Tracking SARS-CoV-2 in Sewage: Evidence of Changes in Virus Variant Predominance during COVID-19 Pandemic

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Wastewater Sample Collection and Processing
2.2. Quantification of SARS-CoV-2 RNA in Wastewater Concentrates by RT-qPCR
2.3. SARS-CoV-2 Whole-Genome Sequences Used for Nucleotide Sequence Analyses
2.4. Nested RT-PCR (nPCR) Amplification
2.5. Sanger Sequencing Analysis of RT-PCR Products
2.6. NGS Analysis of RT-PCR Products
2.7. Generation of SARS-CoV-2 Sequence Contigs and Identification of SNPs
3. Results
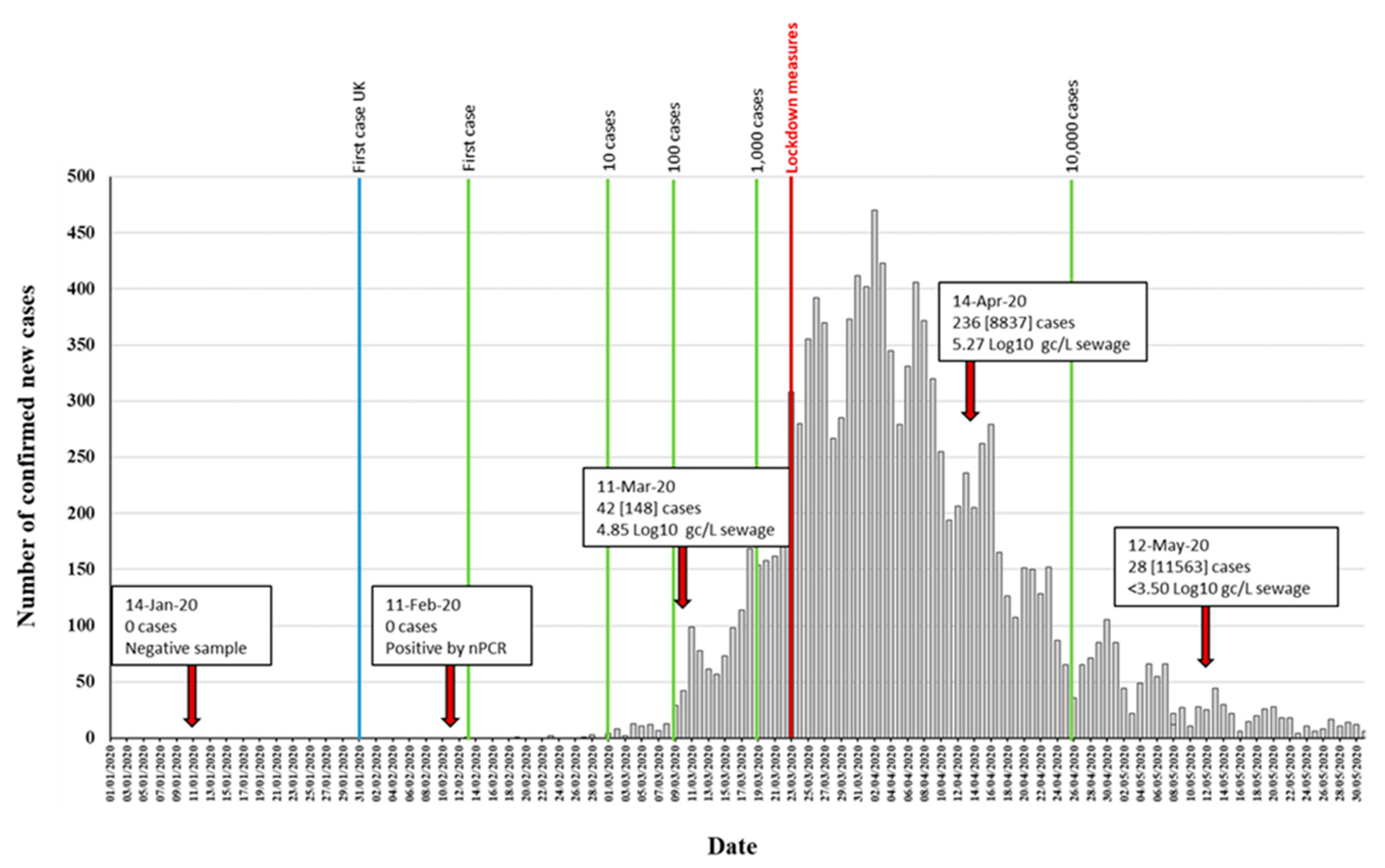
3.1. Detection of SARS-CoV-2 RNA in Wastewater Samples
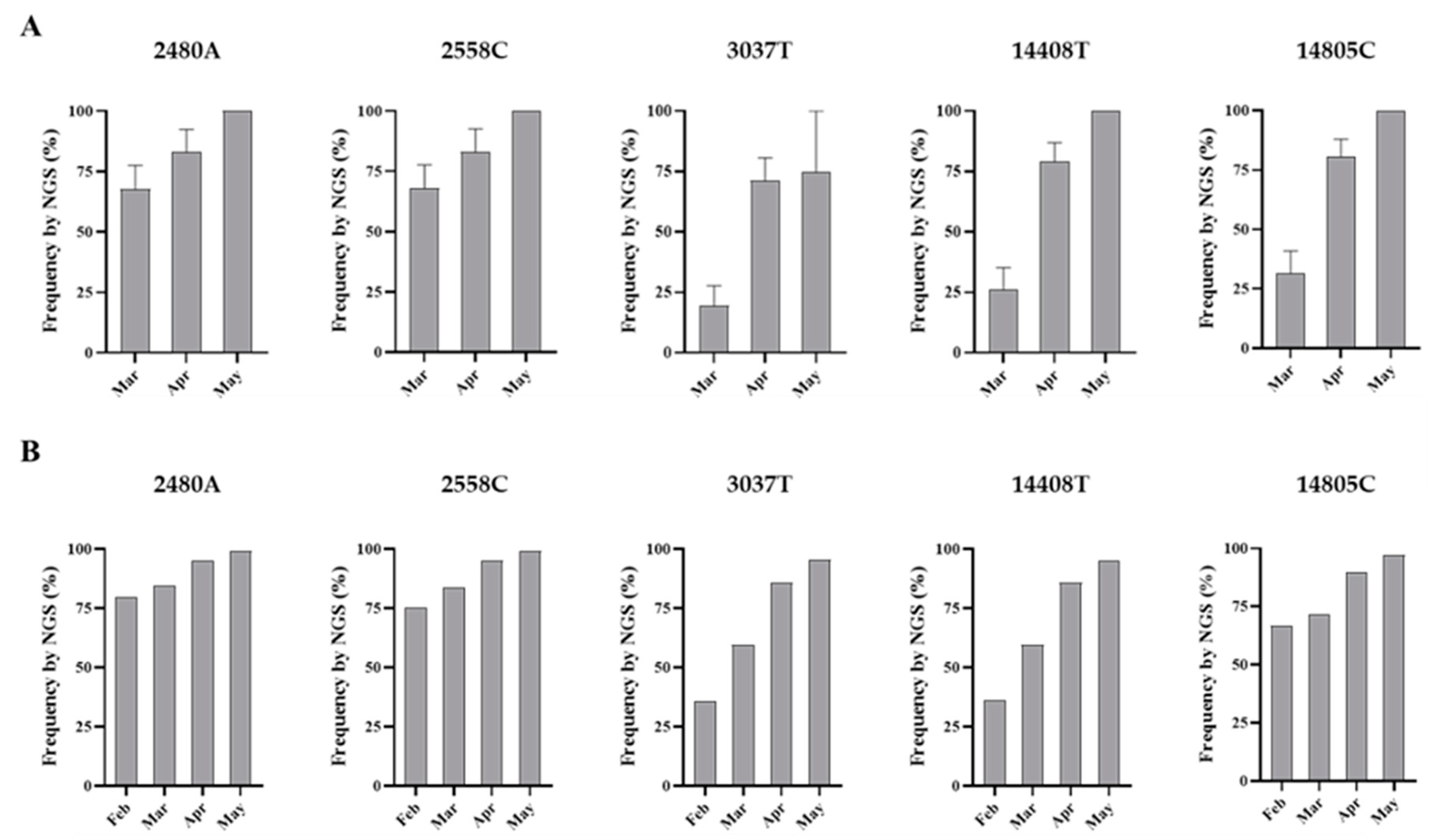
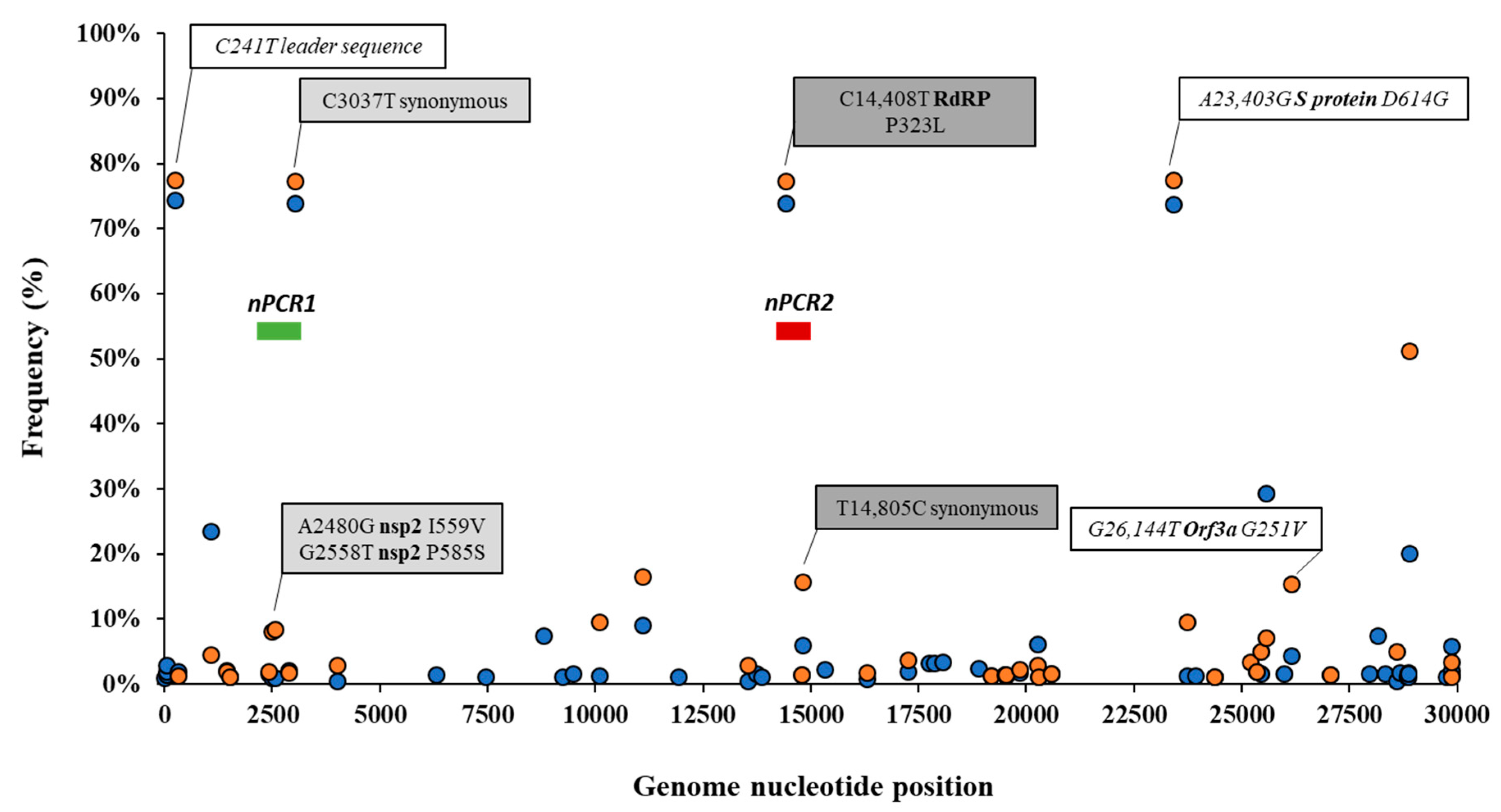
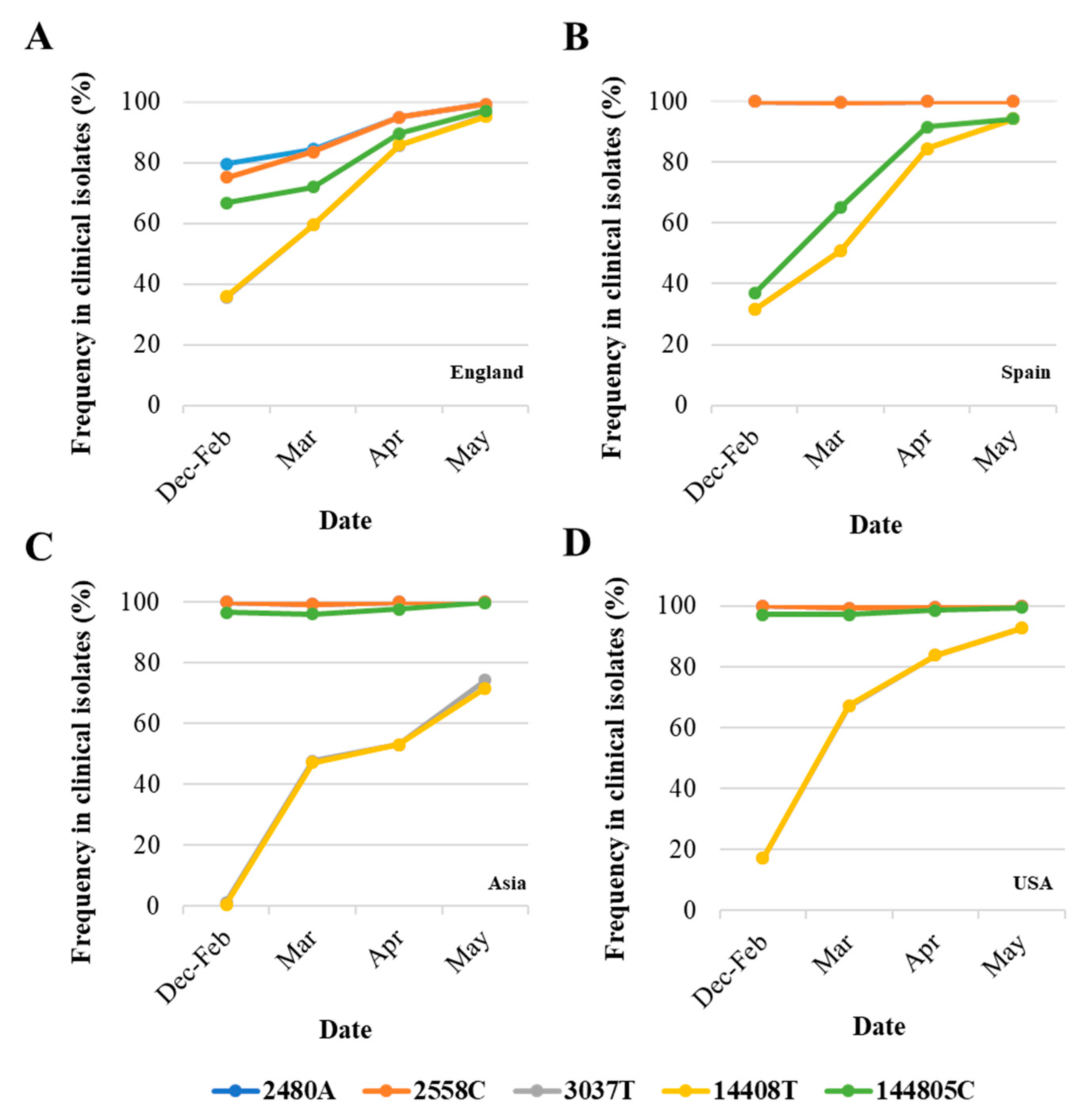
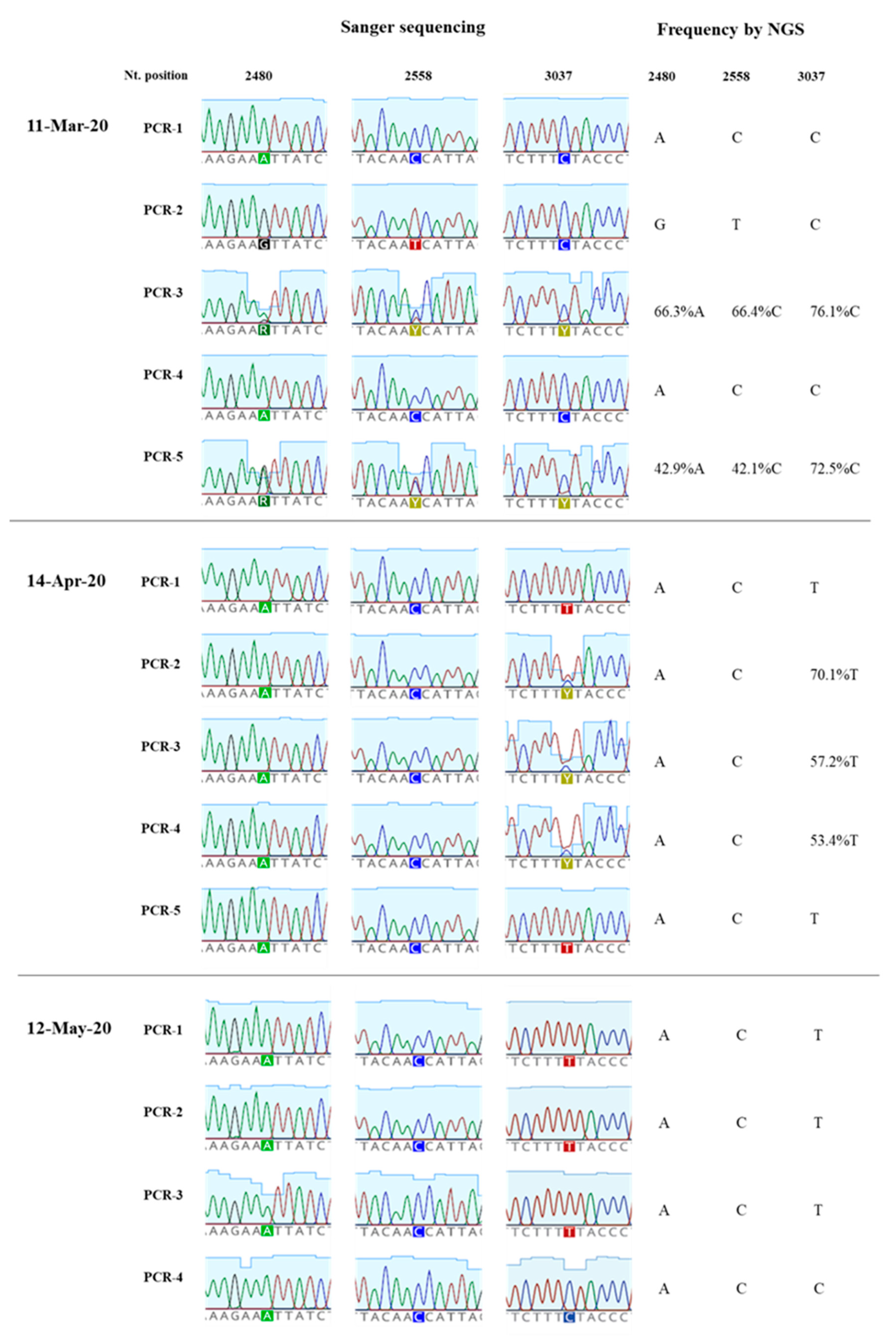
3.2. Analysis of Nucleotide Sequence Variation among SARS-CoV-2 RNA Sequences from Clinical Samples
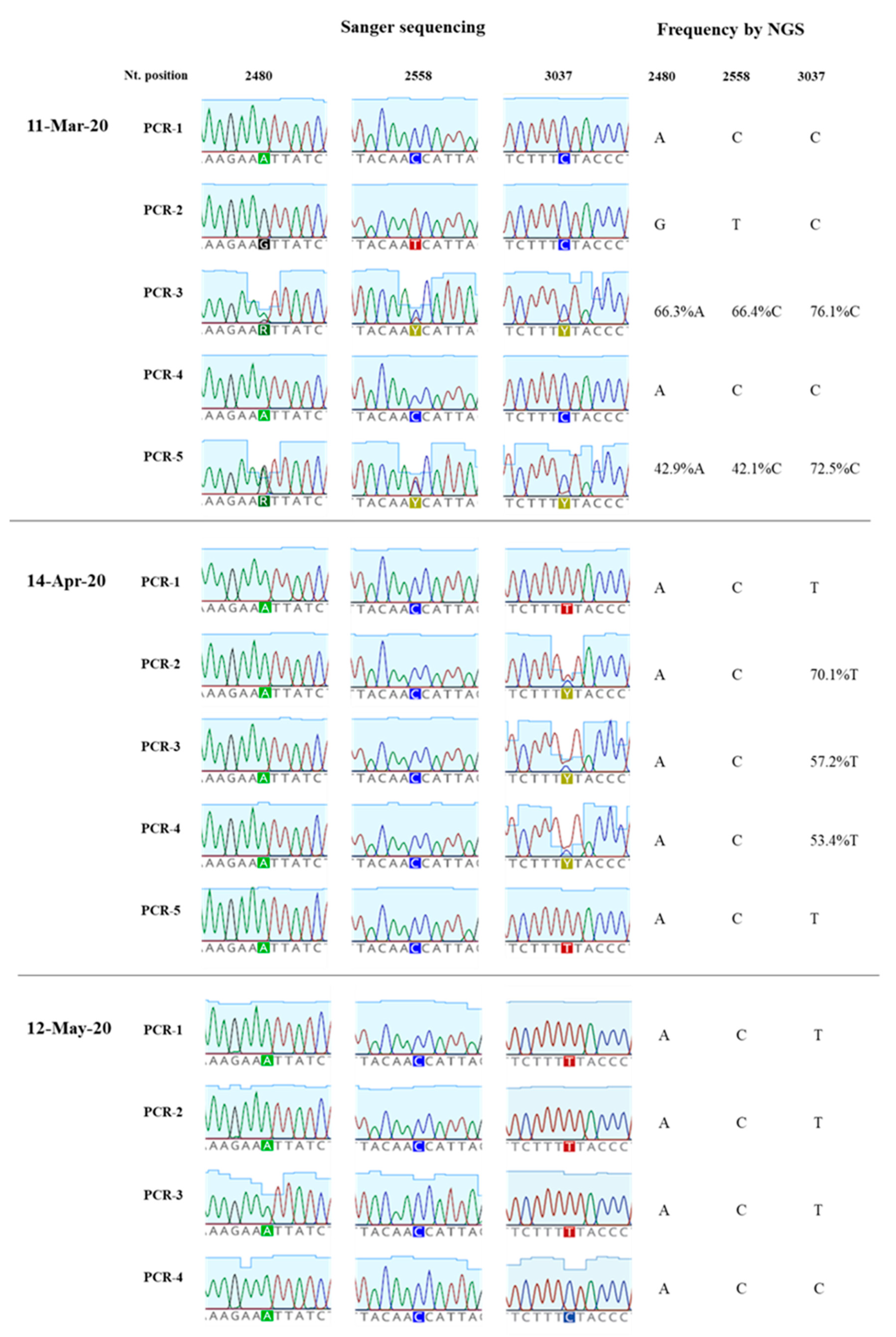
3.3. Generation of nPCR Products for Nucleotide Sequence Analyses
3.4. Nucleotide Sequence Analysis of nPCR Products from Wastewater Concentrates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 10 July 2020).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, X.; Zhu, B.; Liang, H.; Fang, C.; Gong, Y.; Guo, Q.; Sun, X.; Zhao, D.; Shen, J.; et al. Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat. Med. 2020, 26, 502–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Guo, C.; Tang, L.; Hong, Z.; Zhou, J.; Dong, X.; Yin, H.; Xiao, Q.; Tang, Y.; Qu, X.; et al. Prolonged presence of SARS-CoV-2 viral RNA in faecal samples. Lancet Gastroenterol. Hepatol. 2020, 5, 434–435. [Google Scholar] [CrossRef]
- Zhang, N.; Gong, Y.; Meng, F.; Bi, Y.; Yang, P.; Wang, F. Virus shedding patterns in nasopharyngeal and fecal specimens of COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Lescure, F.X.; Bouadma, L.; Nguyen, D.; Parisey, M.; Wicky, P.H.; Behillil, S.; Gaymard, A.; Bouscambert-Duchamp, M.; Donati, F.; Le Hingrat, Q.; et al. Clinical and virological data of the first cases of COVID-19 in Europe: A case series. Lancet Infect. Dis. 2020, 20, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, W.; Angel, N.; Edson, J.; Bibby, K.; Bivins, A.; O’Brien, J.W.; Choi, P.M.; Kitajima, M.; Simpson, S.L.; Li, J.; et al. First confirmed detection of SARS-CoV-2 in untreated wastewater in Australia: A proof of concept for the wastewater surveillance of COVID-19 in the community. Sci. Total Environ. 2020, 728, 138764. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, J.; Xiao, A.; Gu, X.; Lee, W.L.; Armas, F.; Kauffman, K.; Hanage, W.; Matus, M.; Ghaeli, N.; et al. SARS-CoV-2 Titers in Wastewater Are Higher than Expected from Clinically Confirmed Cases. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Randazzo, W.; Truchado, P.; Cuevas-Ferrando, E.; Simon, P.; Allende, A.; Sanchez, G. SARS-CoV-2 RNA in wastewater anticipated COVID-19 occurrence in a low prevalence area. Water Res. 2020, 181, 115942. [Google Scholar] [CrossRef]
- Wurtzer, S.; Marechal, V.; Mouchel, J.-M.; Maday, Y.; Teyssou, R.; Richard, E.; Almayrac, J.L.; Moulin, L. Evaluation of lockdown impact on SARS-CoV-2 dynamics through viral genome quantification in Paris wastewaters. medRxiv 2020. [Google Scholar] [CrossRef]
- Medema, G.; Heijnen, L.; Elsinga, G.; Italiaander, R.; Brouwer, A. Presence of SARS-Coronavirus-2 in sewage. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, G.; Iaconelli, M.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Bonadonna, L.; Lucentini, L.; Suffredini, E. First detection of SARS-CoV-2 in untreated wastewaters in Italy. Water Res. 2020, 736. [Google Scholar] [CrossRef] [PubMed]
- Lodder, W.; de Roda Husman, A.M. SARS-CoV-2 in wastewater: Potential health risk, but also data source. Lancet Gastroenterol. Hepatol. 2020, 5, 533–534. [Google Scholar] [CrossRef]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Surya, K.; Buyukyoruk, M.; Cicha, C.; Vanderwood, K.K.; Wilkinson, R.; Wiedenheft, B. Temporal Detection and Phylogenetic Assessment of SARS-CoV-2 in Municipal Wastewater. Cell Rep. Med. 2020, 1, 100098. [Google Scholar] [CrossRef]
- Kitajima, M.; Ahmed, W.; Bibby, K.; Carducci, A.; Gerba, C.P.; Hamilton, K.A.; Haramoto, E.; Rose, J.B. SARS-CoV-2 in wastewater: State of the knowledge and research needs. Sci. Total Environ. 2020, 739, 139076. [Google Scholar] [CrossRef]
- Foladori, P.; Cutrupi, F.; Segata, N.; Manara, S.; Pinto, F.; Malpei, F.; Bruni, L.; La Rosa, G. SARS-CoV-2 from faeces to wastewater treatment: What do we know? A review. Sci. Total Environ. 2020, 743, 140444. [Google Scholar] [CrossRef]
- Flaxman, S.; Mishra, S.; Gandy, A.; Unwin, H.J.T.; Mellan, T.A.; Coupland, H.; Whittaker, C.; Zhu, H.; Berah, T.; Eaton, J.W.; et al. Estimating the effects of non-pharmaceutical interventions on COVID-19 in Europe. Nature 2020, 584, 257–261. [Google Scholar] [CrossRef]
- Walker, P.G.T.; Whittaker, C.; Watson, O.J.; Baguelin, M.; Winskill, P.; Hamlet, A.; Djafaara, B.A.; Cucunuba, Z.; Olivera Mesa, D.; Green, W.; et al. The impact of COVID-19 and strategies for mitigation and suppression in low- and middle-income countries. Science 2020, 369, 413–422. [Google Scholar] [CrossRef]
- Davies, N.G.; Kucharski, A.J.; Eggo, R.M.; Gimma, A.; Edmunds, W.J.; Centre for the Mathematical Modelling of Infectious Diseases COVID-19 Working Group. Effects of non-pharmaceutical interventions on COVID-19 cases, deaths, and demand for hospital services in the UK: A modelling study. Lancet Public Health 2020, 5, e375–e385. [Google Scholar] [CrossRef]
- Kucharski, A.J.; Klepac, P.; Conlan, A.J.K.; Kissler, S.M.; Tang, M.L.; Fry, H.; Gog, J.R.; Edmunds, W.J.; CMMID COVID-19 Working Group. Effectiveness of isolation, testing, contact tracing, and physical distancing on reducing transmission of SARS-CoV-2 in different settings: A mathematical modelling study. Lancet Infect. Dis. 2020, 20, 1151–1160. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control (ECDC). COVID-19 Pandemic. Available online: https://www.ecdc.europa.eu/en/covid-19-pandemic (accessed on 20 September 2020).
- Pollan, M.; Perez-Gomez, B.; Pastor-Barriuso, R.; Oteo, J.; Hernan, M.A.; Perez-Olmeda, M.; Sanmartin, J.L.; Fernandez-Garcia, A.; Cruz, I.; Fernandez de Larrea, N.; et al. Prevalence of SARS-CoV-2 in Spain (ENE-COVID): A nationwide, population-based seroepidemiological study. Lancet 2020, 396, 535–544. [Google Scholar] [CrossRef]
- Xu, X.; Sun, J.; Nie, S.; Li, H.; Kong, Y.; Liang, M.; Hou, J.; Huang, X.; Li, D.; Ma, T.; et al. Seroprevalence of immunoglobulin M and G antibodies against SARS-CoV-2 in China. Nat. Med. 2020, 26, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Stringhini, S.; Wisniak, A.; Piumatti, G.; Azman, A.S.; Lauer, S.A.; Baysson, H.; De Ridder, D.; Petrovic, D.; Schrempft, S.; Marcus, K.; et al. Seroprevalence of anti-SARS-CoV-2 IgG antibodies in Geneva, Switzerland (SEROCoV-POP): A population-based study. Lancet 2020, 396, 313–319. [Google Scholar] [CrossRef]
- Public Health England. Weekly Coronavirus Disease 2019 (COVID-19) Surveillance Report. Week: 22; 2020. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/888254/COVID19_Epidemiological_Summary_w22_Final.pdf (accessed on 4 July 2020).
- Johnson, N.P.; Mueller, J. Updating the accounts: Global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull. Hist Med. 2002, 76, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Bootsma, M.C.; Ferguson, N.M. The effect of public health measures on the 1918 influenza pandemic in U.S. cities. Proc. Natl. Acad. Sci. USA 2007, 104, 7588–7593. [Google Scholar] [CrossRef] [Green Version]
- Asghar, H.; Diop, O.M.; Weldegebriel, G.; Malik, F.; Shetty, S.; El Bassioni, L.; Akande, A.O.; Al Maamoun, E.; Zaidi, S.; Adeniji, A.J.; et al. Environmental surveillance for polioviruses in the Global Polio Eradication Initiative. J. Infect. Dis. 2014, 210, S294–S303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, L.M.; Gavrilin, E.; Jorba, J.; Martin, J.; Burns, C.C.; Manor, Y.; Moran-Gilad, J.; Sofer, D.; Hindiyeh, M.Y.; Gamzu, R.; et al. Molecular epidemiology of silent introduction and sustained transmission of wild poliovirus type 1, Israel, 2013. Eurosurveillance 2014, 19, 20709. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, G.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Iaconelli, M.; Bonadonna, L.; Lucentini, L.; Suffredini, E. SARS-CoV-2 has been circulating in northern Italy since December 2019: Evidence from environmental monitoring. Sci. Total Environ. 2020, 750, 141711. [Google Scholar] [CrossRef]
- MacLean, O.A.; Orton, R.J.; Singer, J.B.; Robertson, D.L. No evidence for distinct types in the evolution of SARS-CoV-2. Virus Evol. 2020, 6. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef] [PubMed]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Li, M.; Ren, R.; Li, L.; Chen, E.Q.; Li, W.; Ying, B. International Expansion of a Novel SARS-CoV-2 Mutant. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.C.; Anderson, D.E.; Young, B.E.; Zhu, F.; Linster, M.; Kalimuddin, S.; Low, J.G.; Yan, Z.; Jayakumar, J.; Sun, L.; et al. Discovery of a 382-nt deletion during the early evolution of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Van Dorp, L.; Richard, D.; Tan, C.C.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Volz, E.M.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, A.; Southgate, J.A.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the effects of SARS-CoV-2 Spike mutation D614G on transmissibility and pathogenicity. medRxiv 2020. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827 e819. [Google Scholar] [CrossRef]
- Majumdar, M.; Klapsa, D.; Wilton, T.; Akello, J.; Anscombe, C.; Allen, D.; Mee, E.T.; Minor, P.D.; Martin, J. Isolation of vaccine-like poliovirus strains in sewage samples from the UK. J. Infect. Dis. 2017. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, M.; Martin, J. Detection by Direct Next Generation Sequencing Analysis of Emerging Enterovirus D68 and C109 Strains in an Environmental Sample from Scotland. Front. Microbiol. 2018, 9, 1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelstein, M.; Obi, C.; Chand, M.; Hopkins, S.; Brown, K.; Ramsay, M. SARS-CoV-2 infection in London, England: Impact of lockdown on community point-prevalence, March-May 2020. medRxiv 2020. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Guidelines for Environmental Surveillance of Poliovirus Circulation; World Health Organization: Geneva, Switzerland, 2003. [Google Scholar]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Westhaus, S.; Weber, F.A.; Schiwy, S.; Linnemann, V.; Brinkmann, M.; Widera, M.; Greve, C.; Janke, A.; Hollert, H.; Wintgens, T.; et al. Detection of SARS-CoV-2 in raw and treated wastewater in Germany—Suitability for COVID-19 surveillance and potential transmission risks. Sci. Total Environ. 2020, 751, 141750. [Google Scholar] [CrossRef]
- Didion, J.P.; Martin, M.; Collins, F.S. Atropos: Specific, sensitive, and speedy trimming of sequencing reads. PeerJ 2017, 5, e3720. [Google Scholar] [CrossRef] [Green Version]
- Mee, E.T.; Minor, P.D.; Martin, J. High resolution identity testing of inactivated poliovirus vaccines. Vaccine 2015, 33, 3533–3541. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Ling, H.; Huang, X.; Li, J.; Li, W.; Yi, C.; Zhang, T.; Jiang, Y.; He, Y.; Deng, S.; et al. Potential spreading risks and disinfection challenges of medical wastewater by the presence of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) viral RNA in septic tanks of Fangcang Hospital. Sci. Total Environ. 2020, 741, 140445. [Google Scholar] [CrossRef]
- O’Reilly, K.M.; Grassly, N.C.; Allen, D.J.; Bannister-Tyrrell, M.; Cameron, A.; Carrion Martin, A.I.; Ramsay, M.; Pebody, R.; Zambon, M. Surveillance optimisation to detect poliovirus in the pre-eradication era: A modelling study of England and Wales. Epidemiol. Infect. 2020, 148, e157. [Google Scholar]
- Foley, B.; Pan, H.; Buchbinder, S.; Delwart, E.L. Apparent founder effect during the early years of the San Francisco HIV type 1 epidemic (1978–1979). AIDS Res. Hum. Retroviruses 2000, 16, 1463–1469. [Google Scholar] [CrossRef]
- Pybus, O.G.; Rambaut, A.; du Plessis, L.; Zarebski, A.E.; Moritz, U.; Kraemer, G.; Raghwani, J.; Gutiérrez, B.; Hill, V.; McCrone, J.; et al. Preliminary Analysis of SARS-CoV-2 Importation & Establishment of UK Transmission Lineages. Available online: https://virological.org/t/preliminary-analysis-of-sars-cov-2-importation-establishment-of-uk-transmission-lineages/507 (accessed on 20 September 2020).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 RTqPCR (No. of Replicates) 1 | Nested RT-PCR Gene Target (PCR Product Size) 2 | Genome Sequenced (No. of Nucleotides) | ||||||
|---|---|---|---|---|---|---|---|---|
| Sampling Date | RdRP Gene (log10 gc/L Sewage) | E Gene (log10 gc/L Sewage) | nsp2-PLPro Gene nPCR1 (714 nt) | RdRP Gene nPCR2 (523 nt) | RdRP Gene nPCR3 (527 nt) | RdRP Gene nPCR4 (235 nt) | ORF8b-N Gene nPCR5 (612 nt) | |
| 14-Jan-20 | - | - | - | - | - | - | - | - |
| 11-Feb-20 | - | - | - | - | - | + | + | 847 |
| 11-Mar-20 | 4.84 ± 0.45 [4.18–5.52] (n = 10) | 4.98 ± 0.40 [4.63–5.41] (n = 3) | + | + | + | + | + | 2376 |
| 14-Apr-20 | 5.27 ± 0.30 [4.77–5.91] (n = 11) | 5.78 ± 0.07 [5.71–5.84] (n = 3) | + | + | + | + | + | 2376 |
| 12-May-20 | <3.5 (n = 11) 3 | - | + | + | + | + | + | 2376 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, J.; Klapsa, D.; Wilton, T.; Zambon, M.; Bentley, E.; Bujaki, E.; Fritzsche, M.; Mate, R.; Majumdar, M. Tracking SARS-CoV-2 in Sewage: Evidence of Changes in Virus Variant Predominance during COVID-19 Pandemic. Viruses 2020, 12, 1144. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101144
Martin J, Klapsa D, Wilton T, Zambon M, Bentley E, Bujaki E, Fritzsche M, Mate R, Majumdar M. Tracking SARS-CoV-2 in Sewage: Evidence of Changes in Virus Variant Predominance during COVID-19 Pandemic. Viruses. 2020; 12(10):1144. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101144
Chicago/Turabian StyleMartin, Javier, Dimitra Klapsa, Thomas Wilton, Maria Zambon, Emma Bentley, Erika Bujaki, Martin Fritzsche, Ryan Mate, and Manasi Majumdar. 2020. "Tracking SARS-CoV-2 in Sewage: Evidence of Changes in Virus Variant Predominance during COVID-19 Pandemic" Viruses 12, no. 10: 1144. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101144