Solid-State NMR for Studying the Structure and Dynamics of Viral Assemblies
by
 and
and
Lauriane Lecoq
1, 
Marie-Laure Fogeron
1, 
Beat H. Meier
2,
Michael Nassal
3,* and
Anja Böckmann
1,* 1
Molecular Microbiology and Structural Biochemistry, University of Lyon, 7 Passage du Vercors, CEDEX 07, 69367 Lyon, France
2
Physical Chemistry, ETH Zurich, 8093 Zurich, Switzerland
3
Department of Medicine II/Molecular Biology, Medical Center, University Hospital Freiburg, Albert-Ludwigs-University, 79106 Freiburg, Germany
*
Authors to whom correspondence should be addressed.
Viruses 2020, 12(10), 1069; https://0-doi-org.brum.beds.ac.uk/10.3390/v12101069
Submission received: 28 August 2020
/
Revised: 21 September 2020
/
Accepted: 21 September 2020
/
Published: 24 September 2020
(This article belongs to the Special Issue In Memory of Michael Rossmann)
Abstract
:Structural virology reveals the architecture underlying infection. While notably electron microscopy images have provided an atomic view on viruses which profoundly changed our understanding of these assemblies incapable of independent life, spectroscopic techniques like NMR enter the field with their strengths in detailed conformational analysis and investigation of dynamic behavior. Typically, the large assemblies represented by viral particles fall in the regime of biological high-resolution solid-state NMR, able to follow with high sensitivity the path of the viral proteins through their interactions and maturation steps during the viral life cycle. We here trace the way from first solid-state NMR investigations to the state-of-the-art approaches currently developing, including applications focused on HIV, HBV, HCV and influenza, and an outlook to the possibilities opening in the coming years.

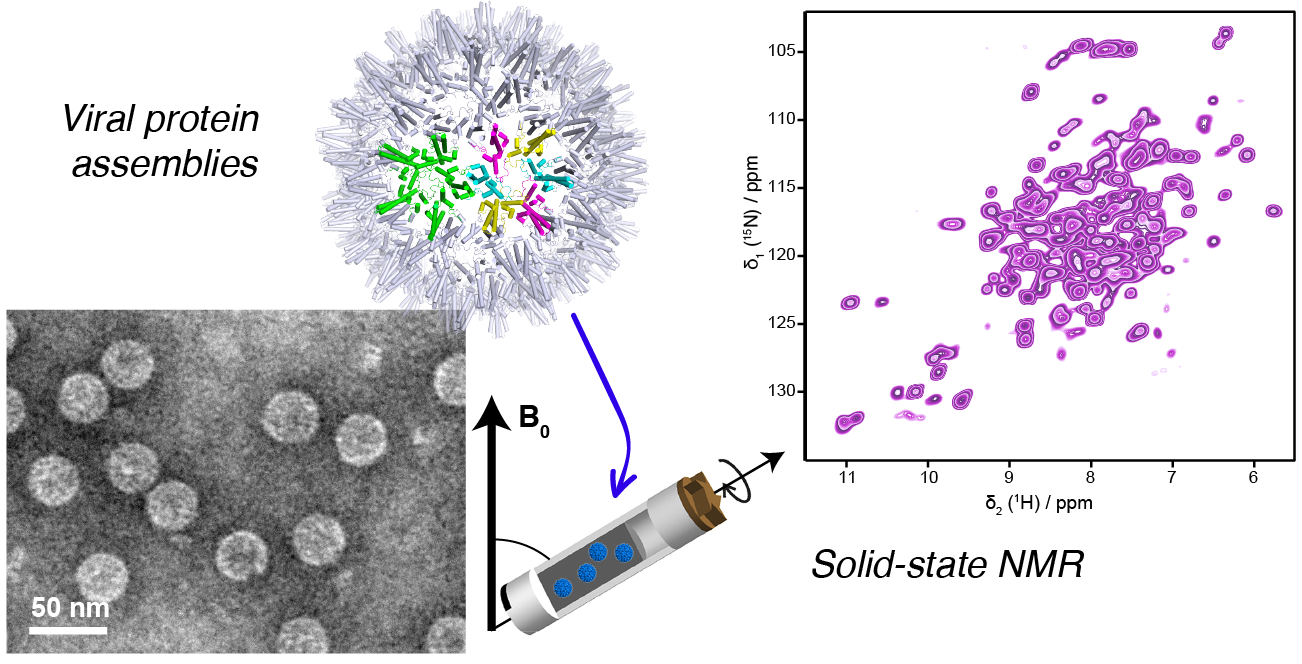{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Solid-state NMR is a structural biology approach that can provide unique information about the molecular assemblies used by viruses to enter host cells, to establish infection and to ensure that progeny virions are released into the environment. Solid-state NMR can contribute to an understanding of how viral proteins and their assemblies function from a structural point of view, revealing weak spots which can be targeted to fight infection through antiviral therapies or vaccines. Solid-state NMR is in this respect complementary to solution NMR, X-ray crystallography and electron cryo-microscopy (cryo-EM), the other atomic-resolution structural biology methods. All can provide images of proteins at resolutions on the order of 1–10 Å. Considering large assemblies, X-ray crystallography was the first approach used to resolve viral capsid and envelope structures at high resolution, and was pioneered by Michael Rossmann and Stephen C. Harrison on plant viruses [1,2], owing to the enormous numbers of progeny viruses per cell that plants produce. Advances in recombinant DNA as well as cell culture technology opened the way to mammalian viruses [3,4]. This approach has subsequently revealed a wide variety of capsid structures, amongst them capsids from rhinovirus [5], poliovirus [6] and hepatitis B virus (HBV) [7], as other early examples of human pathogens. The typical resolution of the structures is around 2–3 Å, but a higher resolution below 1.5 Å has been reached for several viruses (see www.pdb.org). For X-ray structures, at 2.5 Å, the backbone is well-defined and many, though not all, sidechains are visible; at 1.5 Å, most sidechains are well-defined (and in the correct rotameric state) and some hydrogens including water are resolved [8,9]. While the resolution characterizing X-ray, cryo-EM and NMR structures cannot be directly compared, the numbers that they deliver still have a roughly similar meaning.
Resolving capsid structures by X-ray crystallography is, however, frequently impeded by the difficulties in obtaining highly diffracting crystals from these large, often flexible complexes, and by the requirement for milligram amounts of material for crystallization trials. Much of the available structural information has thus been gathered on the soluble subunits of the capsids, called the capsid or core proteins, which might in the crystal assemble into sub-particulate superstructures such as hexamers. This approximates how capsid proteins build hexagonal lattices, which are the foundation of icosahedral capsids [10,11]. Rossmann, besides using crystallography, was one of the first to recognize that a hybrid approach combining the moderate resolution structures from cryo-EM with high-resolution X-ray structures could allow to reconstitute the entire capsid assembly [12]. EM electron density maps, even at lower resolution, are also helpful since they can be used as an initial phasing model for the X-ray structure determination. Today, this follows standard protocols [13], and in addition to the first crystal structures, this highly successful combination approach has largely shaped our view on a large variety of viral capsids. EM thus had, from the beginning, an important share in structural virology, but was for a long time severely limited by the resolution attainable with the available technology. Still, even low resolution maps yielded in some cases structures; the HBV capsid is one of the first examples of a protein whose fold was determined from cryo-EM, although assisted by numerous biochemically derived constraints [14,15].
With the advent of direct detectors, cryo-EM underwent the “resolution revolution” [16], enabling similarly high resolution as X-ray and NMR which, again for the HBV capsid as example, reached 2.8 Å [17]. This step forward was central for structural virology, since it allowed for the first time to look at authentic viruses with atomic resolution, and to investigate structures of virus-like particles in different maturation states, as documented by numerous structures deposited in the PDB. The impressive pictures thus obtained for many viruses, including human pathogens such as Dengue virus (DENV), ZIKA virus, HBV and Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), many of them from Rossmann and coworkers, have shaped our view of infection. Moreover, the high resolution now allows to resolve the arrangement of proteins in challengingly large unit cells, as shown by the impressive 3.5 Å structure of a giant virus capsid, recently reported by Rossmann′s lab, which comprises numerous different proteins which intimately interact to form the capsid shell [18].
However, obtaining homogeneous samples from virus-replicating cells can be very demanding, owing to the often complex mixture of viral particles in different maturation states that coexist in such cells. Single particles can be picked on electron micrographs, allowing to exclude poorly assembled particles and/or to separate clearly different ones, such as the 180 subunit form (triangulation number T = 3) vs. the 240 subunit form (T = 4) of the HBV capsid. This approach has also been applied to capsids of duck HBV (DHBV), which in their larger core protein feature an extra domain that can adopt disordered and folded states [19]. Still, the differences must be large enough to enable separation of the different particle states, otherwise the resulting heterogeneity will manifest itself as a loss of resolution, and tracking different states remains difficult if not impossible.
This is where solid-state NMR comes into play, as it can contribute to aspects where X-ray crystallography and EM have limitations. Two strengths of solid-state NMR are of particular interest. First, conformational changes can be followed with highest sensitivity via the NMR chemical shifts (very small changes in protein conformation lead to considerable changes in resonance positions). Once the sequential assignment of a reference form is established, this allows to rapidly assess conformational variations as they occur, for instance, upon binding of an interaction partner to the reference structure. In fact, no full structure determination is needed to identify such conformational alterations, which makes the direct observation and quantification of structural transitions one of the strengths of NMR. The second major asset of NMR is its unrivaled ability to follow dynamic events over a wide range of experimental timescales that are too fast for cryo-EM and X-ray approaches, i.e., from picoseconds to seconds. NMR works under near-physiological conditions, and does not need the commonly very low temperatures of X-ray or cryo-EM, where mixed states of a sample are frozen out. Hence defining structural dynamics by these techniques often requires the availability of stable mimics of two or more different states.
This review reports on the recent progress in the field, and intends to give an overview on the emerging possibilities and expected insights which can be gained from solid-state NMR.
2. Solid-State NMR
Solid-state NMR is a relative newcomer to the structural virology field, as sufficient spectral resolution and sensitivity to address complex proteins with atomic resolution have only recently been developed. Liquid-state (“in-solution”) and solid-state NMR in fact started out at the same time in the laboratories of Bloch and Purcell [20,21], and the spectra looked remarkably similar [20]. Historically, it was the development of sophisticated pulse techniques and multidimensional spectroscopy in the 1970s and 80s [22,23,24] which set the stage for protein structure determination by solution-state NMR [25]. Then, technical improvements in the homogeneity, stability and strengths of the magnetic fields applied dramatically increased resolution in the spectra of complex biomolecules for liquid-state NMR. Indeed, in solution the anisotropic interactions in the spin system are averaged by molecular tumbling and thus the “natural linewidth” is narrow (a few Hertz). This provides a high enough resolution in order to distinguish hundreds of resonances as they occur in medium-sized proteins. A natural limitation is set by the molecular tumbling which, today, restricts in solution NMR the maximum molecular mass for meaningful analyses of rigid proteins to roughly 50 kDa. Notably, this limit can be significantly pushed up by selective methyl-group protonation, which dilutes the dipolar coupling network; this allows, for example, investigation into the dynamics of proteins with a higher molecular weight, such as the 670 kDa proteasome [26].
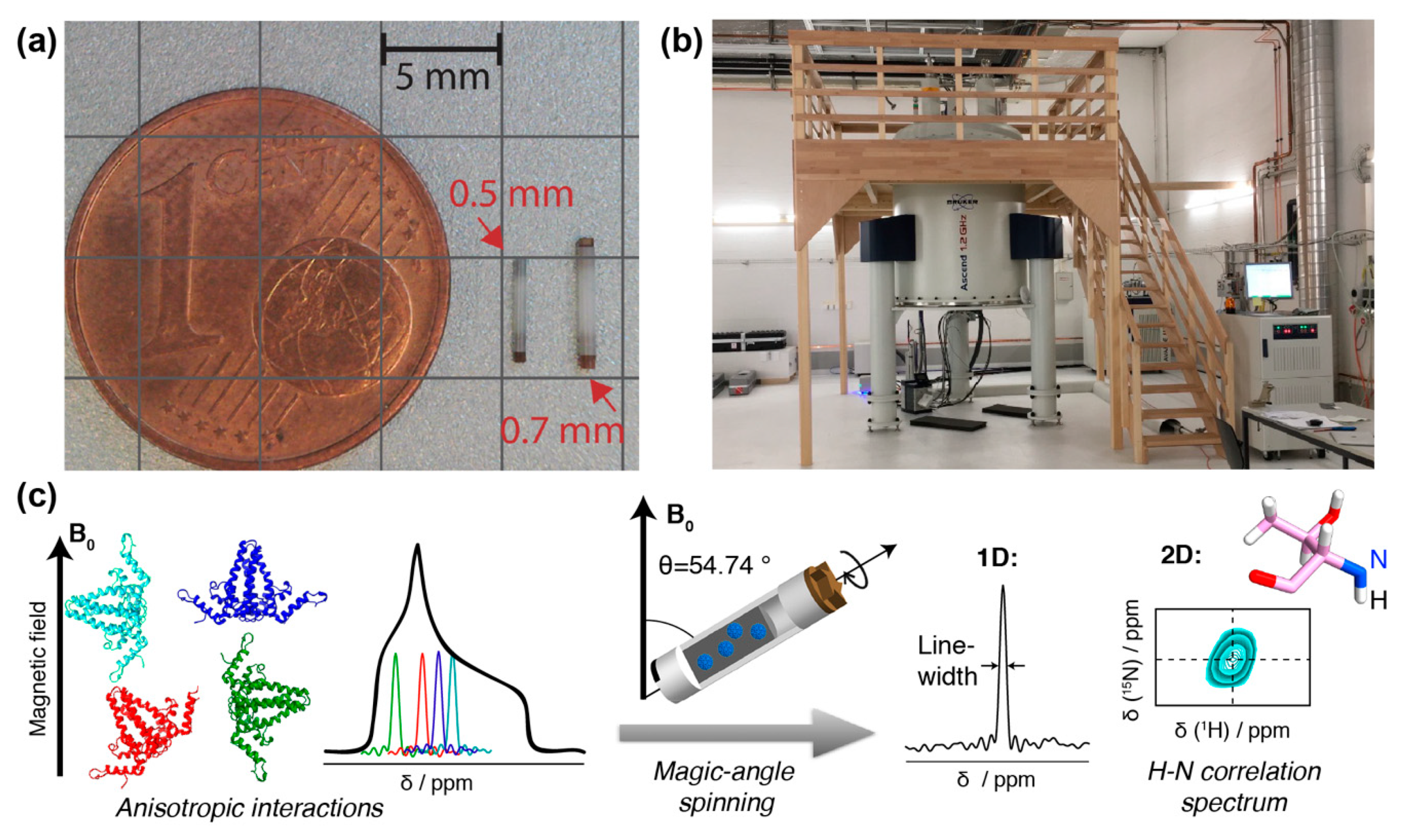
In contrast, protein solid-state NMR was long plagued by much broader lines (around 50 kHz, and thus more than a thousand times broader than in solution), in particular for protons. This is due to the absence of molecular tumbling, which results in orientation-dependent anisotropic contributions to the chemical shifts, and the presence of a strong dipolar-coupling network (see Figure 1). One early conceptual break-through was the observation that mechanical rotation of the sample narrows the resonance lines. The strongest effect is found if the rotation axis is inclined from the magnetic field direction by the “magic” angle of Θ ≈ 54.7° [27] for which the second-order Legendre polynomial, P2(cos θ) equals zero. By this, magic-angle spinning (MAS) removes the anisotropic contribution to the chemical shift and the dipole couplings if the rotation frequency is roughly of the same value as the size of the interaction to be averaged. The spectra then become liquid-like. The strengths of the interactions depends on their type, as well as the nuclei involved. While 13C chemical shift anisotropies are, for instance, on the order of a few kHz, and already very low spinning frequencies are sufficient to average them, it is highly beneficial to spin even faster than 100 kHz to average the strong proton–proton dipolar interactions. These approaches are currently under development [28,29].
Practical realization took a long time until improvements in 13C-detected spectroscopy led to the first high-resolution protein spectra that finally could be assigned [30] and be used for the first protein structure determination by solid-state NMR [31]. Early, proton detection had been applied at moderate spinning frequencies (40–60 kHz) to proteins using extensive deuteration to reduce proton–proton dipolar interactions [32,33]. Only in 2014, MAS frequencies finally exceeded 100 kHz in protein spectroscopy [34]. Indeed, this increase in MAS frequencies substantially increased resolution in deuterated proteins, and allowed for high-resolution 1H-detected spectroscopy also on fully protonated solid proteins, through more efficient averaging of the strong 1H-1H dipolar couplings. Detecting on 1H spins (as is routinely done in solution-state NMR) is more sensitive than 13C detection, mainly through the higher gyromagnetic ratio of the proton when compared to the 13C. This brought an almost 100-fold increase in mass sensitivity [35], reducing sample requirements to below 1 mg of protein and thus opening new possibilities in applications to biomolecules.
Still, the solid-state linewidth is typically somewhat broader than in solution-state NMR, which results in a higher spectral crowding than in solution, and limits the protein size to be investigated. Thus, further improvements in MAS frequency (>100 kHz [29,30,31,32,33,34,35,36]) and magnetic field strength (>800 MHz or 18.8 Tesla; note that magnetic field strength is conventionally expressed in units of the proton resonance frequency) will be important for the study of larger proteins. Typically, going from 800 MHz to 1.2 GHz increases the signal-to-noise ratio by nearly a factor of 2. But already today, the combination of 100 kHz MAS, high magnetic fields (>800 MHz) (Figure 1) and improved sample preparation, e.g., by direct sedimentation [37,38], allows for high-resolution spectroscopy of large biomolecular assemblies like viral capsids or envelopes [35,39,40,41] if they show high local symmetry. If the protein-monomers forming the assemblies are structurally diverse, this results in the observation of multiple sets of NMR signals (see also below), and increases spectral crowding or line widths if the splittings are unresolved.
Solid-state NMR is thus a technique which can address structure and detailed conformation of proteins which are “not soluble”. This term must be understood in the broader sense, often simply meaning that they are involved in molecular interactions leading to larger superstructures, either with lipids for membrane proteins, or with each other, as in molecular assemblies such as molecular machines, protein fibrils, viral capsids or envelopes. “Solid” in this context means that the proteins do not show fast enough molecular tumbling (with correlation times of tens of nanoseconds) to be addressed by solution-state NMR. In a first approximation, therefore, the most attractive targets for solid-state NMR are large assemblies of many relatively small subunits, the latter being important to avoid excessive signal overlap in the spectra.
As mentioned, an additional important feature of solid-state NMR is its ability to characterize dynamic processes by observing relaxation times and exchange effects [42]. For an NMR spectroscopist, dynamics is usually associated with a timescale (the correlation time) and an amplitude (the order parameter), while structural virology dynamics is more associated with the volume of the conformational space for the protein. Dynamics on the pico- and nanosecond timescale (backbone and sidechain motion) can be characterized by relaxation measurements, while microsecond dynamics often leads to very efficient relaxation. In solid-state NMR spectra, dynamic entities (often loops but sometimes entire domains) may completely vanish from the spectrum as a consequence of such dynamics [43]. While limiting the possibilities for structure determination, the ability to assign a defined range of motional timescale to the corresponding residues provides valuable dynamic information.
As in solution-state NMR, the chemical shifts in MAS solid-state NMR give a fingerprint of a protein, which can be used to easily assess structural differences arising, for example, in the presence of interactants. This is of central importance in viral proteins, which often show multiple functions and thus also interactions, during which they change conformation to adapt to the specific role they need to fulfill at a specific point in the viral life cycle.
3. NMR Sample Preparation
3.1. The Need for Recombinant Sample Production
A major challenge, as for all structural techniques, is the isolation of appropriate virus-like or capsid-like particles (VLPs and CLPs, respectively) from cell culture, or to in-vitro reconstruct the viral protein and its interactants one seeks to investigate. As most solid-state NMR approaches rely on the availability of stable-isotope-labeled protein preparations (2H, 13C, 15N) [44], isolation of VLPs or CLPs is often impractical; for HBV capsids, for instance, this would require large-scale cultures of HBV-producing human hepatoma cells growing on isotope-labeled medium, and efficient separation methods. For bacterial viruses, this is much less challenging, and solid-state NMR has successfully been used to investigate Pf1 bacteriophages directly as sediments from ultracentrifugation [45,46]. However, the phage structure is composed mainly of a single protein, and the viral genome is difficult to observe under standard conditions. The DNA signals could be assigned using selective unlabeling of the protein [47,48] or dynamic nuclear polarization (DNP) [49,50], a technique providing signal intensity enhancement using transfer polarization from unpaired electron spins to nuclear spins at low temperatures (typically 100 K) although at the cost of limited spectral resolution (for a review on DNP see [51]).
Another limitation for the investigation of authentic viral particles directly by solid-state NMR is that, while biosafe probes have been designed for the investigation of amyloid proteins [52], they do not reach the safety provided by a biosafety level 3 laboratory often needed for the study of infectious material.
Another challenge is to obtain the amounts of material needed for NMR structural analysis. Until recently, solid-state NMR samples routinely comprised >20 mg of pure protein. With the advent of advanced proton-detection fast MAS techniques, proteins can now be analyzed in 0.7 mm and even 0.5 mm rotors which reach over 150 kHz MAS frequencies (Figure 1a), yielding well-resolved proton-nitrogen correlation spectra [29,34,35,36,53]. The concomitant reduction in sample sizes to the sub-milligram level enables the use of alternative production techniques to bacterial expression. Still, several hundred micrograms of pure protein are usually required, and the isolation of sufficient amounts of authentic viruses, moreover in a single, defined maturation state, would require truly large-scale mammalian cell cultures; complexity in vivo is often further increased by dynamic modifications such as phosphorylation/dephosphorylation events.
Hence NMR specimens are typically not entire virions from patients or cell culture, but rather recombinant purified proteins, or their assembly products such as capsids or envelopes, typically around 500 µg per sample, which are centrifuged directly out of a liquid phase as a sediment into the NMR rotor (Figure 1a). This technique yields good-quality spectra [37,38] and usually keeps the samples stable and hydrated for months if not years [54]. NMR experiments are usually performed near room temperature (−10 °C to 40 °C).
3.2. The Size of the Proteins and Their Assemblies Amenable to “High Resolution” Solid-State NMR
Solid-state NMR has a strength for assemblies that are large but consist of small, optimally identical, repeating units, as is true for many viral capsids and envelopes. Indeed, only a single set of signals is expected for a core protein, even if many equivalent copies (hundreds for helical symmetry, 60 for T = 3 and T = 4 icosahedral symmetry) make up the viral capsid under investigation. If there are N non-equivalent monomers in the unit cell, N peaks may be observed per nucleus, depending on the conformational differences. If they are sizeable, the different peaks are resolved; if they are smaller, they may lead to line broadening or have no effect. While these effects add to the information contents of the spectra [55] (see also below), they also increase spectral overlap, which can be detrimental for large proteins. Solid-state NMR does not require long-range order of the biological assembly, as long as the monomeric subunits feature a similar chemical environment at the atomic level. More complex assemblies containing larger and/or more different proteins however become increasingly difficult to study in native forms, since the complexity of NMR spectra increases with the sheer number of amino-acid residues present. Although solid-state NMR does not have the intrinsic size limitations of liquid-state NMR, where larger proteins give ever broader NMR lines, spectral overlap complicates analysis, such that objects of more than ~500 inequivalent amino acids are difficult to resolve.
3.3. Labeled Protein Production
An important point to be considered is that most NMR techniques rely on labeling of the proteins with NMR-active stable isotopes such as 13C and 15N, in some cases also 2H (2H is an NMR-active nucleus with spin 1, but is here not detected; rather it dilutes the dense 1H network to further reduce the distance-dependent 1H-1H dipolar couplings, in order to further narrow 1H lines). Stable isotope labeling can indeed be achieved in mammalian cell cultures [56], but at larger scales (see above) this rapidly becomes very costly, and is thus best achieved by employing high-yield systems for protein production.
As a result, solid-state NMR of viral proteins relies largely on recombinantly produced material, which under appropriate conditions can be a valid, high homogeneity model, as is often confirmed by EM and X-ray crystallography. E. coli expression is the major workhorse here, offering straightforward isotope labeling of proteins. Genuine E. coli toolboxes have been developed for several viruses, able to produce assemblies mimicking defined in vivo states, e.g., residue-specific phosphorylation (for example [57]) thus allowing the exploration of the conformational space of viral proteins. As demands in sample quantity are diminishing (see above), other production approaches have recently been developed, and a highly attractive compromise between expression in eukaryotic cells vs. E. coli is the use of eukaryotic cell-free systems, optimally one that combines high expression yields with an increased folding capacity and access to a range of post-translational modifications [58]. One such system is based on wheat germ cell-free protein synthesis (WG-CFPS) [40,41,59,60]. The high efficiency and selectivity of isotopic labeling constitutes a major asset of this system for NMR studies. Importantly, and in contrast to cell-based production, only the protein of interest is isotopically labeled [61], enabling sample preparation without extensive purification. This is of particular interest, for example, for the study of ribonucleoprotein complexes which form directly during the cell-free synthesis reaction. In addition, metabolic scrambling of labels between different amino acids is much lower than in E. coli, and can further be minimized by using transaminase inhibitors and glutamine synthase inhibitors [62,63]. WG-CFPS is particularly attractive for deuteration, often used to reduce proton linewidths [64,65], as in contrast to bacterial expression it does not require proton back-exchange, circumventing denaturation and refolding steps which could compromise the native folding of proteins. WG-CFPS indeed allows for the simple addition of deuterated amino acids to the translation reaction performed in H2O medium. Selective labeling, by adding only the desired labeled amino acid to the translation reaction, can efficiently be performed in WG-CFPS [59,62,63,66], and help to significantly reduce the complexity of NMR spectra, as shown, for instance, by the successful application to the NS4B protein of hepatitis C virus [60] (see below). Another complexity-reducing approach is segmental labeling, whereby only part of a protein sequence is isotopically labeled while the remaining parts remain invisible. This was successfully applied to HIV-1 in tubular assemblies [67] and in spherical CLPs formed by the capsid protein [68]. Other schemes can be used, with those where only one partner is labeled particularly relevant for interaction studies, where the non-isotopically-labeled partner(s) remain(s) invisible, and all information on changes is contained in the target protein spectra [69]. Alternatively, labeling protocols using selectively 13C-labeled glucose (1-13C or 2-13C) as a carbon source during protein expression [70] can be used to improve spectral resolution and determine intermolecular restraints, as demonstrated on molecular machines such as the type III secretion system needle [71]. This technique has not yet been employed for viral assemblies but could be useful to study contacts between capsids subunits.
High sample purity is required to reach maximal NMR signal strength, yet low-level contaminants (<5–10% of the total sample) will typically be invisible in the NMR spectra.
4. NMR Protocols
4.1. Sequential Resonance Assignments
Once the proteins are labeled, 2D and 3D NMR spectra are recorded at an MAS frequency that depends on the rotor size used (typically 17 kHz for 3.2 mm rotors to 100 kHz for 0.7 mm ones). In multi-dimensional spectra, each dimension corresponds to the recording of an 1H, 15N or 13C magnetization’s free induction decay over time, which is then Fourier-transformed in order to yield a frequency-domain spectrum, ideally containing a peak for each 1H, 15N or 13C nucleus in the protein. Multi-dimensional spectra help to resolve overlap which would be very high in one-dimensional spectra. In these spectra, a residue is characterized by a “douple” and triple of resonance frequencies in the 2 or 3 dimensions, respectively, e.g., 15N,13CA,13CO intra-residual correlations. A combination of those nuclei with 3D spectra enables to create inter-residual connections by correlating, for example, the 15N of one residue with the 13CO/13CA of the preceding one. Such schemes thus connect the amide 15N to both directions of the protein backbone, and allow for sequential resonance assignments [53,72,73,74,75]. This eventually enables to associate an NMR signal or frequency to every atom in the protein.
4.2. Chemical Shifts Reveal Structural Information
Once the NMR fingerprint is assigned (i.e., all signals and thus frequencies in the spectra are assigned to a nucleus), it is easy to follow conformational changes induced by the addition of a ligand or any other event, by simply following the chemical-shift differences and localizing them on a known 3D structure. Important examples are protein-nucleic acid complexes, where structural changes can in principle even be followed via the chemical shifts from both sides. Protein and RNA or DNA can either be labeled at the same time, e.g., when preparing a viral capsid in E. coli in media containing labeled amino acids and nucleic-acid building blocks, or be labeled separately if one can reassemble the capsid in vitro with labeled or unlabeled RNA, as it is the case for HBV capsids [76].
4.3. Structure Determination
Protein structure determination by solid-state NMR involves combining dihedral angle restraints derived from chemical shifts [77] with distance restraints derived from dipolar couplings. These restraints can then be used to computationally find a solution that fulfills as many of them as possible. The detailed outcome of this optimization depends on the initial condition [78,79,80]. The best solutions constitute the NMR “bundle”, which characterizes the possible range of conformations for the protein. Due to signal overlap, de novo structure determination from solid-state NMR data is currently limited to proteins of up to about 200 amino acids in length. Only a few viral protein structures have been determined de novo using solid-state NMR today.
4.4. Investigating Interactions
NMR can efficiently assess interactions with small molecules, which is extensively used in drug screening by solution NMR [81,82]. For solid-state NMR, high-throughput screening is not yet routinely available, but first compatible sample changers have been developed. Screening can thus be done, though with limited efficiency, providing access to the rich information of NMR chemical shifts and allowing a detailed analysis of interactions with small molecules by similar approaches as in solution. A particular strength of solid-state NMR in such applications is the availability of side-chain information directly from 2D 13C correlation spectra, which indicates which 13C nuclei are spatially nearby the interacting site.
5. Examples of Solid-State NMR Studies on Viral Proteins
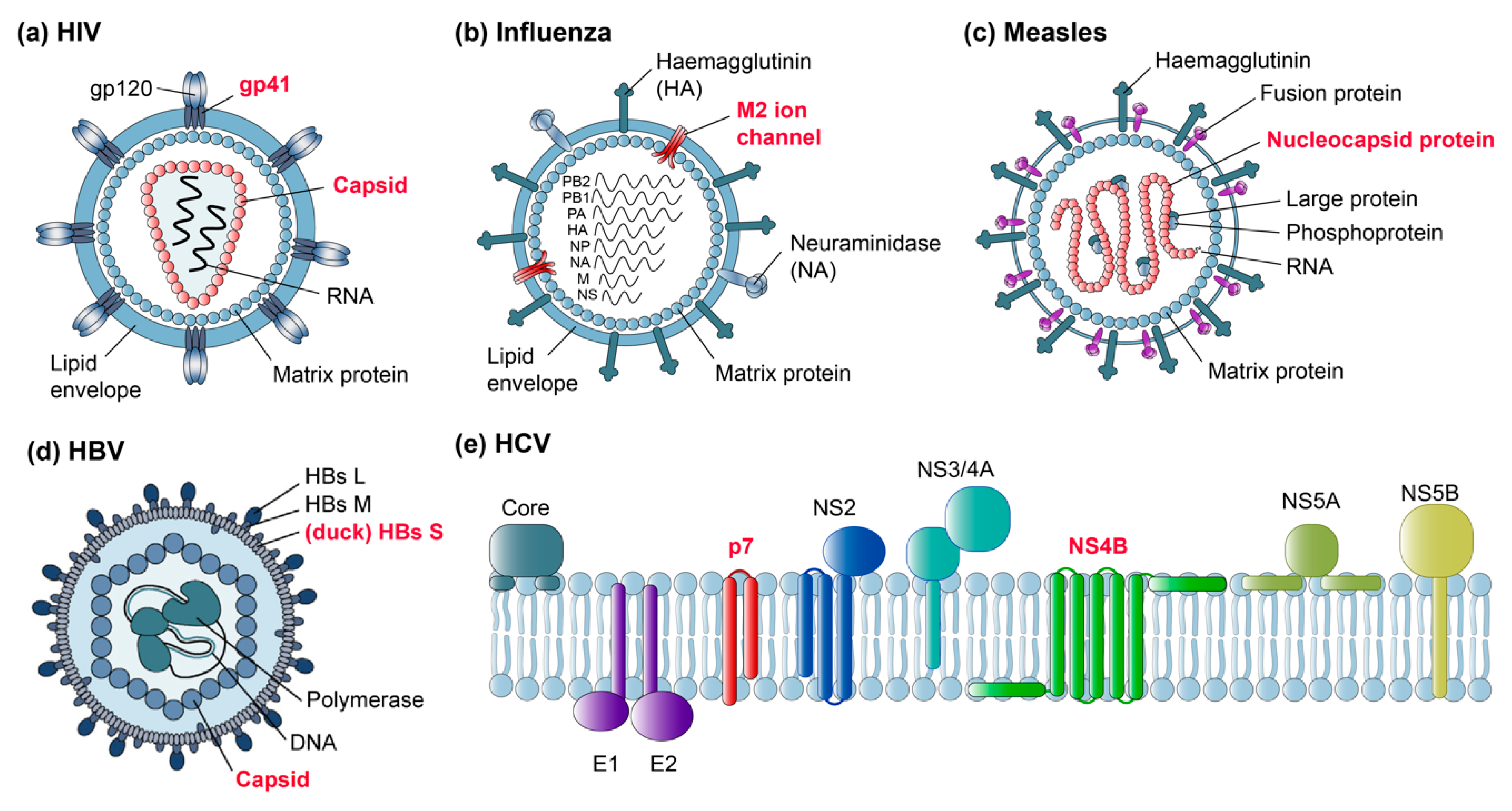
Some of the first viral targets in MAS solid-state NMR were bacteriophages, serving as relatively simple model systems for NMR methods development. Major advantages were their often small coat proteins, which can be readily prepared in stable-isotope-labeled form, and the absence of an envelope, presenting a system containing just one principal structural element (reviewed by [83] and more recently by [84]). Studies of intact filamentous bacteriophages were described recently [50]. In parallel, solid-state NMR on membrane proteins progressed further, first on oriented samples [85] and then by MAS methods, and the study of small viral membrane peptides set the stage for the emerging analyses of much more complex samples such as membrane proteins from human viruses. Figure 2 shows a selection of human viruses and their proteins studied by solid-state NMR today (with red labels), aiming to gain an insight into their dynamics and structure, and their interactions with viral genomes as well as cellular membranes.
Several studies have addressed the human immunodeficiency virus 1 (HIV-1), with an emphasis on the capsid and gp41 fusion proteins (Figure 2a). Influenza virus, notably its M2 ion channel, has been the topic of several studies (Figure 2b), studies on measles virus nucleocapsids have also been initiated (Figure 2c), and more recently hepatitis B virus (HBV) proteins, both from the duck HBV model and the human virus (Figure 2d), have been addressed. In addition, non-structural membrane proteins from the hepatitis C virus (HCV), such as the p7 ion channel and NS4B, are challenging current subjects (Figure 2e). Below we summarize first the successes and then the remaining challenges, with a focus on proteins from pathogenic human viruses.
5.1. Viral Capsids
Viral capsids are superstructures formed by core protein(s) in complex with the viral genome. They usually represent assemblies with high local symmetries, often icosahedral, but they can also have irregular shapes. Their obvious function is to protect the viral genome, but they have often multiple roles in the viral life cycle. To fulfill these diverse functions, e.g., intracellular transport and trafficking, entry and egress from the host cell, they have structures that can change over time and on demand. Their overall shapes can best be addressed by cryo-EM, but the dynamics of their functional domains let them frequently escape detection by this method. Similarly, structural details can go undetected, since even the resolution achievable nowadays may be insufficient to clearly pinpoint subtle changes between states.
5.1.1. HIV Capsids
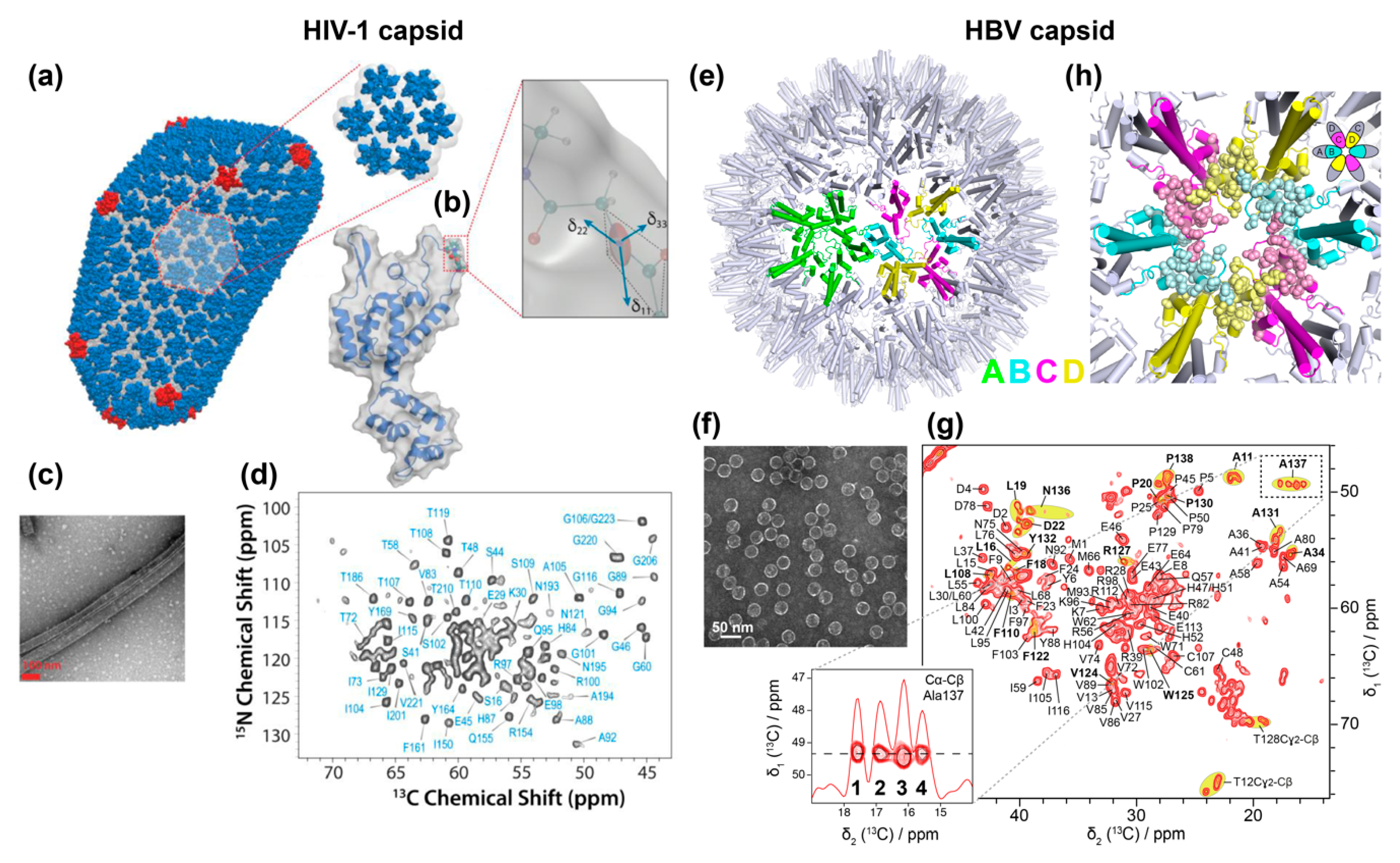
Solid-state NMR investigations of the HIV-1 capsid were initiated by Polenova and coworkers some years ago. The viral capsid comprises 1500 copies of the 26 kDa capsid protein (CA) and forms a characteristic cone-shaped cage around two copies of the viral genomic RNA (Figure 2a). The structural organization of the mature capsid was defined as a “fullerene cone” based on cryo-EM images [88] (Figure 3a,b). CA comprises an N-terminal domain (NTD) and a C-terminal domain (CTD), connected by a flexible linker, the hinge region. NMR spectra of HIV-1 CA, obtained and assigned using sediments of conical and spherical assemblies, formed the basis for structural and dynamic investigations by this and other groups (reviewed in [89] and [90]). NMR revealed that, despite the very different morphologies of the investigated particles, the individual CA proteins adopt very similar structures therein [91]. Probing dynamic behavior on the nano- to microsecond time scales through measurements of chemical-shift anisotropy (CSA) tensors revealed that the CA hinge region is flexible on the millisecond time scale (the chemical shift is not an isotropic quantity described by a number, but is mathematically described by a second-rank tensor, characterized by a 3 × 3 matrix. In the present context a tensorial interaction leads to a characteristic intensity pattern for each NMR resonance line describing the anisotropy and asymmetry of the interaction. The anisotropic part is, however, spun out under MAS to yield only the isotropic average, but can be reintroduced by carefully designed pulse sequences and reveals information on dynamics.)
This conformational flexibility was suggested to be essential for the CA protein to adopt multiple and slightly different conformations in the assembled capsid [92]. In a similar approach, Polenova and coworkers identified the interaction of CA with the host protein Cyclophilin A (CypA), which regulates HIV-1 viral infectivity through direct interactions with the viral capsid. 1H-15N and 1H-13C dipolar tensors and NMR peak intensities revealed that dynamics on the microsecond-to-nanosecond timescale in the CypA binding loop are important for HIV-1′s escape from CypA dependence [93] (again, the dipolar interaction is described by a second rank tensor, which is normally averaged but can be re-introduced to reveal dynamic averaging). More recently, NMR chemical shift and dipolar tensors were measured in HIV-1 CA tubular assemblies (Figure 3c,d). This revealed, in combination with molecular dynamics and theoretical calculations, a drastic decrease in dynamics for residues in the CypA-binding loop of the tubular assemblies upon interaction with CypA [94]. Moreover, the interaction of CA with the restriction factor TRIM5α has recently been described by such a combined approach [95]. In parallel, solid-state NMR studies in combination with EM by Tycko and coworkers have shown that CTD and NTD of CA tubular assembly samples are largely rigid and that they immobilize their general secondary and tertiary structure upon formation of tubes [96]. Later, the authors suggested a curvature generation mechanism [97,98], which would be determined by structural variations in three regions of CA: around the inter-domain linker region, in the segments involved in intermolecular NTD–CTD interactions, and in the C-terminal tail. The effects of an HIV-1 maturation inhibitor on CA was revealed to be rather subtle through the analysis of NMR chemical shifts and relaxation parameters [68].
5.1.2. HBV Capsids
The HBV core protein has been accessible in recombinant form from early on, and its structure has been extensively investigated by EM and X-ray approaches [7,14,100,101,102]. Still, it needed advanced biochemistry to develop expression systems adequate for the production of the full-length form, Cp183 [76], which in contrast to the truncated Cp149 used in most structural studies, packs nucleic acids. The HBV capsid structure is dynamic, and is believed to change/adapt according to the different roles the protein has at different points in its viral life cycle. Its main function is to serve as the building block of the nucleocapsid shell surrounding the pgRNA and viral polymerase. Cp expressed in bacteria assembles into icosahedral particles closely resembling those seen in infected liver [101]. The predominant icosahedral T = 4 capsid contains 240 copies of Cp in 120 dimeric subunits, and comprises four groups of symmetrically non-equivalent subunits arranged as 60 AB and CD homodimers (Figure 3e). The 20 kDa core protein contains an N-terminal domain of 140 residues, required for the assembly into capsid, joined by a 9-residues linker to an arginine-rich C-terminal domain (CTD, residues 150 to 183) responsible for several essential functions, including pgRNA binding [103]. The dynamic structure of the CTD and its interactions with the viral RNA remains one of the blind spots in capsid investigations today. Furthermore, the hypothesized conformational changes of the capsid believed to signal readiness for envelopment [104], or alternatively the predicted single-strand blocking signal [105], have escaped detection as of today.
The HBV capsid has recently been subject to solid-state NMR studies on capsids isolated from E. coli cells where they auto-assemble (Figure 3f). The sequential 1H/13C/15N assignments have been established as a basis for further studies [35,106]. The NMR spectra yielded high-resolution NMR fingerprints (Figure 3g), with linewidths similar to those obtained from the HIV capsids, confirming that this type of symmetrical object is well in the scope of solid-state NMR. A comparison between the chemical shifts of the core protein in its dimeric and capsid states revealed the residues which structurally adapt upon shell assembly [106]. The slight non-symmetry (“quasiequivalence”) between the four subunits was reflected in the NMR spectra [55], in which multiple peaks are observed for ~20% of residues located at the dimer–dimer interfaces at the five- and six-fold symmetry axes (Figure 3g,h), as shown at the example of Ala137. Hence the high sensitivity of NMR chemical-shifts to structural variations allowed the exact pinpointing of the residues where subtle local conformational adaptations are needed for lattice formation, i.e., the molecular hinges [55].
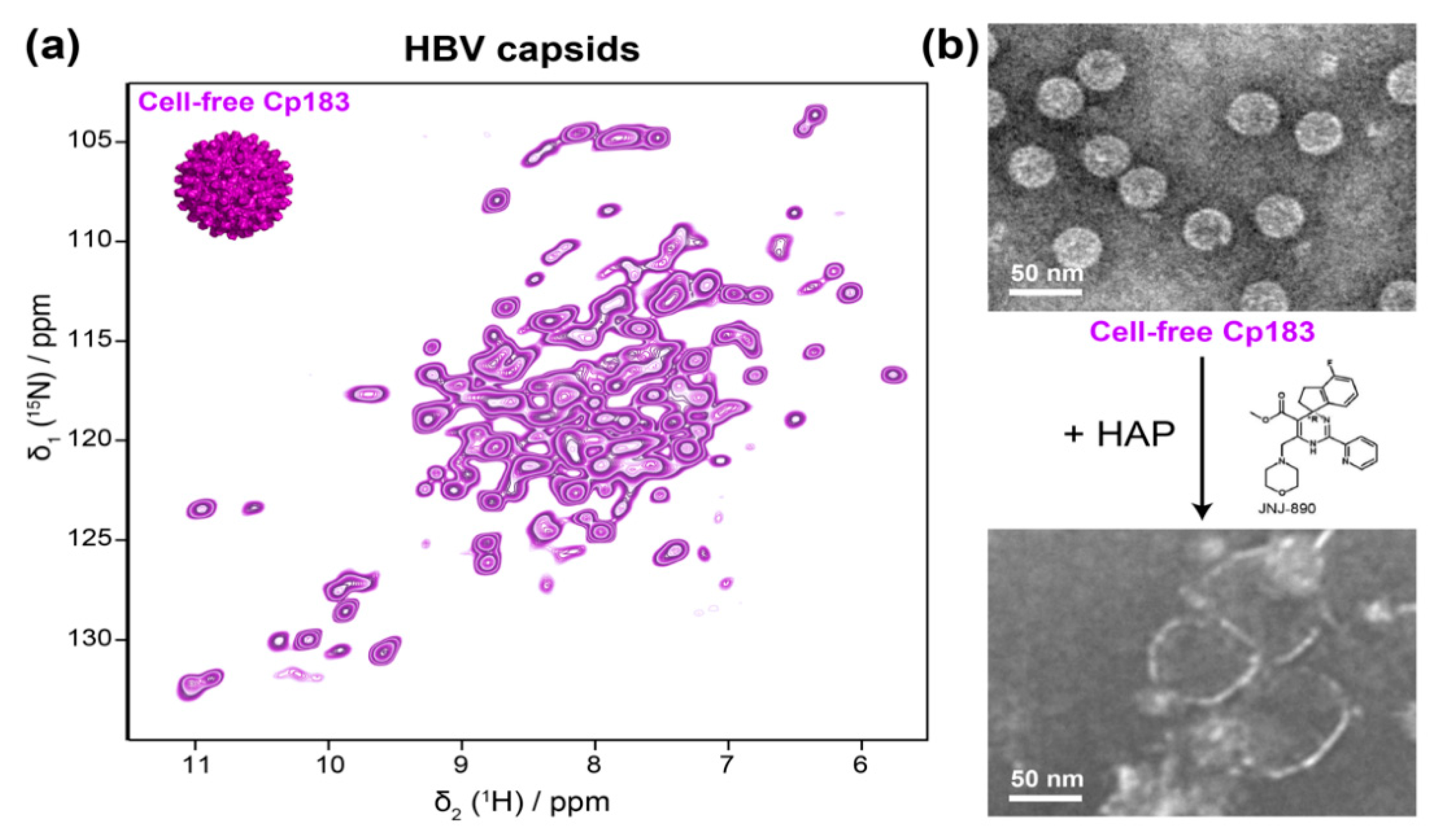
As described above, the development of proton-detected fast MAS NMR spectroscopy beyond 100 kHz sample rotation has recently made it possible to study sub-milligram amounts of protein samples, and enabled proton assignments of HBV Cp, using less than 500 μg of capsids [35]. With the proton resonances assigned, this approach can now be used for further interaction studies, with cellular partners that are available in small quantities, as well as with antivirals like the capsid assembly modulators (CAMs, also called core protein allosteric modulators, CpAMs). CAMs are currently under pharmaceutical development for targeting viral nucleocapsid assembly [107,108]; they act by either promoting assembly of regularly appearing but genome-less capsids, or they induce the formation of aberrantly shaped irregular multimers, which are not functional in viral replication; notably, recent data suggest that this distinction is not always strict in that the concentration of a compound and the time it is in contact can affect the morphological outcome [109]. In this context, the use of cell-free synthesis systems for sample preparation is particularly interesting as assembly modulators can act directly on the capsid protein exiting from the ribosome, and hence in a preassembly state. The sub-milligram Cp183 capsids obtained through cell-free protein synthesis are fully sufficient for the smaller sample requirements of fast MAS NMR, and they can simply be sedimented into a 0.7 mm NMR rotor after purification on a sucrose gradient (Figure 4a) [40]. The capsids directly pack nucleic acids during cell-free synthesis, and only the full-length protein successfully assembles in the system. In the presence of a heteroaryldihydropyrimidine (HAP) CAM, the cell-free Cp183 capsids opened up completely (Figure 4b) [40], indicating suitability of this system for solid-state NMR analysis. These data show how the combination of WG-CFPS and solid-state NMR can open the way for a better understanding of the action of CAMs under conditions that more closely mimic the situation in a cell.
5.1.3. Measles Virus Nucleocapsids
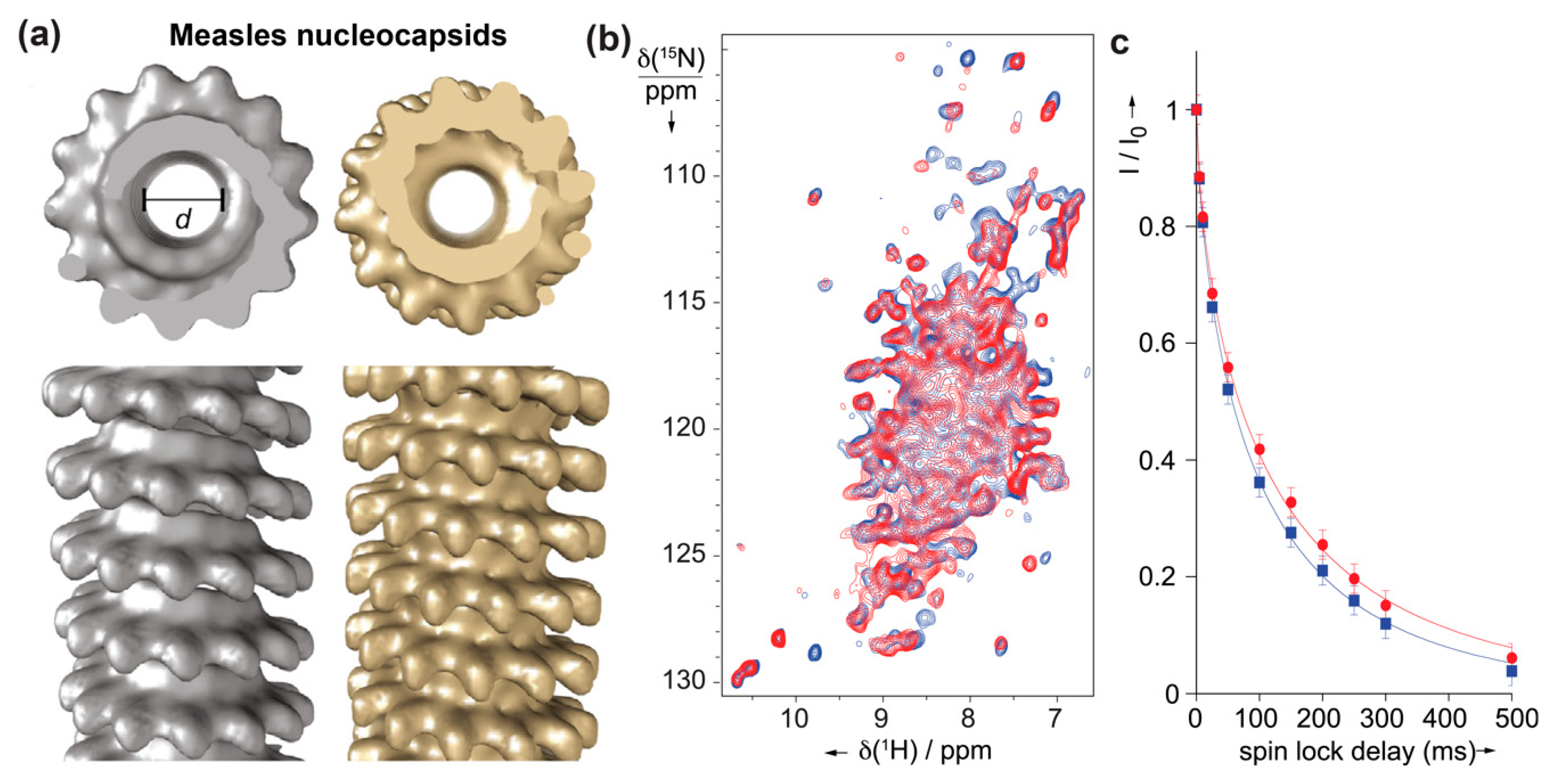
Solid-state NMR studies have also been well-initiated on the helical assemblies of Measles virus (MeV) nucleocapsids (Figure 5) formed by multiple copies of the nucleoprotein (N). N is composed of two globular domains, the N- and C-terminal domains, which together form a stable peanut-shaped nucleoprotein core of 400 residues (NCORE), responsible for self-assembly and RNA binding. In addition, N contains an intrinsically disordered tail domain of 125 residues (NTAIL) required for the interaction with the polymerase complex. For the study, NCORE was also studied with NTAIL removed to obtain cleaved nucleocapsids, which are more compact and regular than the intact nucleocapsids (Figure 5a). 1H-detected solid-state NMR experiments were recorded to compare intact and trypsin-cleaved MeV nucleocapsids [110], which showed no major structural reorganization as both spectra look similar (Figure 5b).
Sequential assignments for site-specific information were not obtained on this large protein. Still, comparing bulk T1ρ relaxation times for both assemblies showed longer relaxation times for the cleaved capsid, indicative of higher local dynamics on a submicrosecond timescale in NCORE of the intact capsids [110]. This dynamic information provides an interesting complement to the meanwhile solved near-atomic structure of the NCORE nucleocapsids [112].
5.2. Viral Envelopes
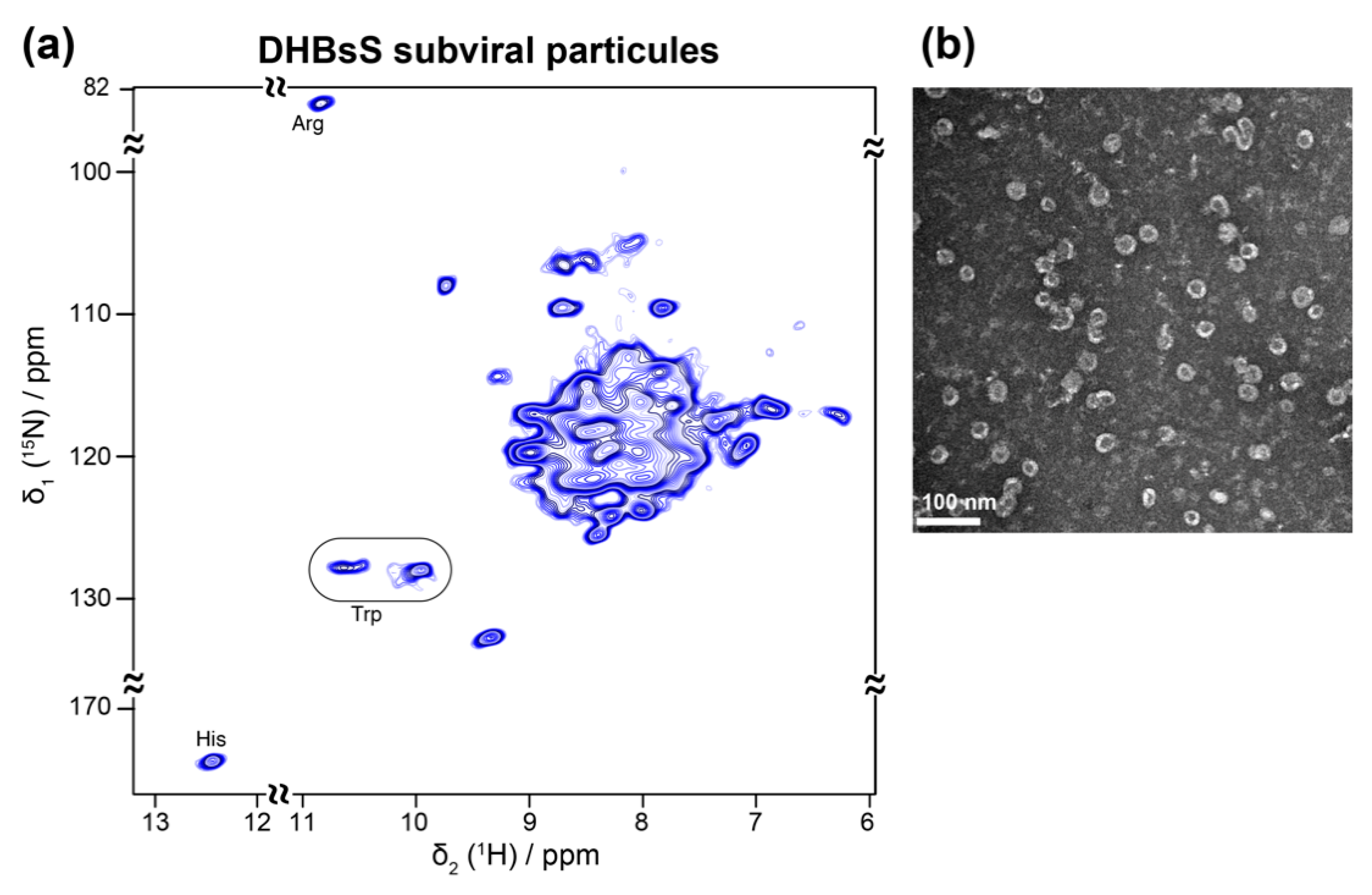
Viral envelopes are often highly complex assemblies of lipid-embedded glycoproteins. Their structure remains difficult to access, as they often only exist in multimeric forms, or even keep their native structure exclusively in the assembled envelope. As bacterial expression is most often not possible, alternative expression methods have been developed. An interesting and medically relevant example are recombinant HBV subviral particles (SVPs) comprising just the small envelope protein S, which can efficiently be produced in yeast and serves as the active constituent of the HBV vaccine. During HBV infection, similar non-infectious SVPs, containing all three envelope proteins and host phospholipids, are secreted by infected cells in huge excess over virions [113,114]. Yeast expression of the SVPs is not compatible with NMR analyses, since isotope labeling is too costly as 13C methanol must also be fed during the long maturation steps. It has been shown early-on that the small HBV envelope protein (HBsS) can assemble in the wheat-germ cell-free system [115]. This system was recently used to assemble SVPs from the S envelope protein of the related duck hepatitis B virus (DHBV) [41] (Figure 6a,b), in a similar manner to the HBV capsids [40] (Figure 4a,b). The addition of mild detergent allowed for the production of milligram amounts of these integral membrane protein assemblies, and a simple sedimentation after sucrose gradient isolation enabled NMR sample preparation [41]. The SVPs showed a rather narrow distribution of diameters, close to those formed during HBV infection [116], and similar antigenic properties as SVPs isolated from DHBV-infected ducks. First two-dimensional 1H-15N solid-state NMR hNH spectra showed a number of isolated peaks with linewidths comparable with model membrane proteins (Figure 6a). Analysis of the chemical-shift distribution allowed the conclusion that their constituent subunits displayed mainly α-helical secondary structure [41]. With such suitable resolution and sensitivity, the duck envelope protein thus represents an attractive pilot model for the structural study of its more complex human homolog, for which no conditions for assembly have yet been reported.
5.3. Other Viral Membrane Proteins
Membrane proteins in general are challenging systems for structural studies because of their hydrophobic nature, and the phospholipid bilayer which forms their native environment. While solution-state NMR and X-ray crystallography was widely used on small viral membrane proteins in detergent, solid-state NMR has the advantage that the proteins can be investigated in lipids, which are closer mimics of native membranes. Solid-state NMR investigations were first initiated on small viral membrane proteins, also called “viral membrane-spanning mini-proteins” [117], often using oriented samples to obtain spectra [118]. In this context, HIV-1, influenza and parainfluenza virus fusion peptides of ~20 amino acids have been studied early on [119,120,121,122] to reveal the structure and orientation behavior of these fragments in lipid bilayers under conditions mimicking membrane fusion. Later, technical advances in sample preparation and NMR equipment enabled larger fusion peptides to be studied.
5.3.1. HIV gp41 and Vpu
Several domains from the HIV gp41 protein were thus investigated [123,124,125,126]. From these data, a topology model of gp41 could be built, with the transmembrane domain well inserted into the lipid bilayer, and the other domains lying on the surface of one bilayer, such that gp41 bridges two lipid bilayers in a hemifusion-like state [126].
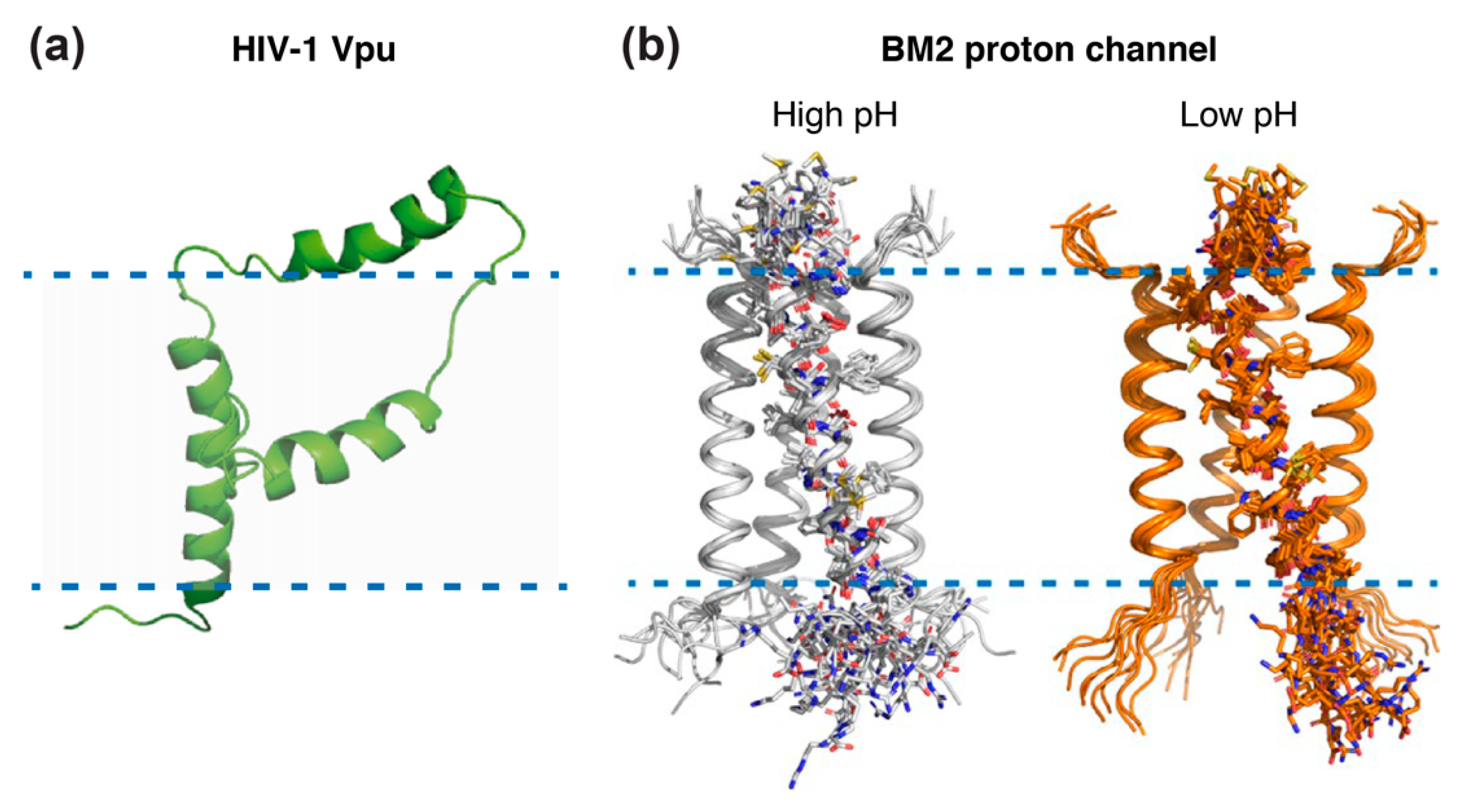
Beyond gp41, the 81 residue HIV-1 specific virus protein “u” (Vpu) accessory protein has been addressed by solid-state NMR [127,128,129]. Vpu represents an oligomeric integral membrane phosphoprotein with numerous biological functions. The structure of full-length Vpu was solved in proteoliposomes and displayed two helices in the cytoplasmic domain forming a U-shape (Figure 7a). NMR data also revealed that the length of the inter-helical loop and the orientation of the third helix vary with the lipid composition, demonstrating its flexibility and accessibility for potential interactions [127].
5.3.2. Influenza M2 Channel
Other well-studied examples of viral membrane proteins using solid-state NMR are Matrix-2 proteins of Influenza A (AM2) and B (BM2) viruses, which are multifunctional proteins that act at several stages of the virus life cycle and have a proton channel function essential for viral replication [131]. Both proteins are activated by low pH to mediate virus uncoating. AM2 conducts protons with strong inward rectification, while BM2 conducts protons both inward and outward. The structure of their trans-membrane channel domain has been extensively characterized by NMR spectroscopy, X-ray crystallography, and other physical methods [131]. Solid-state NMR added important information on the structure of the AM2 proton channel, thanks to seven successive studies: the structure of the monomeric transmembrane region was solved in 2001 [132], followed by the structure of the full transmembrane domain in lipid bilayers, in free form [133] and in complex with amantadine [134,135,136], an antiviral able to block the M2 ion channel pore. Additional information was gained from the structures of the drug-resistant AM2 mutant S31N [137,138], showing that amantadine resistance of mutant S31N is caused by steric hindrance. More recently, the solid-state NMR structures of the BM2 channel in its open and closed conformation were solved in a phospholipid environment [130] (Figure 7b), revealing that the channel is more accessible to water at a low pH and that side chain dynamics are the essential driver of proton shuttling. Solid-state NMR structures were all solved in lipid bilayers, whereas most X-ray structures, e.g., of the transmembrane domain of AM2 in 2008 [139], were solved in detergents. Notably, for the transmembrane domain of BM2, it was shown that the binding to micelles may cause structural perturbations that do not reflect the structure in native lipid bilayers [140].
In addition to the resolution of protein structures at the atomic level, solid-state NMR can be used to measure membrane curvature and determine the binding site of a protein in the membrane using magnetically oriented bicelles and off-magic-angle spinning. This approach was successfully applied to the M2 protein [141], revealing that it mediates virus assembly and induces high membrane curvature. These experiments can be applicable to other proteins inducing membrane-curvature, including those involved in membrane trafficking, membrane fusion and cell division.
5.3.3. HCV p7 and NS4B
All proteins from viruses in the Flaviviridae family, including HCV and Dengue, are membrane-bound. Not only the structural proteins, but also the non-structural proteins thus represent an important challenge to analyze their conformations. Two HCV proteins have been subject to solid-state NMR studies, p7 and NS4B.
P7 protein is a small, hydrophobic transmembrane protein that participates in viral assembly and release. This 63-amino acid protein is generally categorized as a viroporin because it can oligomerize as hexamers to form ion channels in host cell membranes. Its structure is made up of seven different sections, amongst which four are helical segments. Early solid-state NMR studies of p7 aligned in phospholipid bilayers, providing the tilt angles of two of these segments [142] and a topology model of the protein in this environment [143]. p7 contains two trans-membrane helices, while HIV-1 Vpu has only one. Comparison of the solid-state NMR structures of both viroporins revealed significant differences in the structures and dynamics of their internal loop and terminal regions [144]. In both cases, solid-state NMR was compared to solution-state NMR data obtained in micelles, and Opella and coworkers first identified the localization of the transmembrane domains of p7 and Vpu in lipid bilayers. Unfortunately, the full p7 structure in lipids could not yet be solved, as this would help to resolve contrasting data between different detergent-based solution NMR studies [145,146,147,148,149]; the membrane might actually play a central role in determining p7 structure and oligomerization.
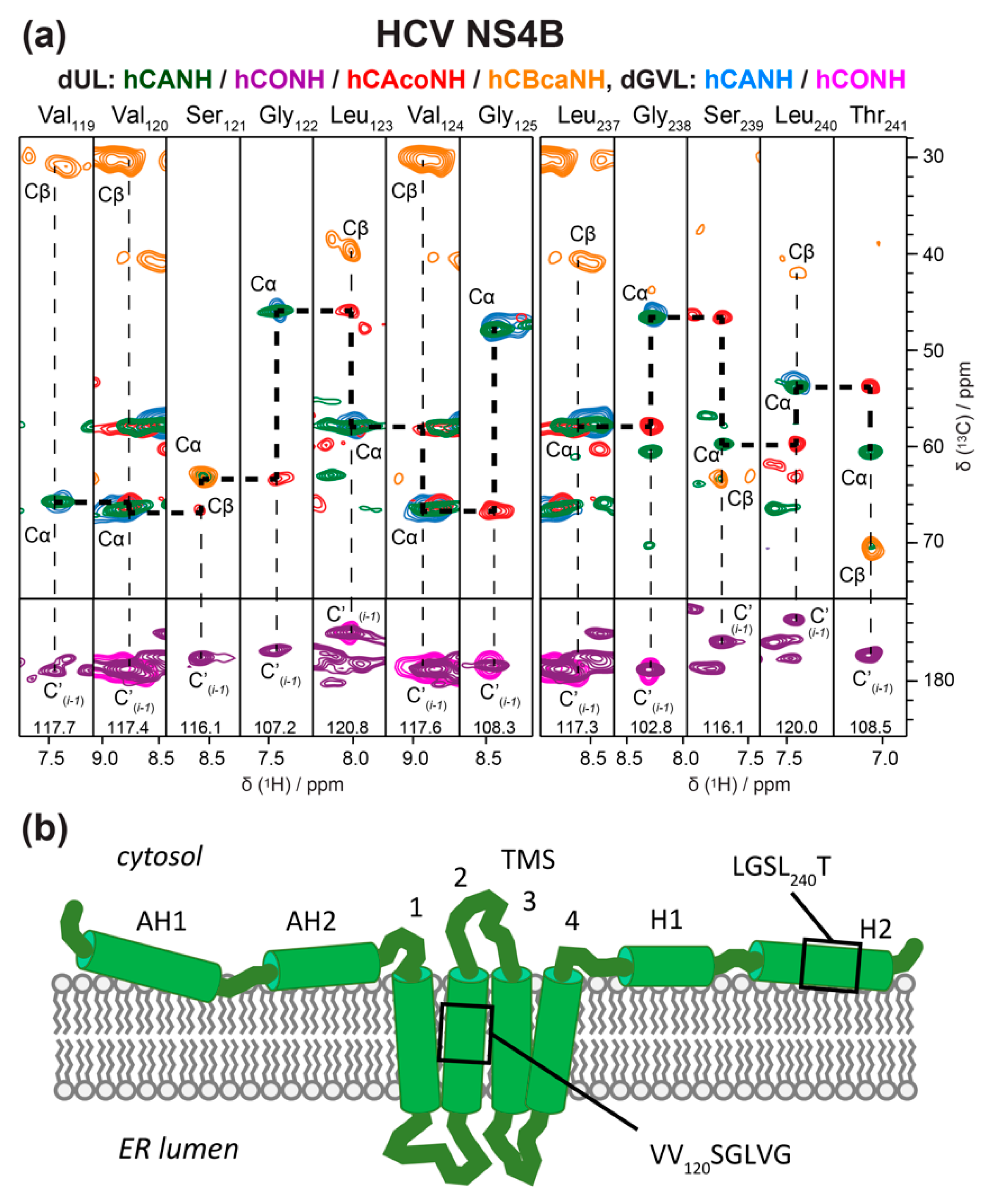
Another HCV protein on which studies in lipids are underway is the nonstructural protein 4B (NS4B). NS4B is a 27 kDa alpha-helical integral membrane protein of 261 amino acid residues. While the NS4B 3D structure has not been solved yet, a topology model is available [150]; it proposes that membrane association of NS4B is mediated by both transmembrane domains in its central parts, and determinants for membrane association in the N- and C-terminal regions [151]. However, given its important role for HCV, the need for more detailed structural information on NS4B is evident. A solid-state NMR study combined with molecular dynamic calculations looked at the second amphipathic helix of NS4B (AH2), known to promote remodeling events that are essential in viral replication [152]. Using a synthetic peptide reconstituted into lipids, the authors showed that AH2 promotes the clustering of negatively charged lipids within the bilayer, facilitating the remodeling of the latter, and increasing the disassociation of AH2 oligomers. Yet, insight into the multiple functions of NS4B which might be governed by distinct membrane topologies and/or interactions with other viral and cellular proteins [151] would be greatly supported by detailed structural information on the full-length protein. Again, WG-CFPS represents an attractive alternative to the poor expression of NSB4 in bacteria. Indeed, allowing for the addition of various additives, including detergents, this system enabled the production of full-length, well-folded and soluble NS4B in milligram amounts, compatible with structural studies [153,154]. The detergent-solubilized NS4B protein was affinity-purified, and then reinserted into lipids to mimic the native membranous environment [59,67,155]. A technical issue in such a reconstitution experiment is finding the optimal lipid-to-protein ratio (LPR) for NMR samples, because the LPR needs to be high enough to ensure a well-folded protein, but as low as possible to maximize protein amounts in the NMR rotor [44]. Adapting recent fast reconstitution schemes [156] has allowed to screen different lipid compositions directly by NMR to optimize resolution [67]. This enabled the recording of 2D and 3D 1H-detected NMR spectra, both on fully and selectively labeled NS4B and to initiate sequential assignments [67] (Figure 8). Two protein segments could be assigned, one validated the structural model, while the other did not confirm the predicted helix. For further work, higher magnetic fields will be an essential ingredient to push the structural understanding of this complex protein.
6. Conclusions and Outlook
Solid-state NMR is advancing in the analysis of viral proteins, concentrating on targets and aspects thereof which are complementary to other high-resolution approaches. It can typically provide important information on dynamic aspects, and also efficiently screen multiple forms of proteins created under different conditions, be it polymorphism, multimerization state, or interaction with partners. A uniquely strong point is the detection of conformational differences with very high sensitivity yet without the need for full structure determination. The recent development of fast MAS and the concomitant use of high-sensitivity proton detection has reduced sample amounts by a spectacular factor of a hundred, paving the way towards studying difficult (membrane) proteins which are only accessible in small amounts. The sensitivity gain is also essential to characterize the amplitude and correlation times of motional processes by NMR relaxation measurements. Solid-state NMR is also able to fill in the missing pieces in the dynamic structures protein access in the context of the viral life cycle, through its high tolerance towards sample preparation conditions, without the need for crystals or symmetry. It can thus reveal unique information on dynamics, membrane-orientation or interactions with partners. As strikingly illustrated for electron microscopy by the resolution revolution made possible by new detector techniques, many advances in solid-state NMR are also linked to technological innovation. Key developments under way are even faster magic-angle spinning, and ever higher field magnets, with the concomitant enabling a gain in resolution and sensitivity. Together, they are bringing many viral proteins and their assemblies into the focus of NMR investigations. Particularly novel and promising is the emerging opportunity to now include viruses that do not form ordered symmetrical capsids, but rather less regular nucleoprotein structures, like for example the ribonucleoprotein complexes from the dengue virus or the hepatitis D virus. Another interesting new option is the combination of solid-state NMR with cryo-EM in a hybrid structural approach, as established, for example, for amyloid fibrils [157,158]. These developments open exciting new research avenues in the future.
Author Contributions
Writing, L.L., M.-L.F., B.H.M., M.N. and A.B.; figure preparation, L.L., B.H.M. and A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the CNRS (CNRS-Momentum 2018) and the French Agence Nationale de Recherches sur le Sida et les hépatites virales (ANRS, ECTZ71388 & ECTZ100488).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hopper, P.; Harrison, S.C.; Sauer, R.T. Structure of tomato bushy stunt virus. V. Coat protein sequence determination and its structural implications. J. Mol. Biol. 1984, 177, 701–713. [Google Scholar] [CrossRef]
- Silva, A.M.; Rossmann, M.G. Refined structure of southern bean mosaic virus at 2.9 A resolution. J. Mol. Biol. 1987, 197, 69–87. [Google Scholar] [CrossRef]
- Krishnaswamy, S.; Rossmann, M.G. Structural refinement and analysis of Mengo virus. J. Mol. Biol. 1990, 211, 803–844. [Google Scholar] [CrossRef]
- Arnold, E.; Rossmann, M.G. The use of molecular-replacement phases for the refinement of the human rhinovirus 14 structure. Acta Crystallogr. A Found. Crystallogr. 1988, 44, 270–282. [Google Scholar] [CrossRef]
- Kim, S.S.; Smith, T.J.; Chapman, M.S.; Rossmann, M.C.; Pevear, D.C.; Dutko, F.J.; Felock, P.J.; Diana, G.D.; McKinlay, M.A. Crystal structure of human rhinovirus serotype 1A (HRV1A). J. Mol. Biol. 1989, 210, 91–111. [Google Scholar] [CrossRef]
- Filman, D.J.; Syed, R.; Chow, M.; Macadam, A.J.; Minor, P.D.; Hogle, J.M. Structural factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO J. 1989, 8, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Wynne, S.A.; Crowther, R.A.; Leslie, A.G. The crystal structure of the human hepatitis B virus capsid. Mol. Cell 1999, 3, 771–780. [Google Scholar] [CrossRef]
- Elias, M.; Liebschner, D.; Koepke, J.; Lecomte, C.; Guillot, B.; Jelsch, C.; Chabriere, E. Hydrogen atoms in protein structures: High-resolution X-ray diffraction structure of the DFPase. BMC Res. Notes 2013, 6, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen atoms can be located accurately and precisely by x-ray crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef] [Green Version]
- Packianathan, C.; Katen, S.P.; Dann, C.E.; Zlotnick, A. Conformational Changes in the Hepatitis B Virus Core Protein Are Consistent with a Role for Allostery in Virus Assembly. J. Virol. 2010, 84, 1607–1615. [Google Scholar] [CrossRef] [Green Version]
- Klumpp, K.; Lam, A.M.; Lukacs, C.; Vogel, R.; Ren, S.; Espiritu, C.; Baydo, R.; Atkins, K.; Abendroth, J.; Liao, G.; et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc. Natl. Acad. Sci. USA 2015, 112, 15196–15201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, B.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray crystallographic structure of the Norwalk virus capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Guu, T.S.Y.; Cao, J.; Li, Y.; Cheng, L.; Tao, Y.J.; Zhang, J. Structure determination of a human virus by the combination of cryo-EM and X-ray crystallography. Biophys Rep. 2016, 2, 55–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttcher, B.; Wynne, S.A.; Crowther, R.A. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 1997, 386, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.F.; Cheng, N.; Zlotnick, A.; Wingfield, P.T.; Stahl, S.J.; Steven, A.C. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 1997, 386, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, B.; Nassal, M. Structure of Mutant Hepatitis B Core Protein Capsids with Premature Secretion Phenotype. J. Mol. Biol. 2018, 430, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Zhu, D.; Agarkova, I.; Adhikari, J.; Klose, T.; Liu, Y.; Chen, Z.; Sun, Y.; Gross, M.L.; Van Etten, J.L.; et al. Near-atomic structure of a giant virus. Nat. Commun. 2019, 10, 388. [Google Scholar] [CrossRef] [Green Version]
- Makbul, C.; Nassal, M.; Böttcher, B. Slowly folding surface extension in the prototypic avian hepatitis B virus capsid governs stability. eLife 2020, 9. [Google Scholar] [CrossRef]
- Bloch, F.; Hansen, W.W.; Packard, M. The Nuclear Induction Experiment. Phys. Rev. 1946, 70, 474–485. [Google Scholar] [CrossRef]
- Purcell, E.M.; Torrey, H.C.; Pound, R.V. Resonance Absorption by Nuclear Magnetic Moments in a Solid. Phys. Rev. 1946, 69, 37–38. [Google Scholar] [CrossRef]
- Aue, W.P.; Bartholdi, E.; Ernst, R.R. Two-dimensional spectroscopy. Application to nuclear magnetic resonance. J. Chem. Phys. 1976, 64, 2229–2246. [Google Scholar] [CrossRef] [Green Version]
- Jeener, J.; Meier, B.H.; Bachmann, P.; Ernst, R.R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- Jeener, J.; Alewaeters, G. “Pulse pair technique in high resolution NMR” a reprint of the historical 1971 lecture notes on two-dimensional spectroscopy. Prog. Nucl. Mag. Res. Spectrosc. 2016, 94–95, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, K. NMR studies of structure and function of biological macromolecules (Nobel lecture). Angew. Chem. Int. Ed. Engl. 2003, 42, 3340–3363. [Google Scholar] [CrossRef] [PubMed]
- Religa, T.L.; Ruschak, A.M.; Rosenzweig, R.; Kay, L.E. Site-directed methyl group labeling as an NMR probe of structure and dynamics in supramolecular protein systems: Applications to the proteasome and to the ClpP protease. J. Am. Chem. Soc. 2011, 133, 9063–9068. [Google Scholar] [CrossRef]
- Andrew, E.R.; Bradbury, A.; Eades, R.G. Removal of Dipolar Broadening of Nuclear Magnetic Resonance Spectra of Solids by Specimen Rotation. Nature 1959, 183, 1802–1803. [Google Scholar] [CrossRef]
- Zhang, Z.; Oss, A.; Org, M.-L.; Samoson, A.; Li, M.; Tan, H.; Su, Y.; Yang, J. Selectively Enhanced 1H-1H Correlations in Proton-Detected Solid-State NMR under Ultrafast MAS Conditions. J. Phys. Chem. Lett. 2020, 11, 8077–8083. [Google Scholar] [CrossRef]
- Schledorn, M.; Malär, A.A.; Torosyan, A.; Penzel, S.; Klose, D.; Oss, A.; Org, M.-L.; Wang, S.; Lecoq, L.; Cadalbert, R.; et al. Protein NMR Spectroscopy at 150 kHz Magic-Angle Spinning Continues To Improve Resolution and Mass Sensitivity. ChemBioChem. Biol. 2020, 115, 11519. [Google Scholar] [CrossRef]
- McDermott, A.E.; Polenova, T.; Böckmann, A.; Zilm, K.W.; Paulsen, E.K.; Martin, R.W.; Montelione, G.T. Partial NMR assignments for uniformly (13C, 15N)-enriched BPTI in the solid state. J. Biomol. NMR 2000, 16, 209–219. [Google Scholar] [CrossRef]
- Castellani, F.; van Rossum, B.; Diehl, A.; Schubert, M.; Rehbein, K.; Oschkinat, H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature 2002, 420, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Linser, R.; Bardiaux, B.; Andreas, L.B.; Hyberts, S.G.; Morris, V.K.; Pintacuda, G.; Sunde, M.; Kwan, A.H.; Wagner, G. Solid-State NMR Structure Determination from Diagonal-Compensated, Sparsely Nonuniform-Sampled 4D Proton–Proton Restraints. J. Am. Chem. Soc. 2014, 136, 11002–11010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbet-Massin, E.; Pell, A.J.; Retel, J.S.; Andreas, L.B.; Jaudzems, K.; Franks, W.T.; Nieuwkoop, A.J.; Hiller, M.; Higman, V.; Guerry, P.; et al. Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J. Am. Chem. Soc. 2014, 136, 12489–12497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Penzel, S.; Szekely, K.; Cadalbert, R.; Testori, E.; Oss, A.; Past, J.; Samoson, A.; Ernst, M.; Böckmann, A.; et al. De novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angew. Chem. Int. Ed. 2014, 53, 12253–12256. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, L.; Schledorn, M.; Wang, S.; Smith-Penzel, S.; Malär, A.A.; Callon, M.; Nassal, M.; Meier, B.H.; Böckmann, A. 100 kHz MAS Proton-Detected NMR Spectroscopy of Hepatitis B Virus Capsids. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Penzel, S.; Oss, A.; Org, M.-L.; Samoson, A.; Böckmann, A.; Ernst, M.; Meier, B.H. Spinning faster: Protein NMR at MAS frequencies up to 126 kHz. J. Biomol. NMR 2019, 73, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardiennet, C.; Schütz, A.K.; Hunkeler, A.; Kunert, B.; Terradot, L.; Böckmann, A.; Meier, B.H. A Sedimented Sample of a 59 kDa Dodecameric Helicase Yields High-Resolution Solid-State NMR Spectra. Angew. Chem. Int. Ed. 2012, 51, 7855–7858. [Google Scholar] [CrossRef]
- Bertini, I.; Luchinat, C.; Parigi, G.; Ravera, E.; Reif, B.; Turano, P. Solid-state NMR of proteins sedimented by ultracentrifugation. Proc. Natl. Acad. Sci. USA 2011, 108, 10396–10399. [Google Scholar] [CrossRef] [Green Version]
- Andreas, L.B.; Jaudzems, K.; Stanek, J.; Lalli, D.; Bertarello, A.; Le Marchand, T.; Cala-De Paepe, D.; Kotelovica, S.; Akopjana, I.; Knott, B.; et al. Structure of fully protonated proteins by proton-detected magic-angle spinning NMR. Proc. Natl. Acad. Sci. USA 2016, 113, 9187–9192. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Fogeron, M.-L.; Schledorn, M.; Dujardin, M.; Penzel, S.; Burdette, D.; Berke, J.M.; Nassal, M.; Lecoq, L.; Meier, B.H.; et al. Combining Cell-Free Protein Synthesis and NMR Into a Tool to Study Capsid Assembly Modulation. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- David, G.; Fogeron, M.-L.; Schledorn, M.; Montserret, R.; Haselmann, U.; Penzel, S.; Badillo, A.; Lecoq, L.; André, P.; Nassal, M.; et al. Structural Studies of Self-Assembled Subviral Particles: Combining Cell-Free Expression with 110 kHz MAS NMR Spectroscopy. Angew. Chem. Int. Ed. 2018, 57, 4787–4791. [Google Scholar] [CrossRef] [PubMed]
- Schanda, P.; Ernst, M. Studying dynamics by magic-angle spinning solid-state NMR spectroscopy: Principles and applications to biomolecules. Prog. Nucl. Mag. Res. Sp. 2016, 96, 1–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid Fibrils of the HET-s(218-289) Prion Form a Solenoid with a Triangular Hydrophobic Core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Lacabanne, D.; Fogeron, M.-L.; Wiegand, T.; Cadalbert, R.; Meier, B.H.; Böckmann, A. Protein sample preparation for solid-state NMR investigations. Prog. Nucl. Mag. Res. Spectrosc. 2019, 110, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Thiriot, D.S.; Nevzorov, A.A.; Zagyanskiy, L.; Wu, C.H.; Opella, S.J. Structure of the Coat Protein in Pf1 Bacteriophage Determined by Solid-state NMR Spectroscopy. J. Mol. Biol. 2004, 341, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Goldbourt, A.; Gross, B.J.; Day, L.A.; McDermott, A.E. Filamentous Phage Studied by Magic-Angle Spinning NMR: Resonance Assignment and Secondary Structure of the Coat Protein in Pf1. J. Am. Chem. Soc. 2007, 129, 2338–2344. [Google Scholar] [CrossRef]
- Morag, O.; Abramov, G.; Goldbourt, A. Complete Chemical Shift Assignment of the ssDNA in the Filamentous Bacteriophage fd Reports on Its Conformation and on Its Interface with the Capsid Shell. J. Am. Chem. Soc. 2014, 136, 2292–2301. [Google Scholar] [CrossRef]
- Abramov, G.; Morag, O.; Goldbourt, A. Magic-angle spinning NMR of intact bacteriophages: Insights into the capsid, DNA and their interface. J. Magn. Reson. 2015, 253, 80–90. [Google Scholar] [CrossRef]
- Sergeyev, I.V.; Day, L.A.; Goldbourt, A.; McDermott, A.E. Chemical Shifts for the Unusual DNA Structure in Pf1 Bacteriophage from Dynamic-Nuclear-Polarization-Enhanced Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2011, 133, 20208–20217. [Google Scholar] [CrossRef]
- Sergeyev, I.V.; Itin, B.; Rogawski, R.; Day, L.A.; McDermott, A.E. Efficient assignment and NMR analysis of an intact virus using sequential side-chain correlations and DNP sensitization. Proc. Natl. Acad. Sci. USA 2017, 114, 5171–5176. [Google Scholar] [CrossRef] [Green Version]
- Maly, T.; Debelouchina, G.T.; Bajaj, V.S.; Hu, K.-N.; Joo, C.-G.; Mak-Jurkauskas, M.L.; Sirigiri, J.R.; van der Wel, P.C.A.; Herzfeld, J.; Temkin, R.J.; et al. Dynamic nuclear polarization at high magnetic fields. J. Chem. Phys. 2008, 128, 052211. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, T.; Hunkeler, A.; Däpp, A.; Verasdonck, J.; Cadalbert, R.; Bousset, L.; Melki, R.; Böckmann, A.; Meier, B.H. CONFINE-MAS: A magic-angle spinning NMR probe that confines the sample in case of a rotor explosion. J. Biomol. NMR 2018, 72, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Penzel, S.; Smith, A.A.; Agarwal, V.; Hunkeler, A.; Org, M.-L.; Samoson, A.; Böckmann, A.; Ernst, M.; Meier, B.H. Protein resonance assignment at MAS frequencies approaching 100 kHz: A quantitative comparison of J-coupling and dipolar-coupling-based transfer methods. J. Biomol. NMR 2015, 63, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, T.; Lacabanne, D.; Torosyan, A.; Boudet, J.; Cadalbert, R.; Allain, F.H.T.; Meier, B.H.; Böckmann, A. Sedimentation Yields Long-Term Stable Protein Samples as Shown by Solid-State NMR. Front. Mol. Biosci. 2020, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecoq, L.; Wang, S.; Wiegand, T.; Bressanelli, S.; Nassal, M.; Meier, B.H.; Böckmann, A. Localizing Conformational Hinges by NMR: Where Do Hepatitis B Virus Core Proteins Adapt for Capsid Assembly? ChemPhysChem 2018, 19, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Saxena, K.; Schwalbe, H.; Klein-Seetharaman, J. Isotope labeling in mammalian cells. Methods Mol. Biol. 2012, 831, 55–69. [Google Scholar]
- Heger-Stevic, J.; Zimmermann, P.; Lecoq, L.; Böttcher, B.; Nassal, M. Hepatitis B virus core protein phosphorylation: Identification of the SRPK1 target sites and impact of their occupancy on RNA binding and capsid structure. PLoS Pathog 2018, 14, e1007488-31. [Google Scholar] [CrossRef] [Green Version]
- David, G.; Fogeron, M.-L.; Montserret, R.; Lecoq, L.; Page, A.; Delolme, F.; Nassal, M.; Böckmann, A. Phosphorylation and Alternative Translation on Wheat Germ Cell-Free Protein Synthesis of the DHBV Large Envelope Protein. Front. Mol. Biosci. 2019, 6, 138. [Google Scholar] [CrossRef]
- Fogeron, M.-L.; Jirasko, V.; Penzel, S.; Paul, D.; Montserret, R.; Danis, C.; Lacabanne, D.; Badillo, A.; Gouttenoire, J.; Moradpour, D.; et al. Cell-free expression, purification, and membrane reconstitution for NMR studies of the nonstructural protein 4B from hepatitis C virus. J. Biomol. NMR 2016, 65, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Jirasko, V.; Lakomek, N.-A.; Penzel, S.; Fogeron, M.-L.; Bartenschlager, R.; Meier, B.H.; Böckmann, A. Proton-Detected Solid-State NMR of the Cell-Free Synthesized α-Helical Transmembrane Protein NS4B from Hepatitis C Virus. ChemBioChem. Biol. 2020, 1, 5–9. [Google Scholar] [CrossRef]
- Morita, E.-H.; Sawasaki, T.; Tanaka, R.; Endo, Y.; Kohno, T. A wheat germ cell-free system is a novel way to screen protein folding and function. Protein Sci. 2003, 12, 1216–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, E.-H.; Shimizu, M.; Ogasawara, T.; Endo, Y.; Tanaka, R.; Kohno, T. A novel way of amino acid-specific assignment in (1)H-(15)N HSQC spectra with a wheat germ cell-free protein synthesis system. J. Biomol. NMR 2004, 30, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Singarapu, K.K.; Makino, S.-I.; Sahu, S.C.; Matsubara, Y.; Endo, Y.; Kainosho, M.; Markley, J.L. Hydrogen exchange during cell-free incorporation of deuterated amino acids and an approach to its inhibition. J. Biomol. NMR 2011, 51, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevelkov, V.; Rehbein, K.; Diehl, A.; Reif, B. Ultrahigh resolution in proton solid-state NMR spectroscopy at high levels of deuteration. Angew. Chem. Int. Ed. Engl. 2006, 45, 3878–3881. [Google Scholar] [CrossRef] [PubMed]
- Asami, S.; Szekely, K.; Schanda, P.; Meier, B.H.; Reif, B. Optimal degree of protonation for 1H detection of aliphatic sites in randomly deuterated proteins as a function of the MAS frequency. J. Biomol. NMR 2012, 54, 155–168. [Google Scholar] [CrossRef]
- Kohno, T.; Endo, Y. Production of protein for nuclear magnetic resonance study using the wheat germ cell-free system. Methods Mol. Biol. 2007, 375, 257–272. [Google Scholar]
- Gupta, S.; Tycko, R. Segmental isotopic labeling of HIV-1 capsid protein assemblies for solid state NMR. J. Biomol. NMR 2018, 70, 103–114. [Google Scholar] [CrossRef]
- Gupta, S.; Louis, J.M.; Tycko, R. Effects of an HIV-1 maturation inhibitor on the structure and dynamics of CA-SP1 junction helices in virus-like particles. Proc. Natl. Acad. Sci. USA 2020, 117, 10286–10293. [Google Scholar] [CrossRef]
- Etzkorn, M.; Böckmann, A.; Lange, A.; Baldus, M. Probing molecular interfaces using 2D magic-angle-spinning NMR on protein mixtures with different uniform labeling. J. Am. Chem. Soc. 2004, 126, 14746–14751. [Google Scholar] [CrossRef]
- Lundström, P.; Teilum, K.; Carstensen, T.; Bezsonova, I.; Wiesner, S.; Hansen, D.F.; Religa, T.L.; Akke, M.; Kay, L.E. Fractional 13C enrichment of isolated carbons using [1-13C]- or [2- 13C]-glucose facilitates the accurate measurement of dynamics at backbone Calpha and side-chain methyl positions in proteins. J. Biomol. NMR 2007, 38, 199–212. [Google Scholar] [CrossRef]
- Loquet, A.; Habenstein, B.; Lange, A. Structural Investigations of Molecular Machines by Solid-State NMR. Acc. Chem. Res. 2013, 46, 2070–2079. [Google Scholar] [CrossRef] [PubMed]
- Rienstra, C.M.; Hohwy, M.; Hong, M.; Griffin, R.G. 2D and 3D 15N− 13C− 13C NMR Chemical Shift Correlation Spectroscopy of Solids: Assignment of MAS Spectra of Peptides. J. Am. Chem. Soc. 2000, 122, 10979–10990. [Google Scholar] [CrossRef]
- Pauli, J.; Baldus, M.; van Rossum, B.; de Groot, H.; Oschkinat, H. Backbone and side-chain 13C and 15N signal assignments of the alpha-spectrin SH3 domain by magic angle spinning solid-state NMR at 17.6 Tesla. ChemBioChem. Biol. 2001, 2, 272–281. [Google Scholar] [CrossRef]
- Habenstein, B.; Wasmer, C.; Bousset, L.; Sourigues, Y.; Schütz, A.; Loquet, A.; Meier, B.H.; Melki, R.; Böckmann, A. Extensive de novo solid-state NMR assignments of the 33 kDa C-terminal domain of the Ure2 prion. J. Biomol. NMR 2011, 51, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuetz, A.; Wasmer, C.; Habenstein, B.; Verel, R.; Greenwald, J.; Riek, R.; Böckmann, A.; Meier, B.H. Protocols for the sequential solid-state NMR spectroscopic assignment of a uniformly labeled 25 kDa protein: HET-s(1-227). ChemBioChem. Biol. 2010, 11, 1543–1551. [Google Scholar] [CrossRef]
- Porterfield, J.Z.; Dhason, M.S.; Loeb, D.D.; Nassal, M.; Stray, S.J.; Zlotnick, A. Full-Length Hepatitis B Virus Core Protein Packages Viral and Heterologous RNA with Similarly High Levels of Cooperativity. J. Virol. 2010, 84, 7174–7184. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Yao, Y.; Marassi, F.M. Membrane protein structure determination in membrana. Acc. Chem. Res. 2013, 46, 2182–2190. [Google Scholar] [CrossRef] [Green Version]
- Güntert, P. Automated structure determination from NMR spectra. Eur. Biophys. J. 2009, 38, 129–143. [Google Scholar] [CrossRef]
- Linge, J.P.; O’Donoghue, S.I.; Nilges, M. Automated assignment of ambiguous nuclear overhauser effects with ARIA. Meth. Enzymol. 2001, 339, 71–90. [Google Scholar]
- Bardiaux, B.; van Rossum, B.-J.; Nilges, M.; Oschkinat, H. Efficient modeling of symmetric protein aggregates from NMR data. Angew. Chem. Int. Ed. 2012, 51, 6916–6919. [Google Scholar] [CrossRef]
- Davis, B. Screening Protein–Small Molecule Interactions by NMR. In Protein NMR; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1008, pp. 389–413. [Google Scholar]
- Dias, D.M.; Ciulli, A. NMR approaches in structure-based lead discovery: Recent developments and new frontiers for targeting multi-protein complexes. Prog. Biophys. Mol. Biol. 2014, 116, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opella, S.J.; Zeri, A.C.; Park, S.H. Structure, Dynamics, and Assembly of Filamentous Bacteriophages by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Phys. Chem. 2008, 59, 635–657. [Google Scholar] [CrossRef] [PubMed]
- Goldbourt, A. Structural characterization of bacteriophage viruses by NMR. Prog. Nucl. Mag. Res. Spectrosc. 2019, 114–115, 192–210. [Google Scholar] [CrossRef] [PubMed]
- Marassi, F.M. NMR of peptides and proteins in oriented membranes. Concepts Magn. Reson. 2002, 14, 212–224. [Google Scholar] [CrossRef]
- Urban, S.; Bartenschlager, R.; Kubitz, R.; Zoulim, F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology 2014, 147, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Hill, C.P.; Sundquist, W.I.; Finch, J.T. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 2000, 407, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.M.; Lu, M.; Suiter, C.L.; Hou, G.; Zhang, H.; Polenova, T. Magic angle spinning NMR of viruses. Prog. Nucl. Mag. Res. Sp. 2015, 86–87, 21–40. [Google Scholar] [CrossRef] [Green Version]
- Suiter, C.L.; Quinn, C.M.; Lu, M.; Hou, G.; Zhang, H.; Polenova, T. MAS NMR of HIV-1 protein assemblies. J. Magn. Reson. 2015, 253, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Ahn, J.; Concel, J.; Byeon, I.-J.L.; Gronenborn, A.M.; Yang, J.; Polenova, T. Solid-State NMR Studies of HIV-1 Capsid Protein Assemblies. J. Am. Chem. Soc. 2010, 132, 1976–1987. [Google Scholar] [CrossRef] [Green Version]
- Byeon, I.-J.L.; Hou, G.; Han, Y.; Suiter, C.L.; Ahn, J.; Jung, J.; Byeon, C.-H.; Gronenborn, A.M.; Polenova, T. Motions on the millisecond time scale and multiple conformations of HIV-1 capsid protein: Implications for structural polymorphism of CA assemblies. J. Am. Chem. Soc. 2012, 134, 6455–6466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Hou, G.; Zhang, H.; Suiter, C.L.; Ahn, J.; Byeon, I.-J.L.; Perilla, J.R.; Langmead, C.J.; Hung, I.; Gor’kov, P.L.; et al. Dynamic allostery governs cyclophilin A-HIV capsid interplay. Proc. Natl. Acad. Sci. USA 2015, 112, 14617–14622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Hou, G.; Lu, M.; Ahn, J.; Byeon, I.-J.L.; Langmead, C.J.; Perilla, J.R.; Hung, I.; Gor’kov, P.L.; Gan, Z.; et al. HIV-1 Capsid Function is Regulated by Dynamics: Quantitative Atomic-Resolution Insights by Integrating Magic-Angle-Spinning NMR, QM/MM, and MD. J. Am. Chem. Soc. 2016, 138, 14066–14075. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.M.; Wang, M.; Fritz, M.P.; Runge, B.; Ahn, J.; Xu, C.; Perilla, J.R.; Gronenborn, A.M.; Polenova, T. Dynamic regulation of HIV-1 capsid interaction with the restriction factor TRIM5α identified by magic-angle spinning NMR and molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2018, 115, 11519–11524. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Tycko, R. Structural and dynamical characterization of tubular HIV-1 capsid protein assemblies by solid state nuclear magnetic resonance and electron microscopy. Protein Sci. 2010, 19, 716–730. [Google Scholar] [CrossRef] [Green Version]
- Bayro, M.J.; Chen, B.; Yau, W.-M.; Tycko, R. Site-Specific Structural Variations Accompanying Tubular Assembly of the HIV-1 Capsid Protein. J. Mol. Biol. 2014, 426, 1109–1127. [Google Scholar] [CrossRef] [Green Version]
- Bayro, M.J.; Ganser-Pornillos, B.K.; Zadrozny, K.K.; Yeager, M.; Tycko, R. Helical Conformation in the CA-SP1 Junction of the Immature HIV-1 Lattice Determined from Solid-State NMR of Virus-like Particles. J. Am. Chem. Soc. 2016, 138, 12029–12032. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef]
- Crowther, R.A.; Kiselev, N.A.; Böttcher, B.; Berriman, J.A.; Borisova, G.P.; Ose, V.; Pumpens, P. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 1994, 77, 943–950. [Google Scholar] [CrossRef]
- Kenney, J.M.; von Bonsdorff, C.H.; Nassal, M.; Fuller, S.D. Evolutionary conservation in the hepatitis B virus core structure: Comparison of human and duck cores. Structure 1995, 3, 1009–1019. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Jin, L.; Jih, J.; Shih, C.; Hong Zhou, Z. 3.5 Å cryo-EM Structure of Hepatitis B Virus Core Assembled from Full-Length Core Protein. PLoS ONE 2013, 8, e69729. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 1992, 66, 4107–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B--like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Ning, X.; Nguyen, D.; Mentzer, L.; Adams, C.; Lee, H.; Ashley, R.; Hafenstein, S.; Hu, J. Secretion of Genome-Free Hepatitis B Virus—Single Strand Blocking Model for Virion Morphogenesis of Para-retrovirus. PLoS Pathog. 2011, 7, e1002255-14. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, L.; Wang, S.; Wiegand, T.; Bressanelli, S.; Nassal, M.; Meier, B.H.; Böckmann, A. Solid-state [13C-15N] NMR resonance assignment of hepatitis B virus core protein. Biomol. NMR Assign. 2018, 12, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, F.; Tong, X.; Hoffmann, D.; Zuo, J.; Lu, M. Treatment of Chronic Hepatitis B Virus Infection Using Small Molecule Modulators of Nucleocapsid Assembly: Recent Advances and Perspectives. ACS Infect Dis. 2019, 5, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Verbinnen, T.; Tan, Y.; Wang, G.; Dehertogh, P.; Vergauwen, K.; Neefs, J.-M.; Jacoby, E.; Lenz, O.; Berke, J.M. Anti-HBV activity of the HBV capsid assembly modulator JNJ-56136379 across full-length genotype A–H clinical isolates and core site-directed mutants in vitro. J. Antimicrob. Chemother. 2020, 75, 2526–2534. [Google Scholar] [CrossRef]
- Schlicksup, C.J.; Laughlin, P.; Dunkelbarger, S.; Wang, J.C.-Y.; Zlotnick, A. Local Stabilization of Subunit–Subunit Contacts Causes Global Destabilization of Hepatitis B Virus Capsids. ACS Chem. Biol. 2020, 15, 1708–1717. [Google Scholar] [CrossRef]
- Barbet-Massin, E.; Felletti, M.; Schneider, R.; Jehle, S.; Communie, G.; Martinez, N.; Jensen, M.R.; Ruigrok, R.W.H.; Emsley, L.; Lesage, A.; et al. Insights into the structure and dynamics of measles virus nucleocapsids by 1H-detected solid-state NMR. Biophys. J. 2014, 107, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Desfosses, A.; Goret, G.; Farias Estrozi, L.; Ruigrok, R.W.H.; Gutsche, I. Nucleoprotein-RNA orientation in the measles virus nucleocapsid by three-dimensional electron microscopy. J. Virol. 2011, 85, 1391–1395. [Google Scholar] [CrossRef] [Green Version]
- Gutsche, I.; Desfosses, A.; Effantin, G.; Ling, W.L.; Haupt, M.; Ruigrok, R.W.H.; Sachse, C.; Schoehn, G. Structural virology. Near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 2015, 348, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, B.S. Australia antigen and the biology of hepatitis B. Science 1977, 197, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heermann, K.H.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W.H. Large Surface Proteins of Hepatitis B Virus containing the Pre-s Sequence. J. Virol. 1984, 52, 396–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eble, B.E.; Lingappa, V.R.; Ganem, D. Hepatitis B surface antigen: An unusual secreted protein initially synthesized as a transmembrane polypeptide. Mol. Cell. Biol. 1986, 6, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.; Lünsdorf, H.; Rinas, U. Assessing stability and assembly of the hepatitis B surface antigen into virus-like particles during down-stream processing. Vaccine 2015, 33, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- Opella, S.J. Relating structure and function of viral membrane-spanning miniproteins. Curr. Opin. Virol. 2015, 12, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opella, S.J. Solid-state NMR and membrane proteins. J. Magn. Reson. 2015, 253, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Prorok, M.; Castellino, F.J.; Weliky, D.P. Oligomeric β-Structure of the Membrane-Bound HIV-1 Fusion Peptide Formed from Soluble Monomers. Biophys. J. 2004, 87, 1951–1963. [Google Scholar] [CrossRef] [Green Version]
- Wasniewski, C.M.; Parkanzky, P.D.; Bodner, M.L.; Weliky, D.P. Solid-state nuclear magnetic resonance studies of HIV and influenza fusion peptide orientations in membrane bilayers using stacked glass plate samples. Chem. Phys. Lipids 2004, 132, 89–100. [Google Scholar] [CrossRef]
- Bodner, M.L.; Gabrys, C.M.; Struppe, J.O.; Weliky, D.P. C13–C13 and N15–C13 correlation spectroscopy of membrane-associated and uniformly labeled human immunodeficiency virus and influenza fusion peptides: Amino acid-type assignments and evidence for multiple conformations. J. Chem. Phys. 2008, 128, 052319. [Google Scholar] [CrossRef]
- Yao, H.; Lee, M.W.; Waring, A.J.; Wong, G.C.L.; Hong, M. Viral fusion protein transmembrane domain adopts β-strand structure to facilitate membrane topological changes for virus–cell fusion. Proc. Natl. Acad. Sci. USA 2015, 112, 10926–10931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, E.P.; Curtis-Fisk, J.; Young, K.M.; Weliky, D.P. Solid-State Nuclear Magnetic Resonance (NMR) Spectroscopy of Human Immunodeficiency Virus gp41 Protein That Includes the Fusion Peptide: NMR Detection of Recombinant Fgp41 in Inclusion Bodies in Whole Bacterial Cells and Structural Characterization of Purified and Membrane-Associated Fgp41. Biochemistry 2011, 50, 10013–10026. [Google Scholar] [PubMed] [Green Version]
- Sackett, K.; Nethercott, M.J.; Zheng, Z.; Weliky, D.P. Solid-State NMR Spectroscopy of the HIV gp41 Membrane Fusion Protein Supports Intermolecular Antiparallel β Sheet Fusion Peptide Structure in the Final Six-Helix Bundle State. J. Mol. Biol. 2014, 426, 1077–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratnayake, P.U.; Sackett, K.; Nethercott, M.J.; Weliky, D.P. pH-dependent vesicle fusion induced by the ectodomain of the human immunodeficiency virus membrane fusion protein gp41: Two kinetically distinct processes and fully-membrane-associated gp41 with predominant β sheet fusion peptide conformation. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Morgan, C.A.; Hong, M. Fully hydrophobic HIV gp41 adopts a hemifusion-like conformation in phospholipid bilayers. J. Biol. Chem. 2019, 294, 14732–14744. [Google Scholar] [CrossRef]
- Zhang, H.; Lin, E.C.; Das, B.B.; Tian, Y.; Opella, S.J. Structural determination of virus protein U from HIV-1 by NMR in membrane environments. Biochim. Biophys. Acta 2015, 1848, 3007–3018. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; De Angelis, A.A.; Nevzorov, A.A.; Wu, C.H.; Opella, S.J. Three-Dimensional Structure of the Transmembrane Domain of Vpu from HIV-1 in Aligned Phospholipid Bicelles. Biophys. J. 2006, 91, 3032–3042. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Mrse, A.A.; Nevzorov, A.A.; Mesleh, M.F.; Oblatt-Montal, M.; Montal, M.; Opella, S.J. Three-dimensional Structure of the Channel-forming Trans-membrane Domain of Virus Protein “u” (Vpu) from HIV-1. J. Mol. Biol. 2003, 333, 409–424. [Google Scholar] [CrossRef]
- Mandala, V.S.; Loftis, A.R.; Shcherbakov, A.A.; Pentelute, B.L.; Hong, M. Atomic structures of closed and open influenza B M2 proton channel reveal the conduction mechanism. Nat. Struct. Mol. Biol. 2020, 27, 160–167. [Google Scholar] [CrossRef]
- Lamb, R.A. The Structure, Function, and Pathobiology of the Influenza A and B Virus Ion Channels. Cold Spring Harbor Perspect. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kim, S.; Kovacs, F.; Cross, T.A. Structure of the transmembrane region of the M2 protein H(+) channel. Protein Sci. 2001, 10, 2241–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, K.; Kim, S.; Zhang, L.; Cross, T.A. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry 2002, 41, 13170–13177. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Asbury, T.; Achuthan, S.; Li, C.; Bertram, R.; Quine, J.R.; Fu, R.; Cross, T.A. Backbone Structure of the Amantadine-Blocked Trans-Membrane Domain M2 Proton Channel from Influenza A Virus. Biophys. J. 2007, 92, 4335–4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cady, S.D.; Mishanina, T.V.; Hong, M. Structure of Amantadine-Bound M2 Transmembrane Peptide of Influenza A in Lipid Bilayers from Magic-Angle-Spinning Solid-State NMR: The Role of Ser31 in Amantadine Binding. J. Mol. Biol. 2009, 385, 1127–1141. [Google Scholar] [CrossRef] [Green Version]
- Cady, S.D.; Schmidt-Rohr, K.; Wang, J.; Soto, C.S.; DeGrado, W.F.; Hong, M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463, 689–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Yi, M.; Dong, H.; Qin, H.; Peterson, E.; Busath, D.D.; Zhou, H.-X.; Cross, T.A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 2010, 330, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Andreas, L.B.; Reese, M.; Eddy, M.T.; Gelev, V.; Ni, Q.Z.; Miller, E.A.; Emsley, L.; Pintacuda, G.; Chou, J.J.; Griffin, R.G. Structure and Mechanism of the Influenza A M218-60 Dimer of Dimers. J. Am. Chem. Soc. 2015, 137, 14877–14886. [Google Scholar] [CrossRef] [Green Version]
- Stouffer, A.L.; Acharya, R.; Salom, D.; Levine, A.S.; Di Costanzo, L.; Soto, C.S.; Tereshko, V.; Nanda, V.; Stayrook, S.; DeGrado, W.F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451, 596–599. [Google Scholar] [CrossRef] [Green Version]
- Mandala, V.S.; Liao, S.Y.; Gelenter, M.D.; Hong, M. The Transmembrane Conformation of the Influenza B Virus M2 Protein in Lipid Bilayers. Sci. Rep. 2019, 9, 3725. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Hong, M. Investigation of the Curvature Induction and Membrane Localization of the Influenza Virus M2 Protein Using Static and Off-Magic-Angle Spinning Solid-State Nuclear Magnetic Resonance of Oriented Bicelles. Biochemistry 2015, 54, 2214–2226. [Google Scholar] [CrossRef] [Green Version]
- Cook, G.A.; Opella, S.J. NMR studies of p7 protein from hepatitis C virus. Eur. Biophys. J. 2010, 39, 1097–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, G.A.; Opella, S.J. Secondary structure, dynamics, and architecture of the p7 membrane protein from hepatitis C virus by NMR spectroscopy. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, G.A.; Zhang, H.; Park, S.H.; Wang, Y.; Opella, S.J. Comparative NMR studies demonstrate profound differences between two viroporins: p7 of HCV and Vpu of HIV-1. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 554–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, G.A.; Dawson, L.A.; Tian, Y.; Opella, S.J. Three-dimensional structure and interaction studies of hepatitis C virus p7 in 1,2-dihexanoyl-sn-glycero-3-phosphocholine by solution nuclear magnetic resonance. Biochemistry 2013, 52, 5295–5303. [Google Scholar] [CrossRef] [Green Version]
- Oestringer, B.P.; Bolivar, J.H.; Claridge, J.K.; Almanea, L.; Chipot, C.; Dehez, F.; Holzmann, N.; Schnell, J.R.; Zitzmann, N. Hepatitis C virus sequence divergence preserves p7 viroporin structural and dynamic features. Sci. Rep. 2019, 9, 8383. [Google Scholar] [CrossRef] [Green Version]
- Oestringer, B.P.; Bolivar, J.H.; Hensen, M.; Claridge, J.K.; Chipot, C.; Dehez, F.; Holzmann, N.; Zitzmann, N.; Schnell, J.R. Re-evaluating the p7 viroporin structure. Nature 2018, 562, E8–E18. [Google Scholar] [CrossRef]
- Chipot, C.; Dehez, F.; Schnell, J.R.; Zitzmann, N.; Pebay-Peyroula, E.; Catoire, L.J.; Miroux, B.; Kunji, E.R.S.; Veglia, G.; Cross, T.A.; et al. Perturbations of Native Membrane Protein Structure in Alkyl Phosphocholine Detergents: A Critical Assessment of NMR and Biophysical Studies. Chem. Rev. 2018, 118, 3559–3607. [Google Scholar] [CrossRef] [Green Version]
- OuYang, B.; Xie, S.; Berardi, M.J.; Zhao, X.; Dev, J.; Yu, W.; Sun, B.; Chou, J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature 2013, 498, 521–525. [Google Scholar] [CrossRef]
- Gouttenoire, J.; Montserret, R.; Paul, D.; Castillo, R.; Meister, S.; Bartenschlager, R.; Penin, F.; Moradpour, D. Aminoterminal Amphipathic α-Helix AH1 of Hepatitis C Virus Nonstructural Protein 4B Possesses a Dual Role in RNA Replication and Virus Production. PLoS Pathog. 2014, 10, e1004501-17. [Google Scholar] [CrossRef]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. Curr. Top. Microbiol. Immunol. 2013, 369, 113–142. [Google Scholar]
- Briggs, E.L.A.; Gomes, R.G.B.; Elhussein, M.; Collier, W.; Findlow, I.S.; Khalid, S.; McCormick, C.J.; Williamson, P.T.F. Interaction between the NS4B amphipathic helix, AH2, and charged lipid headgroups alters membrane morphology and AH2 oligomeric state—Implications for the Hepatitis C virus life cycle. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 1671–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogeron, M.-L.; Badillo, A.; Jirasko, V.; Gouttenoire, J.; Paul, D.; Lancien, L.; Moradpour, D.; Bartenschlager, R.; Meier, B.H.; Penin, F.; et al. Wheat germ cell-free expression: Two detergents with a low critical micelle concentration allow for production of soluble HCV membrane proteins. Protein Expr. Purif. 2015, 105, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Fogeron, M.-L.; Badillo, A.; Penin, F.; Böckmann, A. Wheat Germ Cell-Free Overexpression for the Production of Membrane Proteins. Methods Mol. Biol. 2017, 1635, 91–108. [Google Scholar] [PubMed]
- Fogeron, M.-L.; Paul, D.; Jirasko, V.; Montserret, R.; Lacabanne, D.; Molle, J.; Badillo, A.; Boukadida, C.; Georgeault, S.; Roingeard, P.; et al. Functional expression, purification, characterization, and membrane reconstitution of non-structural protein 2 from hepatitis C virus. Protein Expr. Purif. 2015, 116, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lacabanne, D.; Lends, A.; Danis, C.; Kunert, B.; Fogeron, M.-L.; Jirasko, V.; Chuilon, C.; Lecoq, L.; Orelle, C.; Chaptal, V.; et al. Gradient reconstitution of membrane proteins for solid-state NMR studies. J. Biomol. NMR 2017, 69, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Sborgi, L.; Ravotti, F.; Dandey, V.P.; Dick, M.S.; Mazur, A.; Reckel, S.; Chami, M.; Scherer, S.; Huber, M.; Böckmann, A.; et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 13237–13242. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Ferreira, R.; Taylor, N.M.; Arteni, A.-A.; Kumari, P.; Mona, D.; Ringler, P.; Britschgi, M.; Lauer, M.E.; Makky, A.; Verasdonck, J.; et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. eLife 2019, 8, e48907. [Google Scholar] [CrossRef]
Figure 1.
NMR rotors, spectrometer and resonance signals: (a) NMR rotors for fast spinning: 110 kHz for 0.7 mm rotor, 150 kHz for 0.5 mm (picture courtesy of Susanne Penzel). (b) 1200 MHz magnet for solid-state NMR at ETH Zurich. (c) Schematic representation of anisotropic interactions arising from different orientation of the individual molecules with respect to the magnetic field. The superposition of all possible chemical shifts gives rise to broad peaks with a characteristic shape, the powder pattern. The anisotropic interactions can be averaged out using MAS, which results in a single resonance line centered at the isotropic chemical shift of the spin. In addition, spinning sidebands may appear. This spin can be correlated to a neighboring spin, in the example, the amide 1H to the amide 15N. Two-dimensional spectroscopy then shows peaks which represent, for the present example, the amide proton frequency in one dimension, and the nitrogen frequency in the other. Such a signal will be observed for every NH pair in the protein. The resonance-line position is given by the isotropic part of NMR chemical shift and is usually specified in ppm (parts per million) of the resonance frequency. To obtain high-resolution spectra, the linewidth should be as narrow as possible, as this allows to distinguish (resolve) a maximum of resonances. A narrowing of linewidths can be achieved by (i) using a spectrometer operating at a higher magnetic field, (ii) improving sample homogeneity and symmetry, and (iii) reducing the dipolar coupling interactions by decreasing the density of protons in the system (through protein deuteration, for example) and/or increasing the MAS frequency by using smaller diameter rotors.
Figure 1.
NMR rotors, spectrometer and resonance signals: (a) NMR rotors for fast spinning: 110 kHz for 0.7 mm rotor, 150 kHz for 0.5 mm (picture courtesy of Susanne Penzel). (b) 1200 MHz magnet for solid-state NMR at ETH Zurich. (c) Schematic representation of anisotropic interactions arising from different orientation of the individual molecules with respect to the magnetic field. The superposition of all possible chemical shifts gives rise to broad peaks with a characteristic shape, the powder pattern. The anisotropic interactions can be averaged out using MAS, which results in a single resonance line centered at the isotropic chemical shift of the spin. In addition, spinning sidebands may appear. This spin can be correlated to a neighboring spin, in the example, the amide 1H to the amide 15N. Two-dimensional spectroscopy then shows peaks which represent, for the present example, the amide proton frequency in one dimension, and the nitrogen frequency in the other. Such a signal will be observed for every NH pair in the protein. The resonance-line position is given by the isotropic part of NMR chemical shift and is usually specified in ppm (parts per million) of the resonance frequency. To obtain high-resolution spectra, the linewidth should be as narrow as possible, as this allows to distinguish (resolve) a maximum of resonances. A narrowing of linewidths can be achieved by (i) using a spectrometer operating at a higher magnetic field, (ii) improving sample homogeneity and symmetry, and (iii) reducing the dipolar coupling interactions by decreasing the density of protons in the system (through protein deuteration, for example) and/or increasing the MAS frequency by using smaller diameter rotors.

Figure 2.
Overview of human viruses where solid-state NMR contributed to the understanding of global organization, orientation in the membrane and protein structure and dynamics. Viral proteins investigated by solid-state NMR are labeled in red. (a) HIV-1 virion with envelope-embedded gp41 protein containing the fusion peptide and internal capsid. (b) Influenza virion with M2 ion channel. (c) Measles virion with nucleocapsid protein. (d) HBV virion with small surface protein S and internal capsid. (e) Membrane topology of HCV with p7 and NS4B membrane proteins. Panel (d) is reprinted from [86], Copyright (2014), with permission from Elsevier. Panel (e) is adapted from [87].
Figure 2.
Overview of human viruses where solid-state NMR contributed to the understanding of global organization, orientation in the membrane and protein structure and dynamics. Viral proteins investigated by solid-state NMR are labeled in red. (a) HIV-1 virion with envelope-embedded gp41 protein containing the fusion peptide and internal capsid. (b) Influenza virion with M2 ion channel. (c) Measles virion with nucleocapsid protein. (d) HBV virion with small surface protein S and internal capsid. (e) Membrane topology of HCV with p7 and NS4B membrane proteins. Panel (d) is reprinted from [86], Copyright (2014), with permission from Elsevier. Panel (e) is adapted from [87].
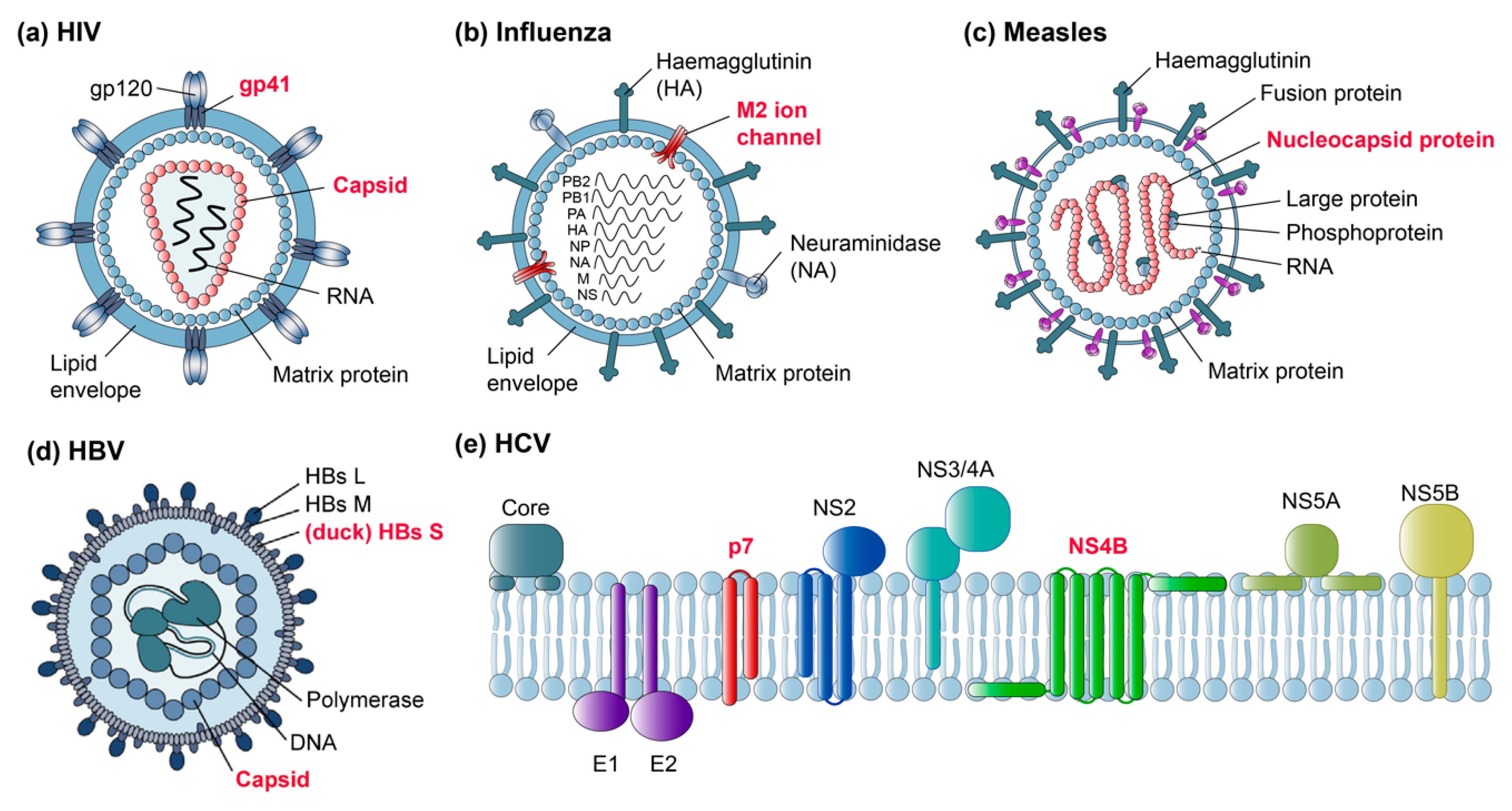
Figure 3.
Solid-state NMR studies on the HIV and HBV capsids. (a) Illustration of the mature HIV-1 capsid (PDB: 3J3Y [99]); and the CA protein in (b), with a zoom on the backbone 15NH CSA tensor and its orientation in the protein molecular frame. (c) Transmission EM image of CA and (d) the corresponding 2D NCA spectrum recorded at 14 kHz MAS. (e) HBV capsid structure (PDB: 1QGT [7]) showing the pentamers formed by A subunits and hexamers formed by B, C and D subunits. (f) Negative-staining EM picture of HBV capsids and (g) corresponding 2D Dipolar Assisted Rotational Resonance (DARR) spectrum recorded at 17.5 kHz MAS. Residues with NMR peak splitting due to the asymmetric subunits are highlighted in yellow and are shown as spheres in (h) on the capsid structure. Pictures from panels (a–d) were reprinted with permission from reference [94], Copyright (2016) American Chemical Society. Pictures from panels (e–h) were taken with permissions from reference [55].
Figure 3.
Solid-state NMR studies on the HIV and HBV capsids. (a) Illustration of the mature HIV-1 capsid (PDB: 3J3Y [99]); and the CA protein in (b), with a zoom on the backbone 15NH CSA tensor and its orientation in the protein molecular frame. (c) Transmission EM image of CA and (d) the corresponding 2D NCA spectrum recorded at 14 kHz MAS. (e) HBV capsid structure (PDB: 1QGT [7]) showing the pentamers formed by A subunits and hexamers formed by B, C and D subunits. (f) Negative-staining EM picture of HBV capsids and (g) corresponding 2D Dipolar Assisted Rotational Resonance (DARR) spectrum recorded at 17.5 kHz MAS. Residues with NMR peak splitting due to the asymmetric subunits are highlighted in yellow and are shown as spheres in (h) on the capsid structure. Pictures from panels (a–d) were reprinted with permission from reference [94], Copyright (2016) American Chemical Society. Pictures from panels (e–h) were taken with permissions from reference [55].

Figure 4.
(a) 2D hNH spectrum of HBV capsids made of full-length core protein (Cp183) expressed and auto-assembled using WG-CFPS, recorded at 100 kHz. (b) Negative-staining EM pictures of the capsids prepared by WG-CFPS in the absence (top) and in the presence (bottom) of a JNJ-890 capsid assembly modulator, leading to opened objects. Figure adapted with permissions from reference [40].
Figure 4.
(a) 2D hNH spectrum of HBV capsids made of full-length core protein (Cp183) expressed and auto-assembled using WG-CFPS, recorded at 100 kHz. (b) Negative-staining EM pictures of the capsids prepared by WG-CFPS in the absence (top) and in the presence (bottom) of a JNJ-890 capsid assembly modulator, leading to opened objects. Figure adapted with permissions from reference [40].
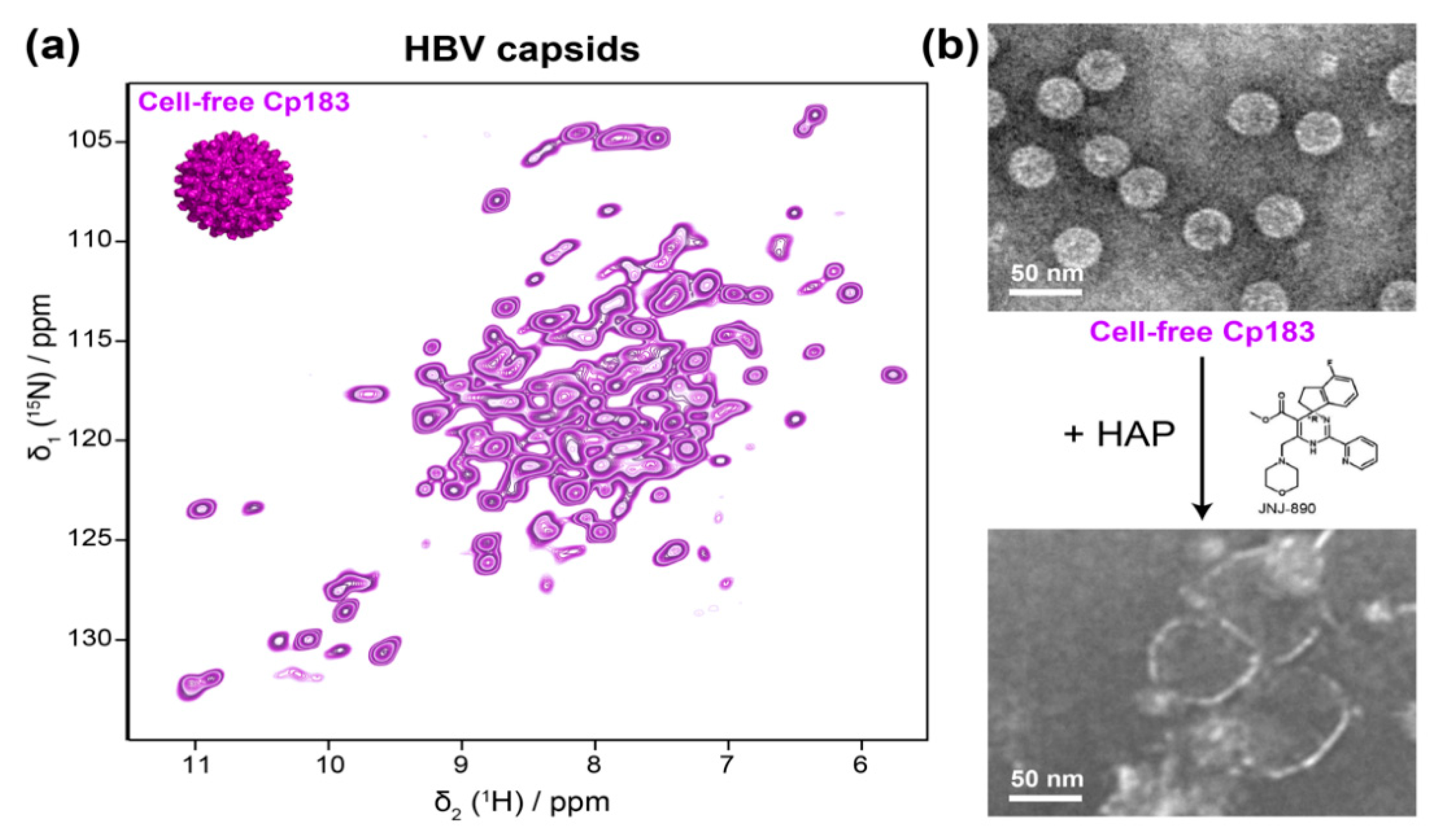
Figure 5.
(a) EM pictures of intact (grey) and cleaved (brown) measle nucleocapsids [111]. (b) 2D hNH spectra recorded at 60 kHz MAS of intact (blue) and cleaved (red) measle nucleocapsids and corresponding bulk 15N R1rho decays in (c), revealing longer life times and thus different dynamics for the cleaved form. Figure reprinted from [110], Copyright (2014), with permission from Elsevier.
Figure 5.
(a) EM pictures of intact (grey) and cleaved (brown) measle nucleocapsids [111]. (b) 2D hNH spectra recorded at 60 kHz MAS of intact (blue) and cleaved (red) measle nucleocapsids and corresponding bulk 15N R1rho decays in (c), revealing longer life times and thus different dynamics for the cleaved form. Figure reprinted from [110], Copyright (2014), with permission from Elsevier.
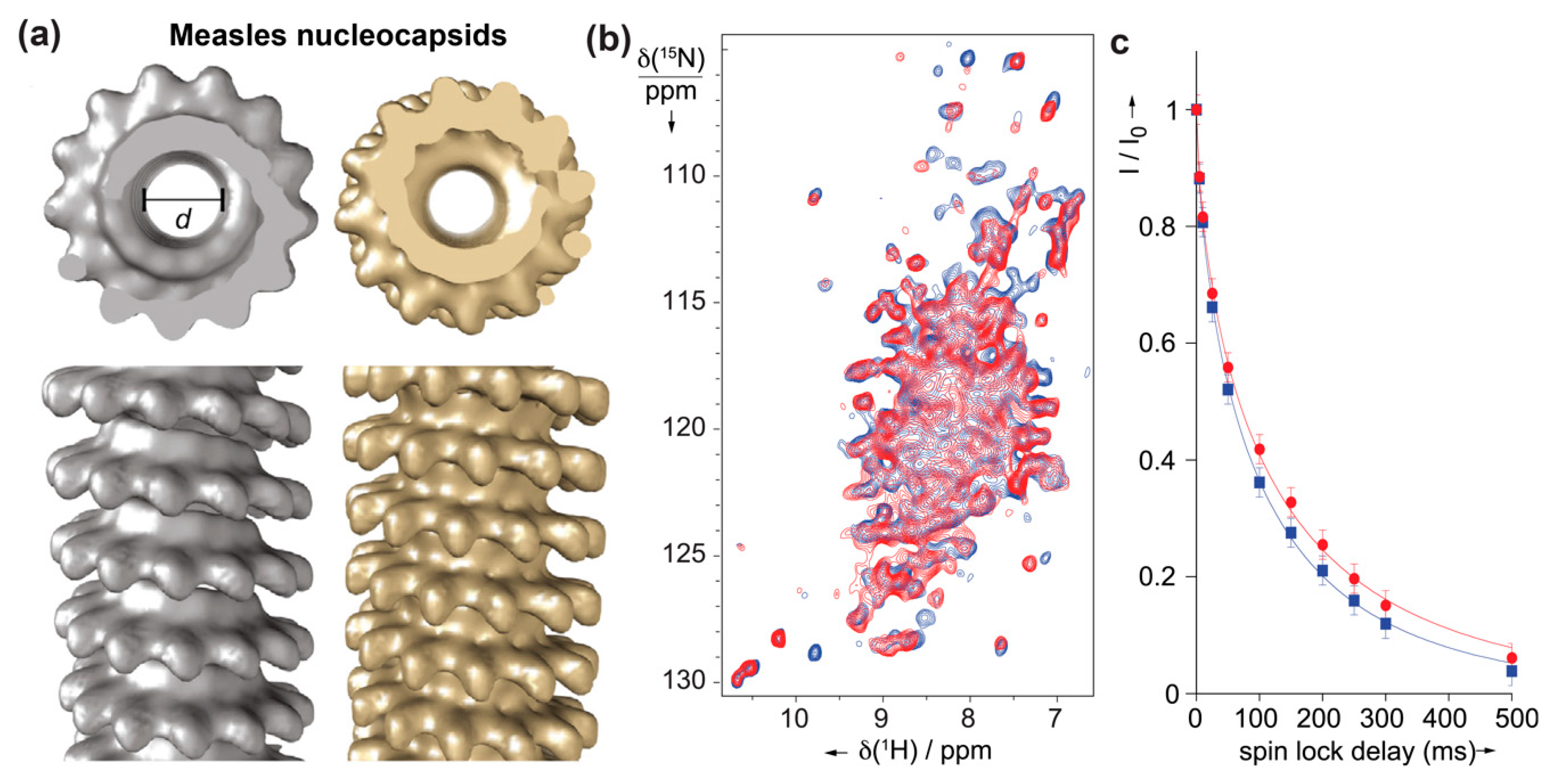
Figure 6.
(a) 2D hNH spectrum of DHBs small envelope protein produced using WG-CFPS, recorded at 110 kHz MAS. (b) Negative-staining EM pictures of the corresponding protein, which self-assembles into spherical particles of ~29 nm. Figures taken with permission from reference [41].
Figure 6.
(a) 2D hNH spectrum of DHBs small envelope protein produced using WG-CFPS, recorded at 110 kHz MAS. (b) Negative-staining EM pictures of the corresponding protein, which self-assembles into spherical particles of ~29 nm. Figures taken with permission from reference [41].
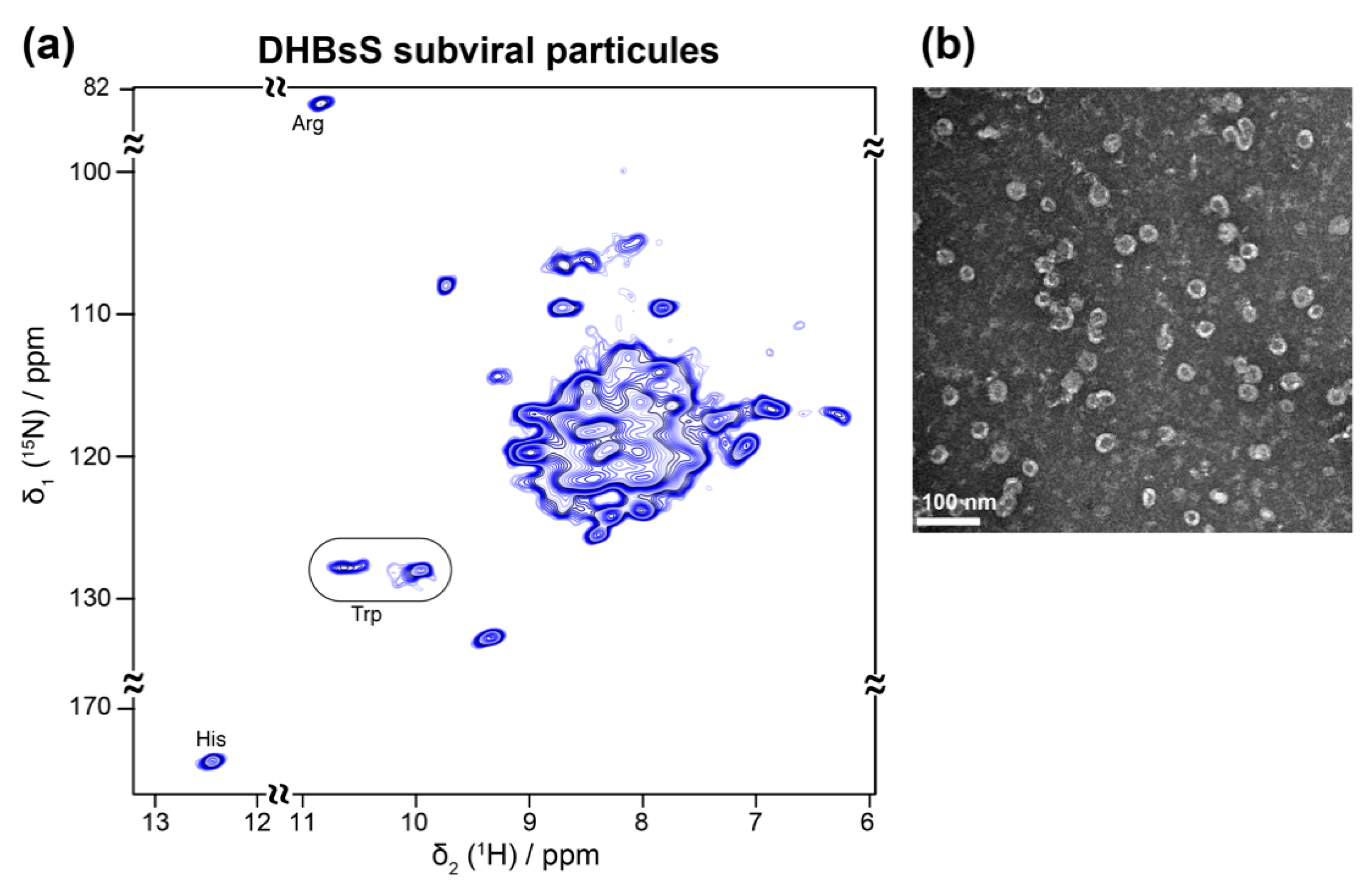
Figure 7.
Solid-state NMR structures of (a) Vpu from HIV-1 (PDB: 2N28 [127]), and (b) closed and open influenza BM2 channels in lipid membranes (PDB: 6pvr (pH 7.5) and 6pvt (pH 4.5) [130]). Panel (a) reprinted from reference [127] Copyright (2015), with permission from Elsevier. Panel (b) reprinted from reference [130] Copyright (2020), with permission from Springer Nature.
Figure 7.
Solid-state NMR structures of (a) Vpu from HIV-1 (PDB: 2N28 [127]), and (b) closed and open influenza BM2 channels in lipid membranes (PDB: 6pvr (pH 7.5) and 6pvt (pH 4.5) [130]). Panel (a) reprinted from reference [127] Copyright (2015), with permission from Elsevier. Panel (b) reprinted from reference [130] Copyright (2020), with permission from Springer Nature.

Figure 8.
(a) 3D correlation spectra of deuterated NS4B from HCV with sequential connectivities. Selected strip plots representing the assignment of amino acid stretches of residues 119 to 125 and 237 to 241 using a set of 6 spectra recorded at 110 kHz MAS on two NS4B samples with different labeling schemes [60]. The orange spectrum was acquired at 60 kHz MAS. (b) A putative topology model of the NS4B protein adapted from [150], in which NS4B is proposed to contain four presumably amphipathic α-helices and four predicted transmembrane segments in the middle. The black boxes indicate the location of the two assigned regions shown in panel a. Figure reproduced with permission from reference [60].
Figure 8.
(a) 3D correlation spectra of deuterated NS4B from HCV with sequential connectivities. Selected strip plots representing the assignment of amino acid stretches of residues 119 to 125 and 237 to 241 using a set of 6 spectra recorded at 110 kHz MAS on two NS4B samples with different labeling schemes [60]. The orange spectrum was acquired at 60 kHz MAS. (b) A putative topology model of the NS4B protein adapted from [150], in which NS4B is proposed to contain four presumably amphipathic α-helices and four predicted transmembrane segments in the middle. The black boxes indicate the location of the two assigned regions shown in panel a. Figure reproduced with permission from reference [60].
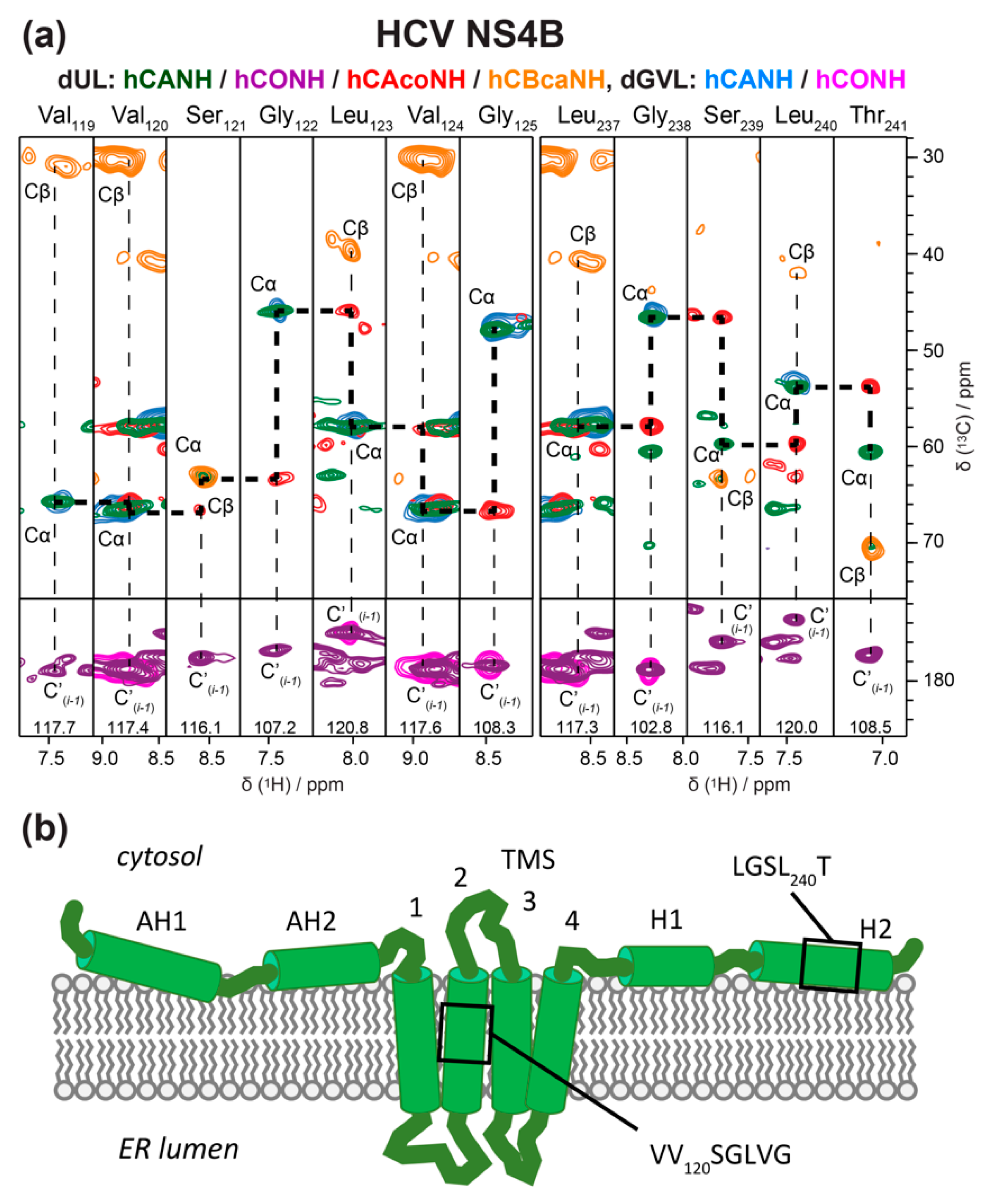
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lecoq, L.; Fogeron, M.-L.; Meier, B.H.; Nassal, M.; Böckmann, A. Solid-State NMR for Studying the Structure and Dynamics of Viral Assemblies. Viruses 2020, 12, 1069. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101069
AMA Style
Lecoq L, Fogeron M-L, Meier BH, Nassal M, Böckmann A. Solid-State NMR for Studying the Structure and Dynamics of Viral Assemblies. Viruses. 2020; 12(10):1069. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101069
Chicago/Turabian StyleLecoq, Lauriane, Marie-Laure Fogeron, Beat H. Meier, Michael Nassal, and Anja Böckmann. 2020. "Solid-State NMR for Studying the Structure and Dynamics of Viral Assemblies" Viruses 12, no. 10: 1069. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101069
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.