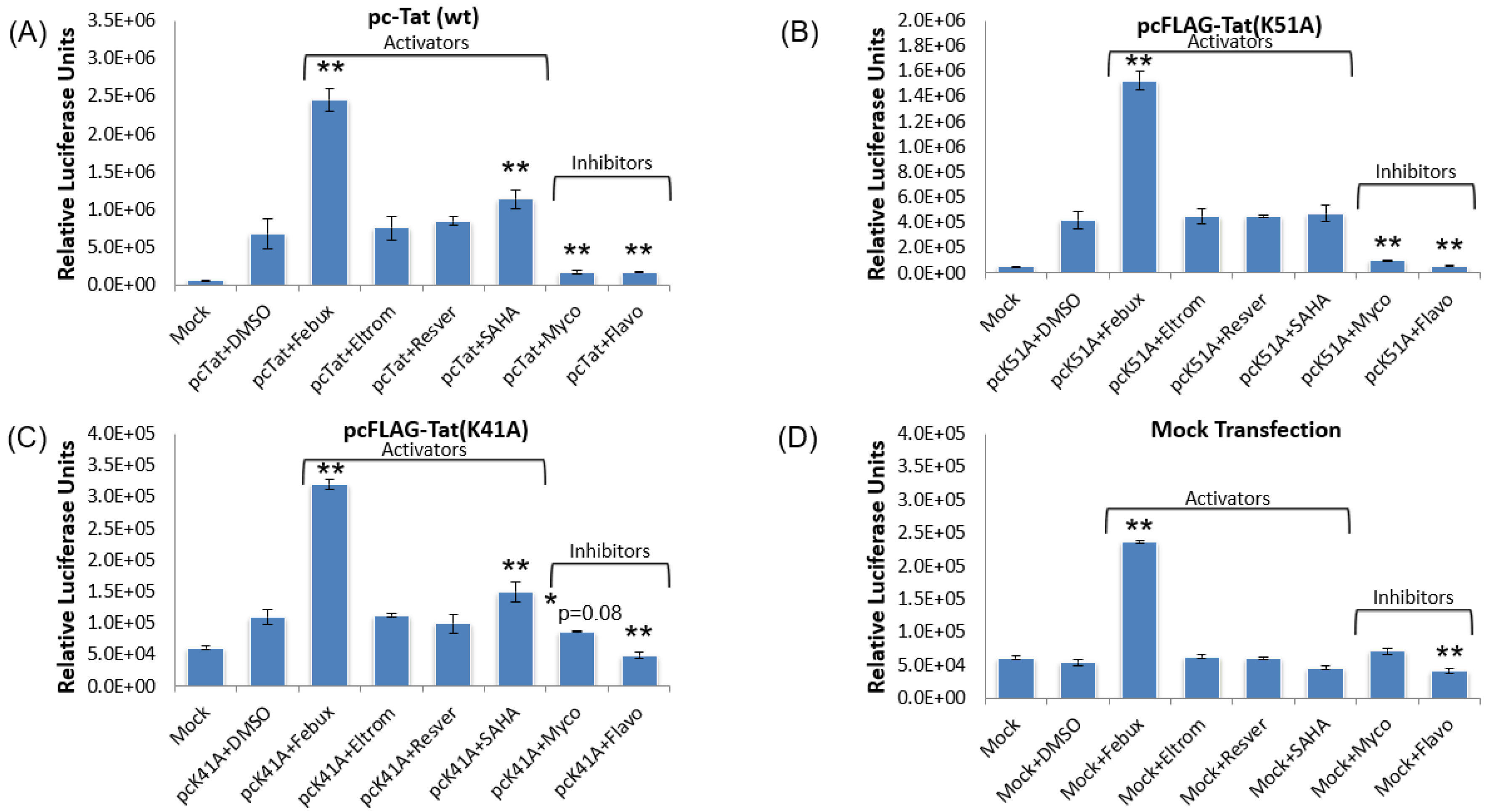
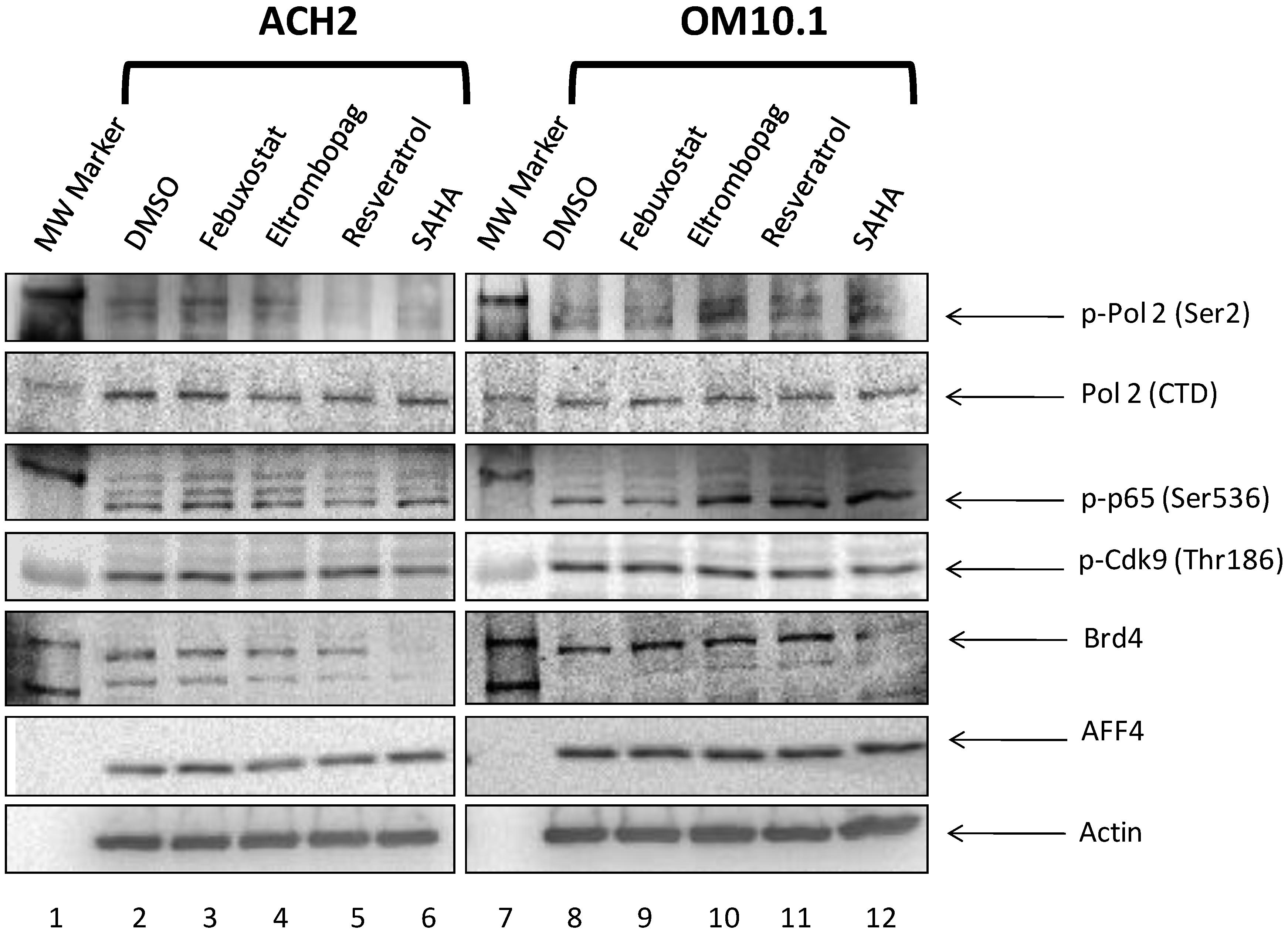
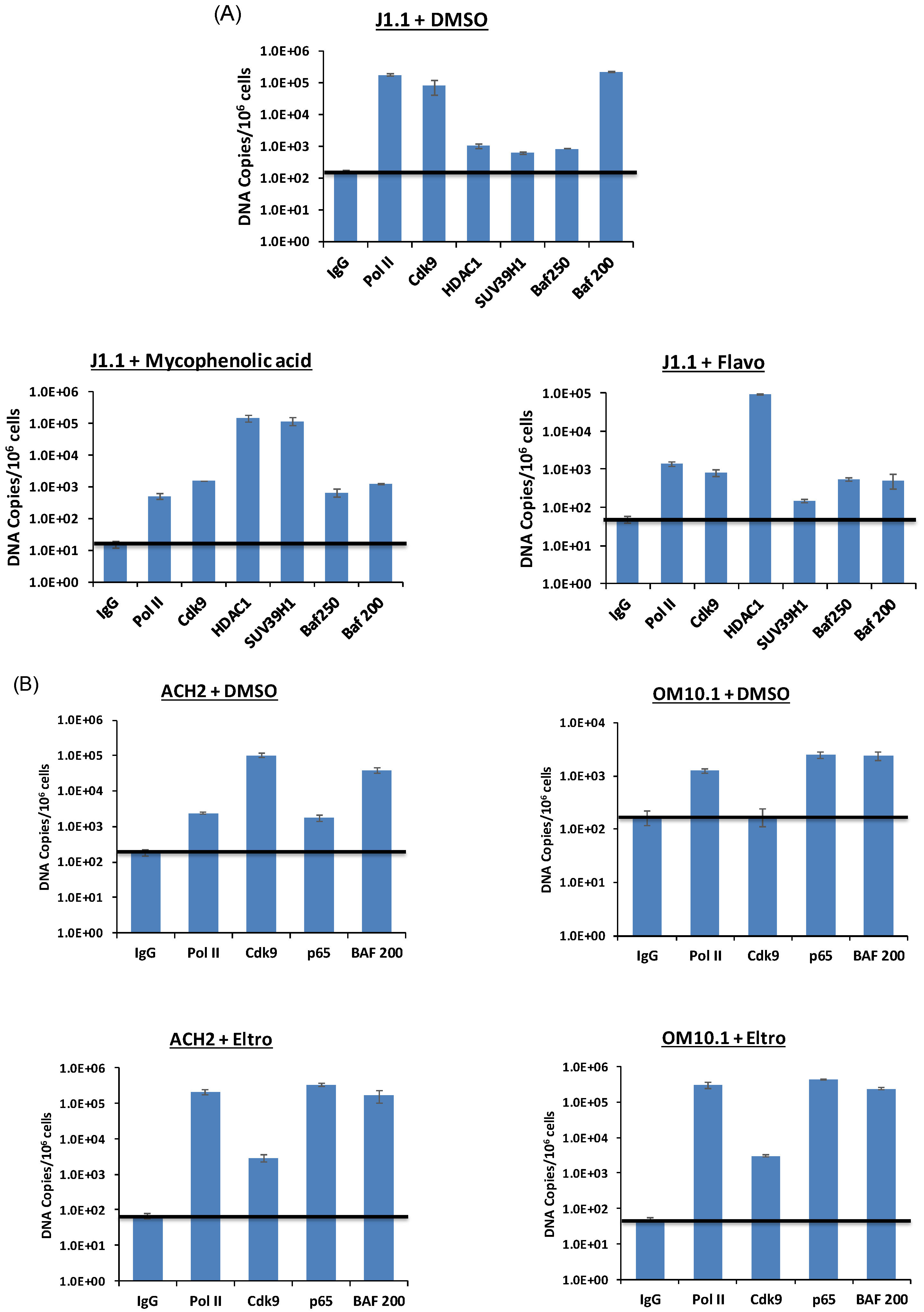
3.9. Elucidation of Mechanism of Action of Lead HIV-1 Activators and Inhibitors
3.10. Levels of Transcription Factors and Occupancy on the HIV-1 Promoter
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Joint United Nations Programme on HIV/AIDS. United Nations Global Report: UNAIDS Report on the Global AIDS Epidemic: 2012; UNAIDS: Geneva, Switzerland, 2012; ISBN 978-92-9173-996-7. [Google Scholar]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- US. Federal Funding for HIV/AIDS. The President’s FY 2016 Budget Request; Health and Human Services: Washington, DC, USA, 2016. [Google Scholar]
- Coquet, I.; Pavie, J.; Palmer, P.; Barbier, F.; Legriel, S.; Mayaux, J.; Molina, J.M.; Schlemmer, B.; Azoulay, E. Survival trends in critically ill HIV-infected patients in the highly active antiretroviral therapy era. Crit. Care 2010, 14, R107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocroft, A.; Ledergerber, B.; Katlama, C.; Kirk, O.; Reiss, P.; d’Arminio Monforte, A.; Knysz, B.; Dietrich, M.; Phillips, A.N.; Lundgren, J.D.; et al. Decline in the AIDS and death rates in the EuroSIDA study: An observational study. Lancet 2003, 362, 22–29. [Google Scholar] [CrossRef]
- Cohen, C.J.; Meyers, J.L.; Davis, K.L. Association between daily antiretroviral pill burden and treatment adherence, hospitalisation risk, and other healthcare utilisation and costs in a US medicaid population with HIV. BMJ Open 2013, 3, e003028. [Google Scholar] [CrossRef] [Green Version]
- Parienti, J.-J.; Bangsberg, D.R.; Verdon, R.; Gardner, E.M. Better adherence with once-daily antiretroviral regimens: A meta-analysis. Clin. Infect. Dis. 2009, 48, 484–488. [Google Scholar] [CrossRef]
- Llibre, J.M.; Clotet, B. Once-daily single-tablet regimens: A long and winding road to excellence in antiretroviral treatment. AIDS Rev. 2012, 14, 168–178. [Google Scholar] [PubMed]
- Bulteel, N.; Bansi-Matharu, L.; Churchill, D.; Dunn, D.; Bibby, D.; Hill, T.; Sabin, C.; Nelson, M.; UK Collaborative HIV Cohort (CHIC) Study Group; UK HIV Drug Resistance Database (UK HDRD) Study Group. The emergence of drug resistant HIV variants at virological failure of HAART combinations containing efavirenz, tenofovir and lamivudine or emtricitabine within the UK Collaborative HIV Cohort. J. Infect. 2014, 68, 77–84. [Google Scholar] [CrossRef]
- Margot, N.A.; Enejosa, J.; Cheng, A.K.; Miller, M.D.; McColl, D.J.; Study 934 Team. Development of HIV-1 drug resistance through 144 weeks in antiretroviral-naïve subjects on emtricitabine, tenofovir disoproxil fumarate, and efavirenz compared with lamivudine/zidovudine and efavirenz in study GS-01-934. J. Acquir. Immune Defic. Syndr. 2009, 52, 209–221. [Google Scholar] [CrossRef]
- Marcelin, A.G.; Charpentier, C.; Wirden, M.; Landman, R.; Valantin, M.A.; Simon, A.; Katlama, C.; Yeni, P.; Descamps, D.; Aubron-Olivier, C.; et al. Resistance profiles of emtricitabine and lamivudine in tenofovir-containing regimens. J. Antimicrob. Chemother. 2012, 67, 1475–1478. [Google Scholar] [CrossRef] [Green Version]
- Cadosch, D.; Bonhoeffer, S.; Kouyos, R. Assessing the impact of adherence to anti-retroviral therapy on treatment failure and resistance evolution in HIV. J. R. Soc. Interface 2012, 9, 2309–2320. [Google Scholar] [CrossRef] [Green Version]
- Ciccarelli, N.; Fabbiani, M.; Colafigli, M.; Trecarichi, E.M.; Silveri, M.C.; Cauda, R.; Murri, R.; De Luca, A.; Di Giambenedetto, S. Revised central nervous system neuropenetration-effectiveness score is associated with cognitive disorders in HIV-infected patients with controlled plasma viraemia. Antivir. Ther. 2013, 18, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [Green Version]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Zhu, T.; Muthui, D.; Holte, S.; Nickle, D.; Feng, F.; Brodie, S.; Hwangbo, Y.; Mullins, J.I.; Corey, L. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J. Virol. 2002, 76, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Sundstrom, J.B.; Ellis, J.E.; Hair, G.A.; Kirshenbaum, A.S.; Metcalfe, D.D.; Yi, H.; Cardona, A.C.; Lindsay, M.K.; Ansari, A.A. Human tissue mast cells are an inducible reservoir of persistent HIV infection. Blood 2007, 109, 5293–5300. [Google Scholar] [CrossRef]
- Loré, K.; Smed-Sörensen, A.; Vasudevan, J.; Mascola, J.R.; Koup, R.A. Myeloid and plasmacytoid dendritic cells transfer HIV-1 preferentially to antigen-specific CD4+ T cells. J. Exp. Med. 2005, 201, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.A.; Cherry, C.L.; Bell, J.E.; McLean, C.A. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am. J. Pathol. 2011, 179, 1623–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.; Zhang, H.; Lopez, P.; Pardo, C.A.; Gartner, S. In vitro modeling of the HIV-macrophage reservoir. J. Leukoc. Biol. 2006, 80, 1127–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Garcia, J.V. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.V.; Staskus, K.; Wietgrefe, S.W.; Rothenberger, M.; Reilly, C.; Chipman, J.G.; Beilman, G.J.; Khoruts, A.; Thorkelson, A.; Schmidt, T.E.; et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Ramratnam, B.; Mittler, J.E.; Zhang, L.; Boden, D.; Hurley, A.; Fang, F.; Macken, C.A.; Perelson, A.S.; Markowitz, M.; Ho, D.D. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 2000, 6, 82–85. [Google Scholar] [CrossRef]
- Tanaka, J.; Ozaki, H.; Yasuda, J.; Horai, R.; Tagawa, Y.; Asano, M.; Saijo, S.; Imai, M.; Sekikawa, K.; Kopf, M.; et al. Lipopolysaccharide-induced HIV-1 expression in transgenic mice is mediated by tumor necrosis factor-alpha and interleukin-1, but not by interferon-gamma nor interleukin-6. AIDS 2000, 14, 1299–1307. [Google Scholar] [CrossRef]
- Finnegan, A.; Roebuck, K.A.; Nakai, B.E.; Gu, D.S.; Rabbi, M.F.; Song, S.; Landay, A.L. IL-10 cooperates with TNF-alpha to activate HIV-1 from latently and acutely infected cells of monocyte/macrophage lineage. J. Immunol. 1996, 156, 841–851. [Google Scholar]
- Warfel, A.H.; Thorbecke, G.J.; Belsito, D.V. Synergism between interferon-gamma and cytokines or lipopolysaccharide in the activation of the HIV-LTR in macrophages. J. Leukoc. Biol. 1995, 57, 469–476. [Google Scholar] [CrossRef]
- Kim, Y.K.; Mbonye, U.; Hokello, J.; Karn, J. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J. Mol. Biol. 2011, 410, 896–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.A.; Kwon, H.; Chen, L.-F.; Greene, W.C. Sustained Induction of NF-κB Is Required for Efficient Expression of Latent Human Immunodeficiency Virus Type 1. J. Virol. 2007, 81, 6043–6056. [Google Scholar] [CrossRef] [Green Version]
- Rosen, C.A.; Sodroski, J.G.; Haseltine, W.A. The location of cis-acting regulatory sequences in the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell 1985, 41, 813–823. [Google Scholar] [CrossRef]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Salemi, M.; Rife, B. Phylogenetics and Phyloanatomy of HIV/SIV Intra-Host Compartments and Reservoirs: The Key Role of the Central Nervous System. Curr. HIV Res. 2016, 14, 110–120. [Google Scholar] [CrossRef]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [CrossRef]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.A.; Schmeltz Søgaard, O.; Brinkmann, C.; Wightman, F.; Lewin, S.R.; Melchjorsen, J.; Dinarello, C.; Østergaard, L.; Tolstrup, M. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Hum. Vaccin. Immunother. 2013, 9, 993–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.-L.; Spector, S.A. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J. Biol. Chem. 2015, 290, 5028–5040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, L.M.; Brannan, J.M.; Delos, S.E.; Shoemaker, C.J.; Stossel, A.; Lear, C.; Hoffstrom, B.G.; Dewald, L.E.; Schornberg, K.L.; Scully, C.; et al. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci. Transl. Med. 2013, 5, 190ra79. [Google Scholar] [CrossRef] [Green Version]
- Madrid, P.B.; Chopra, S.; Manger, I.D.; Gilfillan, L.; Keepers, T.R.; Shurtleff, A.C.; Green, C.E.; Iyer, L.V.; Dilks, H.H.; Davey, R.A.; et al. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS ONE 2013, 8, e60579. [Google Scholar] [CrossRef] [Green Version]
- De Wilde, A.H.; Jochmans, D.; Posthuma, C.C.; Zevenhoven-Dobbe, J.C.; van Nieuwkoop, S.; Bestebroer, T.M.; van den Hoogen, B.G.; Neyts, J.; Snijder, E.J. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob. Agents Chemother. 2014, 58, 4875–4884. [Google Scholar] [CrossRef] [Green Version]
- Platt, E.J.; Bilska, M.; Kozak, S.L.; Kabat, D.; Montefiori, D.C. Evidence that Ecotropic Murine Leukemia Virus Contamination in TZM-bl Cells Does Not Affect the Outcome of Neutralizing Antibody Assays with Human Immunodeficiency Virus Type 1. J. Virol. 2009, 83, 8289–8292. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, Y.; McClure, M.O.; Pizzato, M. Identification of Gammaretroviruses Constitutively Released from Cell Lines Used for Human Immunodeficiency Virus Research. J. Virol. 2008, 82, 12585–12588. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of Resistant Human Immunodeficiency Virus Type 1 in Patients Receiving Fusion Inhibitor (T-20) Monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Wu, X.; O’Brien, W.A.; Ratner, L.; Kappes, J.C.; Shaw, G.M.; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 2000, 74, 8358–8367. [Google Scholar] [CrossRef] [Green Version]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef] [Green Version]
- Perez, V.L.; Rowe, T.; Justement, J.S.; Butera, S.T.; June, C.H.; Folks, T.M. An HIV-1-infected T cell clone defective in IL-2 production and Ca2+ mobilization after CD3 stimulation. J. Immunol. 1991, 147, 3145–3148. [Google Scholar]
- Clouse, K.A.; Powell, D.; Washington, I.; Poli, G.; Strebel, K.; Farrar, W.; Barstad, P.; Kovacs, J.; Fauci, A.S.; Folks, T.M. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J. Immunol. 1989, 142, 431–438. [Google Scholar] [PubMed]
- Butera, S.T.; Perez, V.L.; Wu, B.Y.; Nabel, G.J.; Folks, T.M. Oscillation of the human immunodeficiency virus surface receptor is regulated by the state of viral activation in a CD4+ cell model of chronic infection. J. Virol. 1991, 65, 4645–4653. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Safran, M.; Kim, W.Y.; O’Connell, F.; Flippin, L.; Günzler, V.; Horner, J.W.; Depinho, R.A.; Kaelin, W.G. Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: Assessment of an oral agent that stimulates erythropoietin production. Proc. Natl. Acad. Sci. USA 2006, 103, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Cujec, T.P.; Okamoto, H.; Fujinaga, K.; Meyer, J.; Chamberlin, H.; Morgan, D.O.; Peterlin, B.M. The HIV transactivator TAT binds to the CDK-activating kinase and activates the phosphorylation of the carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997, 11, 2645–2657. [Google Scholar] [CrossRef] [Green Version]
- Fridell, R.A.; Harding, L.S.; Bogerd, H.P.; Cullen, B.R. Identification of a novel human zinc finger protein that specifically interacts with the activation domain of lentiviral Tat proteins. Virology 1995, 209, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D.; Grdina, T.A.; Bogerd, H.P.; Cullen, B.R. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998, 17, 7056–7065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, Y.; Reza, S.M.; Young, T.M.; Mathews, M.B.; Pe’ery, T. Human and rodent transcription elongation factor P-TEFb: Interactions with human immunodeficiency virus type 1 tat and carboxy-terminal domain substrate. J. Virol. 1999, 73, 5448–5458. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; de la Fuente, C.; Fu, P.; Wang, L.; Donnelly, R.; Wade, J.D.; Lambert, P.; Li, H.; Lee, C.G.; Kashanchi, F. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology 2000, 277, 278–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagans, S.; Kauder, S.E.; Kaehlcke, K.; Sakane, N.; Schroeder, S.; Dormeyer, W.; Trievel, R.C.; Verdin, E.; Schnolzer, M.; Ott, M. The Cellular lysine methyltransferase Set7/9-KMT7 binds HIV-1 TAR RNA, monomethylates the viral transactivator Tat, and enhances HIV transcription. Cell Host Microbe 2010, 7, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, A.; Iordanskiy, S.; Das, R.; van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [Green Version]
- Biglione, S.; Byers, S.A.; Price, J.P.; Nguyen, V.T.; Bensaude, O.; Price, D.H.; Maury, W. Inhibition of HIV-1 replication by P-TEFb inhibitors DRB, seliciclib and flavopiridol correlates with release of free P-TEFb from the large, inactive form of the complex. Retrovirology 2007, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.J.; Sweeney, B.; Mainwaring, D.O.; James, D.C. NF-κB, CRE and YY1 elements are key functional regulators of CMV promoter-driven transient gene expression in CHO cells. Biotechnol. J. 2015, 10, 1019–1028. [Google Scholar] [CrossRef]
- Lee, Y.; Sohn, W.-J.; Kim, D.-S.; Kwon, H.-J. NF-kappaB- and c-Jun-dependent regulation of human cytomegalovirus immediate-early gene enhancer/promoter in response to lipopolysaccharide and bacterial CpG-oligodeoxynucleotides in macrophage cell line RAW 264.7. Eur. J. Biochem. 2004, 271, 1094–1105. [Google Scholar] [CrossRef]
- Sambucetti, L.C.; Cherrington, J.M.; Wilkinson, G.W.; Mocarski, E.S. NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J. 1989, 8, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Komoriya, K.; Osada, Y.; Hasegawa, M.; Horiuchi, H.; Kondo, S.; Couch, R.C.; Griffin, T.B. Hypouricemic effect of allopurinol and the novel xanthine oxidase inhibitor TEI-6720 in chimpanzees. Eur. J. Pharmacol. 1993, 250, 455–460. [Google Scholar] [CrossRef]
- Osada, Y.; Tsuchimoto, M.; Fukushima, H.; Takahashi, K.; Kondo, S.; Hasegawa, M.; Komoriya, K. Hypouricemic effect of the novel xanthine oxidase inhibitor, TEI-6720, in rodents. Eur. J. Pharmacol. 1993, 241, 183–188. [Google Scholar] [CrossRef]
- Jenkins, J.M.; Williams, D.; Deng, Y.; Uhl, J.; Kitchen, V.; Collins, D.; Erickson-Miller, C.L. Phase 1 clinical study of eltrombopag, an oral, nonpeptide thrombopoietin receptor agonist. Blood 2007, 109, 4739–4741. [Google Scholar] [CrossRef]
- Frankel, E.N.; Waterhouse, A.L.; Kinsella, J.E. Inhibition of human LDL oxidation by resveratrol. Lancet 1993, 341, 1103–1104. [Google Scholar] [CrossRef]
- Zeng, X.; Pan, X.; Xu, X.; Lin, J.; Que, F.; Tian, Y.; Li, L.; Liu, S. Resveratrol Reactivates Latent HIV through Increasing Histone Acetylation and Activating Heat Shock Factor 1. J. Agric. Food Chem. 2017, 65, 4384–4394. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retrovir. 2009, 25, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Contreras, X.; Schweneker, M.; Chen, C.-S.; McCune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar] [CrossRef] [Green Version]
- Boocock, D.J.; Faust, G.E.S.; Patel, K.R.; Schinas, A.M.; Brown, V.A.; Ducharme, M.P.; Booth, T.D.; Crowell, J.A.; Perloff, M.; Gescher, A.J.; et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1246–1252. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-A.; Park, J.-Y. Comparison of pharmacokinetics and uric acid lowering effect between two different strength febuxostat formulations (80 mg vs. 40 mg) in healthy subjects. Int. J. Clin. Pharmacol. Ther. 2015, 53, 667–673. [Google Scholar] [CrossRef]
- Matthys, G.; Park, J.W.; McGuire, S.; Wire, M.B.; Bowen, C.; Williams, D.; Jenkins, J.; Peng, B. Clinical pharmacokinetics, platelet response, and safety of eltrombopag at supratherapeutic doses of up to 200 mg once daily in healthy volunteers. J. Clin. Pharmacol. 2011, 51, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Priel, E.; Showalter, S.D.; Blair, D.G. Inhibition of human immunodeficiency virus (HIV-1) replication in vitro by noncytotoxic doses of camptothecin, a topoisomerase I inhibitor. AIDS Res. Hum. Retrovir. 1991, 7, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Carteau, S.; Mouscadet, J.F.; Goulaouic, H.; Subra, F.; Auclair, C. Effect of Topoisomerase Inhibitors on the in Vitro HIV DNA Integration Reaction. Biochem. Biophys. Res. Commun. 1993, 192, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Hawley, T.; Spear, M.; Guo, J.; Wu, Y. Inhibition of HIV replication in vitro by clinical immunosuppressants and chemotherapeutic agents. Cell Biosci. 2013, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Allison, A.C.; Eugui, E.M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000, 47, 85–118. [Google Scholar] [CrossRef]
- Margolis, D.; Heredia, A.; Gaywee, J.; Oldach, D.; Drusano, G.; Redfield, R. Abacavir and mycophenolic acid, an inhibitor of inosine monophosphate dehydrogenase, have profound and synergistic anti-HIV activity. J. Acquir. Immune Defic. Syndr. 1999, 21, 362–370. [Google Scholar] [CrossRef]
- Hossain, M.M.; Coull, J.J.; Drusano, G.L.; Margolis, D.M. Dose proportional inhibition of HIV-1 replication by mycophenolic acid and synergistic inhibition in combination with abacavir, didanosine, and tenofovir. Antiviral Res. 2002, 55, 41–52. [Google Scholar] [CrossRef]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected Cells Stimulate Production of Pro-inflammatory Cytokines through Trans-activating Response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [Green Version]
- Guendel, I.; Iordanskiy, S.; Sampey, G.C.; Van Duyne, R.; Calvert, V.; Petricoin, E.; Saifuddin, M.; Kehn-Hall, K.; Kashanchi, F. Role of Bruton’s tyrosine kinase inhibitors in HIV-1-infected cells. J. Neurovirol. 2015, 21, 257–275. [Google Scholar] [CrossRef]
- Guendel, I.; Iordanskiy, S.; van Duyne, R.; Kehn-Hall, K.; Saifuddin, M.; Das, R.; Jaworski, E.; Sampey, G.C.; Senina, S.; Shultz, L.; et al. Novel neuroprotective GSK-3β inhibitor restricts Tat-mediated HIV-1 replication. J. Virol. 2014, 88, 1189–1208. [Google Scholar] [CrossRef] [Green Version]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Cheng, G.; Saleh, M.N.; Psaila, B.; Kovaleva, L.; Meddeb, B.; Kloczko, J.; Hassani, H.; Mayer, B.; Stone, N.L.; et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N. Engl. J. Med. 2007, 357, 2237–2247. [Google Scholar] [CrossRef] [PubMed]
- Mahady, G.B.; Pendland, S.L.; Chadwick, L.R. Resveratrol and red wine extracts inhibit the growth of CagA+ strains of Helicobacter pylori in vitro. Am. J. Gastroenterol. 2003, 98, 1440–1441. [Google Scholar] [CrossRef] [PubMed]
- Docherty, J.J.; Fu, M.M.; Stiffler, B.S.; Limperos, R.J.; Pokabla, C.M.; DeLucia, A.L. Resveratrol inhibition of herpes simplex virus replication. Antiviral Res. 1999, 43, 145–155. [Google Scholar] [CrossRef]
- Delmas, D.; Lançon, A.; Colin, D.; Jannin, B.; Latruffe, N. Resveratrol as a chemopreventive agent: A promising molecule for fighting cancer. Curr. Drug Targets 2006, 7, 423–442. [Google Scholar] [CrossRef]
- Yao, L.; Wan, J.; Li, H.; Ding, J.; Wang, Y.; Wang, X.; Li, M. Resveratrol relieves gestational diabetes mellitus in mice through activating AMPK. Reprod. Biol. Endocrinol. 2015, 13, 118. [Google Scholar] [CrossRef] [Green Version]
- Sabán-Ruiz, J.; Alonso-Pacho, A.; Fabregate-Fuente, M.; de la Puerta González-Quevedo, C. Xanthine oxidase inhibitor febuxostat as a novel agent postulated to act against vascular inflammation. Antiinflamm. Antiallergy Agents Med. Chem. 2013, 12, 94–99. [Google Scholar] [CrossRef]
- Della Corte, E.; Gozzetti, G.; Novello, F.; Stirpe, F. Properties of the xanthine oxidase from human liver. Biochim. Biophys. Acta 1969, 191, 164–166. [Google Scholar] [CrossRef]
- Hille, R.; Massey, V. Studies on the oxidative half-reaction of xanthine oxidase. J. Biol. Chem. 1981, 256, 9090–9095. [Google Scholar]
- Tsuda, H.; Kawada, N.; Kaimori, J.; Kitamura, H.; Moriyama, T.; Rakugi, H.; Takahara, S.; Isaka, Y. Febuxostat suppressed renal ischemia-reperfusion injury via reduced oxidative stress. Biochem. Biophys. Res. Commun. 2012, 427, 266–272. [Google Scholar] [CrossRef]
- Jones, A.E.; Brown, K.C.; Werner, R.E.; Gotzkowsky, K.; Gaedigk, A.; Blake, M.; Hein, D.W.; van der Horst, C.; Kashuba, A.D.M. Variability in drug metabolizing enzyme activity in HIV-infected patients. Eur. J. Clin. Pharmacol. 2010, 66, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobmeyer, T.S.; Findhammer, S.; Dobmeyer, J.M.; Klein, S.A.; Raffel, B.; Hoelzer, D.; Helm, E.B.; Kabelitz, D.; Rossol, R. Ex vivo induction of apoptosis in lymphocytes is mediated by oxidative stress: Role for lymphocyte loss in HIV infection. Free Radic. Biol. Med. 1997, 22, 775–785. [Google Scholar] [CrossRef]
- Manfredi, R.; Mastroianni, A.; Coronado, O.V.; Chiodo, F. Hyperuricemia and progression of HIV disease. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 12, 318–319. [Google Scholar] [CrossRef]
- Zhang, H.-S.; Li, H.-Y.; Zhou, Y.; Wu, M.-R.; Zhou, H.-S. Nrf2 is involved in inhibiting Tat-induced HIV-1 long terminal repeat transactivation. Free Radic. Biol. Med. 2009, 47, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.; Smirnova, O.; Ivanov, A.V.; Kilpelainen, A.; Kuzmenko, Y.; Petkov, S.; Latanova, A.; Krotova, O.; Engström, G.; Karpov, V.; et al. Oxidative stress induced by HIV-1 reverse transcriptase modulates the enzyme’s performance in gene immunization. Hum. Vaccin. Immunother. 2013, 9, 2111–2119. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-S.; Wu, T.-C.; Sang, W.-W.; Ruan, Z. EGCG inhibits Tat-induced LTR transactivation: Role of Nrf2, AKT, AMPK signaling pathway. Life Sci. 2012, 90, 747–754. [Google Scholar] [CrossRef]
- Drachman, J.G.; Griffin, J.D.; Kaushansky, K. The c-Mpl ligand (thrombopoietin) stimulates tyrosine phosphorylation of Jak2, Shc, and c-Mpl. J. Biol. Chem. 1995, 270, 4979–4982. [Google Scholar] [CrossRef] [Green Version]
- Erhardt, J.A.; Erickson-Miller, C.L.; Aivado, M.; Abboud, M.; Pillarisetti, K.; Toomey, J.R. Comparative analyses of the small molecule thrombopoietin receptor agonist eltrombopag and thrombopoietin on in vitro platelet function. Exp. Hematol. 2009, 37, 1030–1037. [Google Scholar] [CrossRef]
- Rouyez, M.C.; Boucheron, C.; Gisselbrecht, S.; Dusanter-Fourt, I.; Porteu, F. Control of thrombopoietin-induced megakaryocytic differentiation by the mitogen-activated protein kinase pathway. Mol. Cell. Biol. 1997, 17, 4991–5000. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.; de Gunzburg, J.; Eychène, A.; Gisselbrecht, S.; Porteu, F. Thrombopoietin-mediated sustained activation of extracellular signal-regulated kinase in UT7-Mpl cells requires both Ras-Raf-1- and Rap1-B-Raf-dependent pathways. Mol. Cell. Biol. 2001, 21, 2659–2670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, S.; Wang, Z.; Thompson, A.; Kaluzhny, Y.; Zimmet, J.; Ravid, K. Signaling by the Mpl receptor involves IKK and NF-kappaB. J. Cell. Biochem. 2002, 85, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Venkatachari, N.J.; Zerbato, J.M.; Jain, S.; Mancini, A.E.; Chattopadhyay, A.; Sluis-Cremer, N.; Bar-Joseph, Z.; Ayyavoo, V. Temporal transcriptional response to latency reversing agents identifies specific factors regulating HIV-1 viral transcriptional switch. Retrovirology 2015, 12, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wire, M.B.; McLean, H.B.; Pendry, C.; Theodore, D.; Park, J.W.; Peng, B. Assessment of the pharmacokinetic interaction between eltrombopag and lopinavir-ritonavir in healthy adult subjects. Antimicrob. Agents Chemother. 2012, 56, 2846–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [Green Version]
- Pagans, S.; Pedal, A.; North, B.J.; Kaehlcke, K.; Marshall, B.L.; Dorr, A.; Hetzer-Egger, C.; Henklein, P.; Frye, R.; McBurney, M.W.; et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005, 3, e41. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Choi, A.Y.; Oh, Y.T.; Choe, W.; Yeo, E.-J.; Ha, J.; Kang, I. Corrigendum to “AMP-activated protein kinase mediates T cell activation-induced expression of FasL and COX-2 via protein kinase C theta-dependent pathway in human Jurkat T leukemia cells”. Cell. Signal. 2018, 52, 163. [Google Scholar] [CrossRef]
- Franklin, T.J.; Cook, J.M. The inhibition of nucleic acid synthesis by mycophenolic acid. Biochem. J. 1969, 113, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.A.; Herrmann, B.; Keppler, D.O. Selective guanosine phosphate deficiency in hepatoma cells induced by inhibitors of IMP dehydrogenase. Hoppe-Seyler’s Z. Physiol. Chem. 1980, 361, 1503–1510. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Paolo Rizzardi, G.; D’Agostino, C.; Attinger, A.; Knabenhans, C.; Fleury, S.; Acha-Orbea, H.; Pantaleo, G. Effects of mycophenolic acid on human immunodeficiency virus infection in vitro and in vivo. Nat. Med. 2000, 6, 762–768. [Google Scholar] [CrossRef]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müssig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, C.A.; Carlsten, H. Inosine monophosphate dehydrogenase (IMPDH) inhibition in vitro suppresses lymphocyte proliferation and the production of immunoglobulins, autoantibodies and cytokines in splenocytes from MRLlpr/lpr mice. Clin. Exp. Immunol. 2001, 124, 486–491. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | % Luminescence of pcTat + DMSO | Brief Description |
|---|---|---|
| Febuxostat (Uloric) | 459.50 | Febuxostat is a non-purine selective xanthine oxidase inhibitor with an IC50 of 114–210 nM. |
| Nitrofurazone (Nitrofural) | 271.89 | Nitrofurazone is a topical anti-infective agent with an IC50 of 22.83 ± 1.2 µM. (Rat LD50 = 590 mg/kg). |
| Nitazoxanide (Alinia, Annita) | 261.92 | Nitazoxanide (Alinia, Annita) is a synthetic nitrothiazolyl-salicylamide derivative and an antiprotozoal agent. (IC50 for canine influenza virus ranges from 0.17 to 0.21 μM). |
| Eltrombopag (SB-497115-GR) | 205.50 | Eltrombopag (SB-497115-GR, Promacta, Revolade) is a small molecule agonist of the c-mpl (TpoR) receptor with an IC50 of 0.69 μM for the inhibition of hERG K+ channel tail current. |
| Vincristine | 170.52 | Vincristine sulfate is a microtubule function inhibitor with an IC50 of 10.4 ± 1.1, 28.1 ± 3.4, 22.4 ± 2.1 µM for HL-60, Bel7402, HO-8910, respectively. |
| Resveratrol | 168.85 | Resveratrol is a phytoalexin produced naturally by several plants with anti-cancer, anti-inflammatory, blood-sugar-lowering and other beneficial cardiovascular effects. |
| Prednisolone acetate (Omnipred) | 168.32 | Prednisolone acetate is a synthetic corticosteroid drug that is particularly effective as an immunosuppressant agent. |
| Ethinyl estradiol | 157.10 | Ethynyl estradiol is an orally bio-active estrogen used in almost all modern formulations of combined oral contraceptive pills. |
| Econazole nitrate (Spectazole) | 156.49 | Econazole nitrate (Spectazole) is an imidazole class antifungal medication. |
| Budesonide | 154.59 | Budesonide is a glucocorticoid steroid for the treatment of asthma, non-infectious rhinitis. |
| Aztreonam (Azactam, Cayston) | 152.67 | Aztreonam (Azactam, Cayston) is a synthetic monocyclic beta-lactam antibiotic. |
| Vorinostat (SAHA) | 150.70 | Vorinostat also known as SAHA, Zolinza, MK-0683 is an HDAC inhibitor. Vorinostat CAS No 149647-78-9 with purity >99% and solubility DMSO is available. |
| Omeprazole (Prilosec) | 147.83 | Omeprazole (Prilosec) is a proton pump inhibitor used in the treatment of dyspepsia. |
| Minoxidil | 144.63 | Minoxidil is a vasodilator medication known for its ability to slow or stop hair loss and promote hair regrowth. |
| Clorsulon | 139.45 | Clorsulon is a competitive 8-phosphoglycerate kinase and phospho-glyceromutase inhibitor. |
| Leflunomide | 139.23 | Immunosuppressant agent. Its active metabolite is A77 1726 (RS-61980). |
| Amorolfine hydrochloride | 137.65 | Amorolfine hydrochloride is an antifungal reagent. |
| Azelastine hydrochloride (Astelin) | 137.36 | Azelastine is a potent, second-generation, selective, histamine antagonist. |
| Perindopril erbumine (Aceon) | 136.11 | Perindopril erbumine(Aceon) is the tert-butylamine salt of perindopril, the ethyl ester of a non-sulfhydryl angiotensin-converting enzyme (ACE) inhibitor. |
| Fenoprofen calcium | 135.40 | Fenoprofen calcium is a nonsteroidal, anti-inflammatory antiarthritic agent. |
| Drug Name | % Luminescence of pcTat + DMSO | Brief Description |
|---|---|---|
| Idarubicin HCl | 2.58 | Idarubicin is the anthracycline antibiotic and target DNA topoisomerase II (topo II). MCF-7 cells were sensitive to idarubicin, with an IC50 value for growth inhibition of 0.01 μM. |
| Bortezomib (Velcade) | 2.81 | Bortezomib also known as Velcade, MG-341, PS-341 is a proteasome inhibitor, effectively inhibits proteasome activity (Ki-0.6 nM). |
| Camptothecin | 11.15 | Camptothecin (CPT) is a cytotoxic quinoline alkaloid which inhibits the DNA enzyme topoisomerase I (topo I) with an IC50 and IC70 of 50 nM and 0. 225 μM. |
| Topotecan HCl | 11.27 | Topotecan hydrochloride (Hycamtin) is a topoisomerase I inhibitor with an IC50 of 13 and 2 nM for MCF-7 Luc cells and DU-145 Luc cells. |
| Mitoxantrone hydrochloride | 20.59 | Mitoxantrone is a type II topoisomerase inhibitor with an IC50 of 2.0 µM, 0.42 mM for HepG2 and MCF-7/wt cells, respectively. |
| Cladribine | 22.74 | Cladribine (Leustatin) is an adenosine deaminase inhibitor with an IC50 of about 0.2 μM. |
| Mycophenolic (mycophenolate) | 24.00 | Mycophenolic acid (Mycophenolate) is an immunosuppressant drug used to prevent rejection in organ transplantation. |
| Gemcitabine HCl (Gemzar) | 27.23 | Gemcitabine is an antimetabolite-inhibiting DNA synthesis (cell IC50 of 0.06 µM). |
| Floxuridine | 27.41 | Floxuridine (FUDR, FdUrd, Floxuridin) is a pro-drug of floxuridine and an oncology drug with an GI50 of 5.1 μM for the inhibition of MDCK/PEPT1. |
| Clofarabine | 28.18 | Inhibits the enzymatic activities of ribonucleotide reductase (IC50 = 65 nM) and DNA polymerase |
| Epirubicin hydrochloride | 30.77 | Epirubicin hydrochloride (Ellence) is an anthracyclin antibiotic and a DNA topoisomerase II inhibitor with an IC50 of 12 μg/mL on an estrogen-receptor-positive ER (+) hyperdiploid EAT cell line. |
| Sorafenib (Nexavar) | 31.46 | Sorafenib Tosylate is a novel, small molecular inhibitor of several tyrosine protein kinases (VEGFR and PDGFR) and a RAF/MEK/ERK cascade inhibitor with an IC50 of 6, 22, 38 nM for Raf-1, wt BRAF and V599E mutant BRAF. |
| Doxorubicin (Adriamycin) | 32.46 | Doxorubicin is a topoisomerase II inhibitor with an IC50 of 1 and 2 μM for the inhibition of MCF-7 and MDA-MB231. |
| Pemetrexed | 34.97 | Pemetrexed disodium (Alimta) is a thymidylate synthase, DHFR and glycinamideribonucleotide, and formyltransferase inhibitor with IC50s of 25, 34, 220 nM, respectively. |
| Mycophenolate mofetil (CellCept) | 36.45 | Mycophenolate mofetil is an inhibitor of inosine monophosphate dehydrogenase and an immunosuppressant. |
| Axitinib | 36.49 | Axitinib blocks the phosphorylation of VEGFR-2 and VEGFR-3 with average IC50s of 0.2 and 0.1 to 0.3 nM. |
| Teniposide (Vumon) | 36.85 | Teniposide (Vumon) is a chemotherapeutic medication mainly used in the treatment of childhood acute lymphocytic leukemia (ALL). |
| Tretinoin (Aberela) | 46.89 | Tretinoin (Aberela) is a drug commonly used to treat acne vulgaris and keratosis pilaris. |
| Sunitinib malate (Sutent) | 49.15 | Sunitinib malate (Sutent) is a multitargeted FLT3, PDGFRs, VEGFRs, and Kit kinase inhibitor with a Ki of 0.009 and 0.008 µM for Flk-1 and PDGFR. |
| Nilotinib (AMN-107) | 49.44 | Inhibitor of BCR-ABL, IC50 < 30 nM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampey, G.C.; Iordanskiy, S.; Pleet, M.L.; DeMarino, C.; Romerio, F.; Mahieux, R.; Kashanchi, F. Identification of Modulators of HIV-1 Proviral Transcription from a Library of FDA-Approved Pharmaceuticals. Viruses 2020, 12, 1067. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101067
Sampey GC, Iordanskiy S, Pleet ML, DeMarino C, Romerio F, Mahieux R, Kashanchi F. Identification of Modulators of HIV-1 Proviral Transcription from a Library of FDA-Approved Pharmaceuticals. Viruses. 2020; 12(10):1067. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101067
Chicago/Turabian StyleSampey, Gavin C., Sergey Iordanskiy, Michelle L. Pleet, Catherine DeMarino, Fabio Romerio, Renaud Mahieux, and Fatah Kashanchi. 2020. "Identification of Modulators of HIV-1 Proviral Transcription from a Library of FDA-Approved Pharmaceuticals" Viruses 12, no. 10: 1067. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101067