New RNA Structural Elements Identified in the Coding Region of the Coxsackie B3 Virus Genome

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. DNA Templates and RNA Synthesis
2.3. RNA Structure Probing In Vitro
2.4. Sequences of CVB-3 Isolates
2.5. Secondary Structure Modeling
2.6. Prediction of Conserved Structural Regions
2.7. Digestion with Ribonuclease L (RNase L)
2.8. Dimerization Studies
2.9. Predicting RNA Interactions with Proteins Using the ATtRACT Database
2.10. Cell Culture and Cytoplasmic Lysate Preparation
2.11. RNase-Assisted RNA Chromatography
2.12. Mass Spectrometry Analysis
3. Results and Discussion
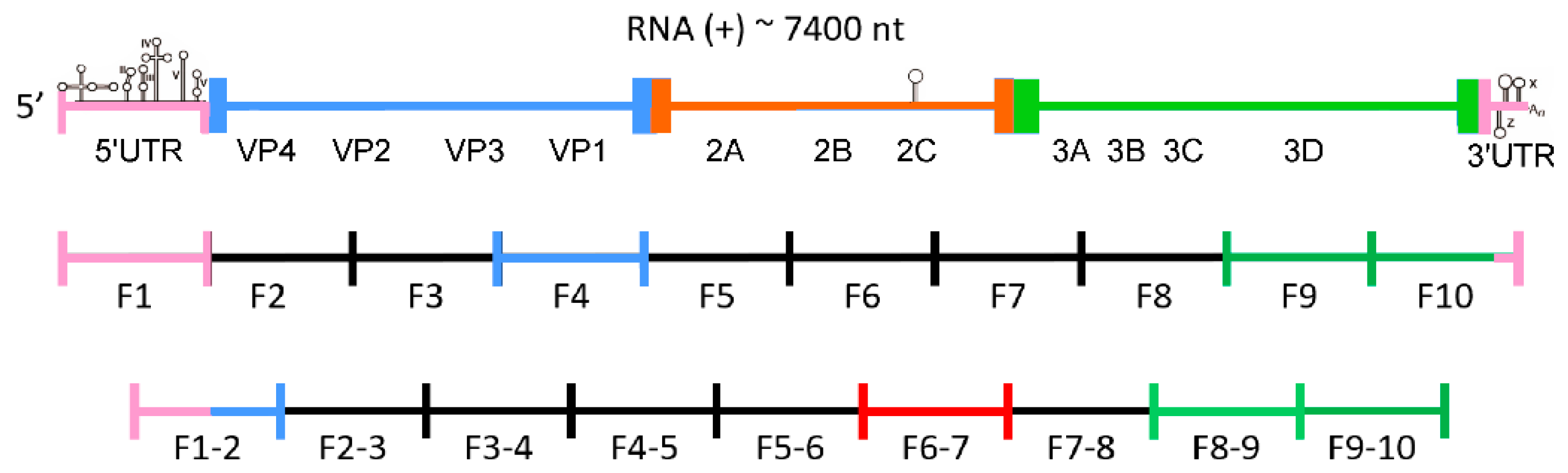
3.1. Modeling of RNA Secondary Structures of RNA Fragments and Searching for the Most Probable Elements, which Remain Unchanged in Multiple Structure Predictions
3.1.1. Secondary Structure of Partly Overlapping Fragments: F1 and F1–2
3.1.2. Secondary Structure of Fragment F4 and Fragment F6–7
3.1.3. Secondary Structure of Partly Overlapping Fragments: F8–9 and F9
3.1.4. Secondary Structure of Partly Overlapping Fragments: F9–10 and F10
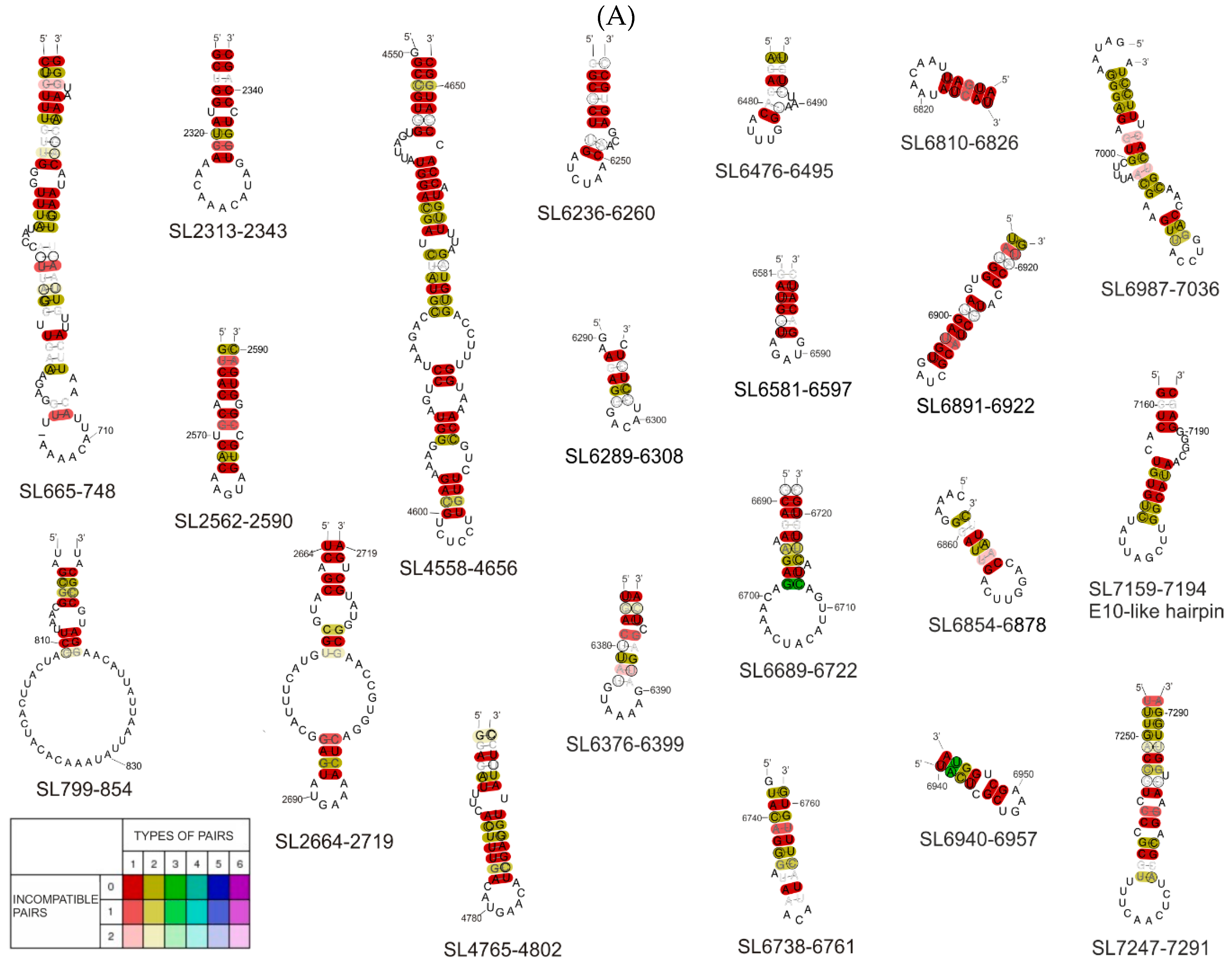
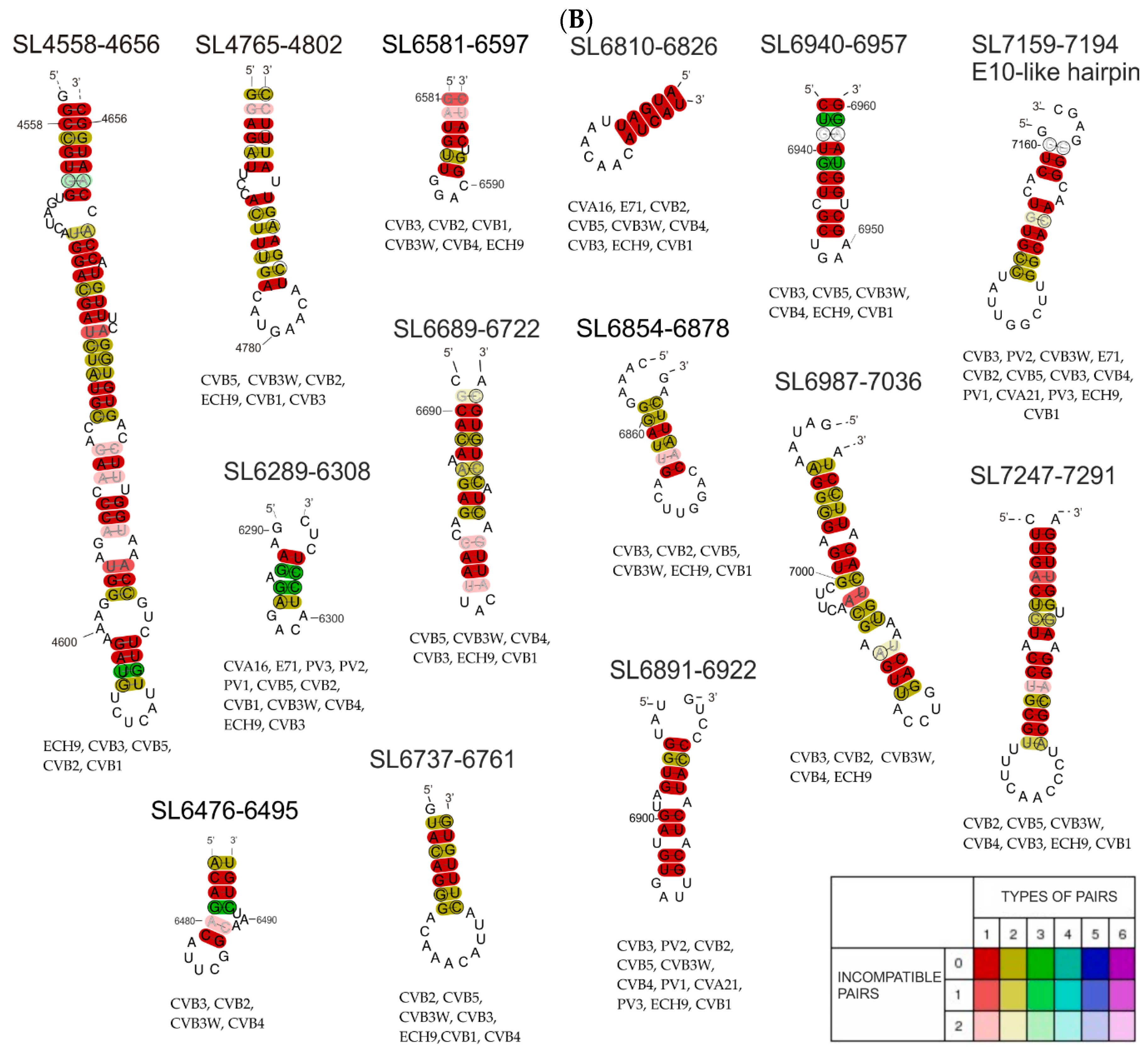
3.2. Phylogenetic Analysis of Selected Structural Motifs Conserved among CVB3 Strains and Other Enteroviruses
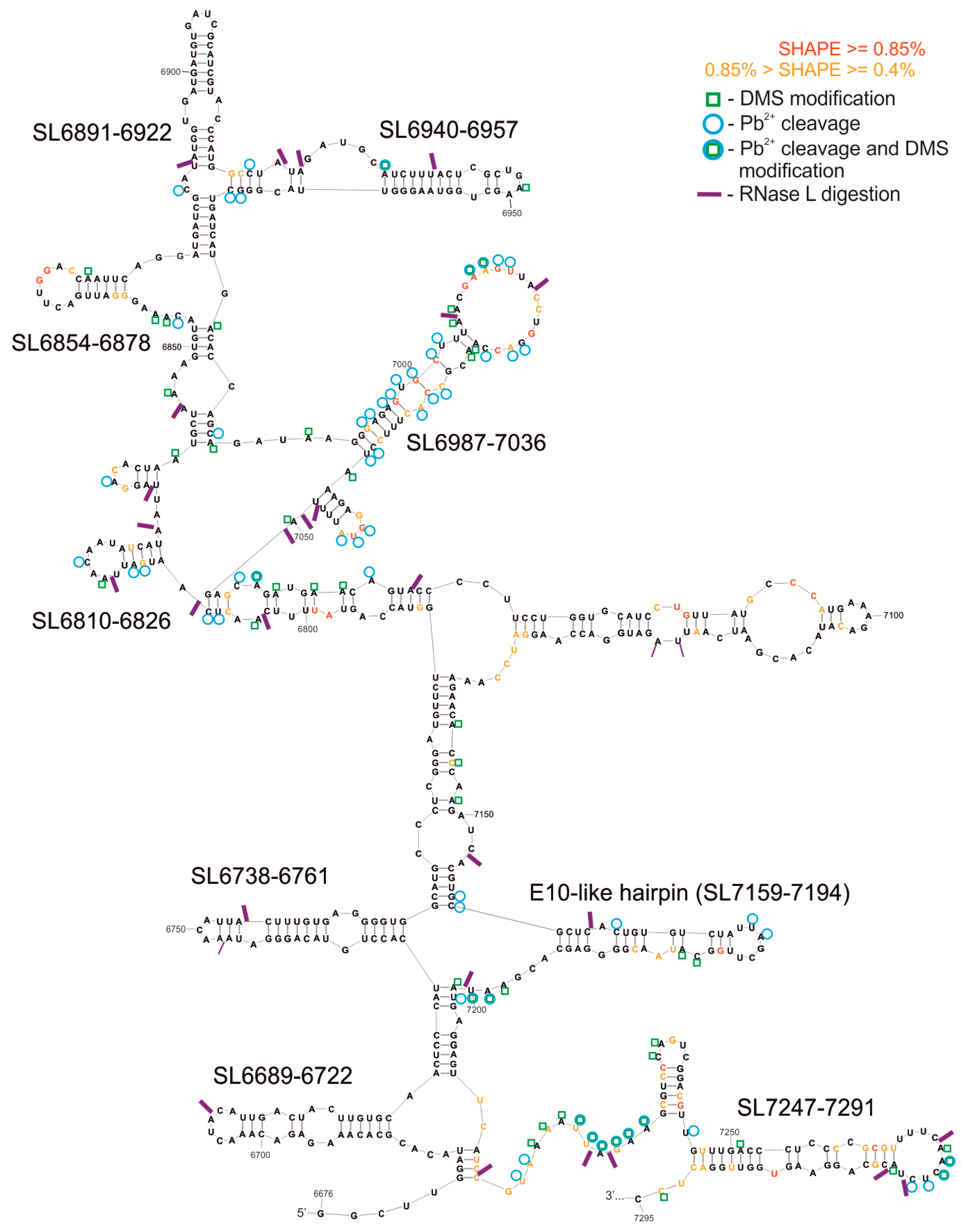
3.3. RNase L Assay-Limited Digestion of 3′-Terminal Fragment (F10) of CVB3 Genome with Ribonuclease L
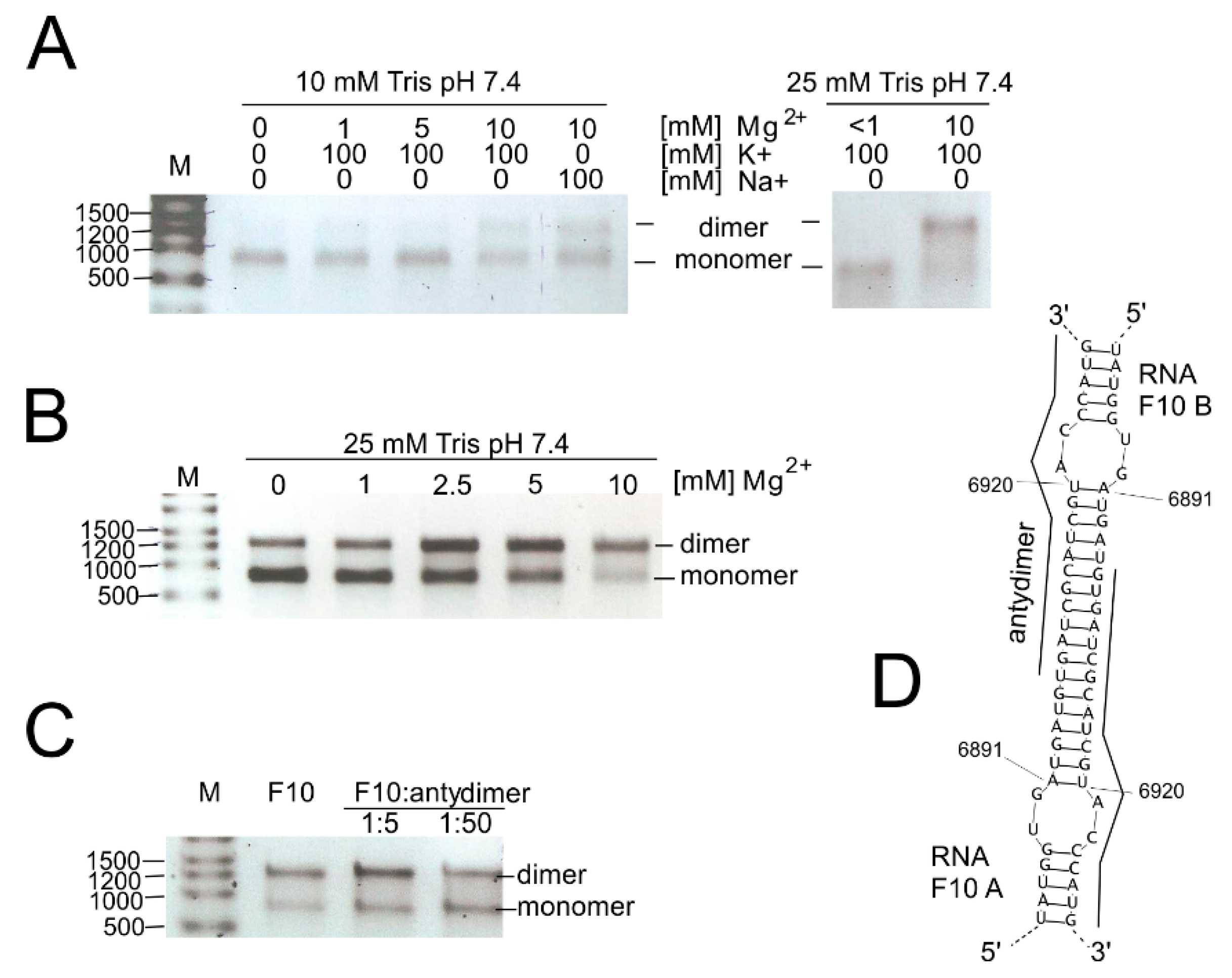
3.4. Investigation of the Dimerization Process of F10 Fragment Representing 3′-Terminal Region of the CVB3 Genome
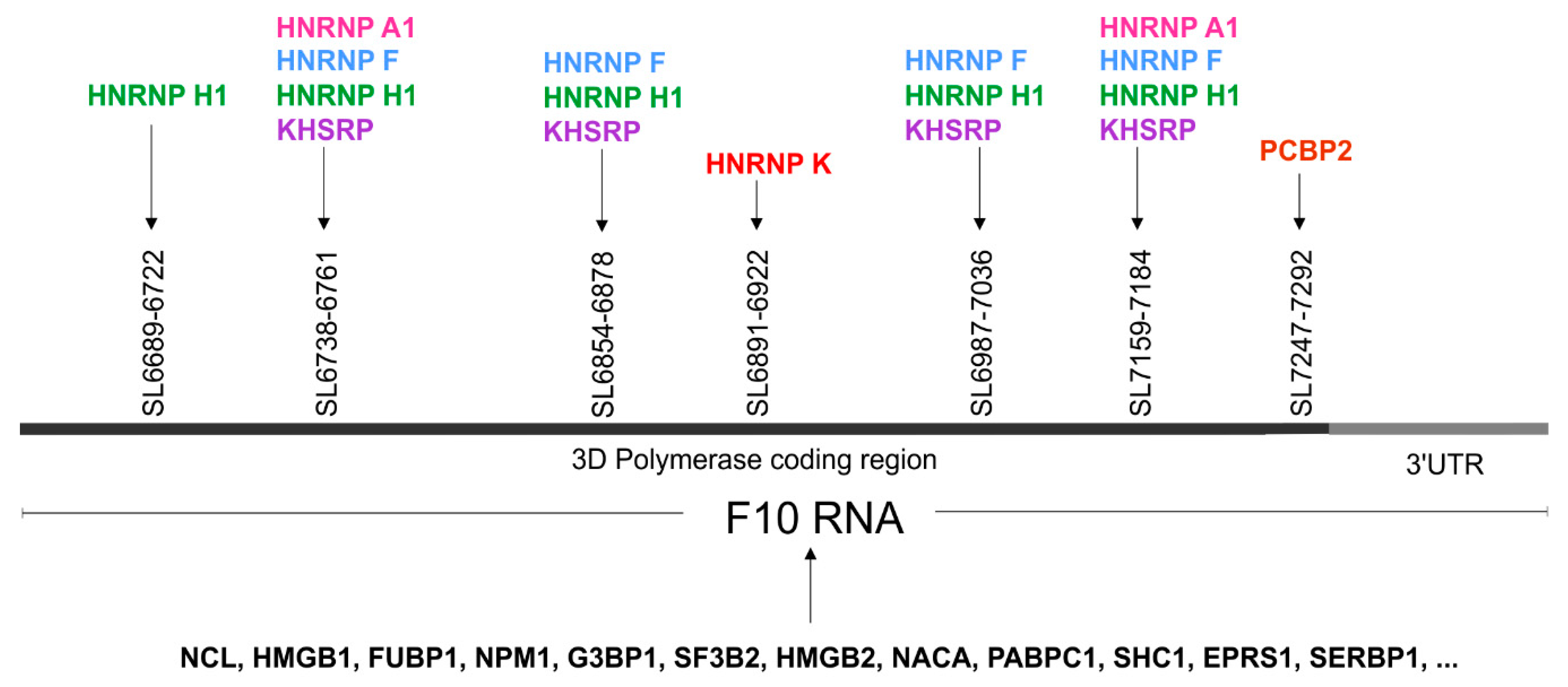
3.5. High Throughput Protein Analysis Reveals Proteins, which are able to Bind to the 3′-Terminal Part of the CVB3 Genomic RNA
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W., Jr.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zheng, Q.; Ryvkin, P.; Dragomir, I.; Desai, Y.; Aiyer, S.; Valladares, O.; Yang, J.; Bambina, S.; Sabin, L.R.; et al. Global analysis of RNA secondary structure in two metazoans. Cell Rep. 2012, 1, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Moss, W.N.; Steitz, J.A. Genome-wide analyses of Epstein-Barr virus reveal conserved RNA structures and a novel stable intronic sequence RNA. BMC Genom. 2013, 14, 543. [Google Scholar] [CrossRef] [Green Version]
- Mauger, D.M.; Golden, M.; Yamane, D.; Williford, S.; Lemon, S.M.; Martin, D.P.; Weeks, K.M. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc. Natl. Acad. Sci. USA 2015, 112, 3692–3697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrill, C.P.; Westesson, O.; Schulte, M.B.; Strings, V.R.; Segal, M.; Andino, R. Global RNA structure analysis of poliovirus identifies a conserved RNA structure involved in viral replication and infectivity. J. Virol. 2013, 87, 11670–11683. [Google Scholar] [CrossRef] [Green Version]
- Friebe, P.; Boudet, J.; Simorre, J.P.; Bartenschlager, R. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 2005, 79, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Mortimer, S.A.; Kidwell, M.A.; Doudna, J.A. Insights into RNA structure and function from genome-wide studies. Nat. Rev. Genet. 2014, 15, 469–479. [Google Scholar] [CrossRef]
- Bartas, M.; Brazda, V.; Bohalova, N.; Cantara, A.; Volna, A.; Stachurova, T.; Malachova, K.; Jagelska, E.B.; Porubiakova, O.; Cerven, J.; et al. In-Depth Bioinformatic Analyses of Nidovirales Including Human SARS-CoV-2, SARS-CoV, MERS-CoV Viruses Suggest Important Roles of Non-canonical Nucleic Acid Structures in Their Lifecycles. Front. Microbiol. 2020, 11, 1583. [Google Scholar] [CrossRef]
- Stein, B.S.; Engleman, E.G. Intracellular processing of the gp160 HIV-1 envelope precursor. Endoproteolytic cleavage occurs in a cis or medial compartment of the Golgi complex. J. Biol. Chem. 1990, 265, 2640–2649. [Google Scholar]
- You, S.; Rice, C.M. 3′ RNA elements in hepatitis C virus replication: Kissing partners and long poly(U). J. Virol. 2008, 82, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Dutkiewicz, M.; Ciesiolka, J. Form confers function: Case of the 3′X region of the hepatitis C virus genome. World J. Gastroenterol. 2018, 24, 3374–3383. [Google Scholar] [CrossRef] [PubMed]
- Witwer, C.; Rauscher, S.; Hofacker, I.L.; Stadler, P.F. Conserved RNA secondary structures in Picornaviridae genomes. Nucleic Acids Res. 2001, 29, 5079–5089. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.; Chaudhry, Y.; Richardson, A.; Meredith, J.; Almond, J.W.; Barclay, W.; Evans, D.J. Identification of a cis-acting replication element within the poliovirus coding region. J. Virol. 2000, 74, 4590–4600. [Google Scholar] [CrossRef] [PubMed]
- Van Ooij, M.J.; Vogt, D.A.; Paul, A.; Castro, C.; Kuijpers, J.; van Kuppeveld, F.J.; Cameron, C.E.; Wimmer, E.; Andino, R.; Melchers, W.J. Structural and functional characterization of the coxsackievirus B3 CRE(2C): Role of CRE(2C) in negative- and positive-strand RNA synthesis. J. Gen. Virol. 2006, 87, 103–113. [Google Scholar] [CrossRef]
- Song, Y.; Liu, Y.; Ward, C.B.; Mueller, S.; Futcher, B.; Skiena, S.; Paul, A.V.; Wimmer, E. Identification of two functionally redundant RNA elements in the coding sequence of poliovirus using computer-generated design. Proc. Natl. Acad. Sci. USA 2012, 109, 14301–14307. [Google Scholar] [CrossRef] [Green Version]
- Han, J.Q.; Townsend, H.L.; Jha, B.K.; Paranjape, J.M.; Silverman, R.H.; Barton, D.J. A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J. Virol. 2007, 81, 5561–5572. [Google Scholar] [CrossRef] [Green Version]
- Townsend, H.L.; Jha, B.K.; Han, J.Q.; Maluf, N.K.; Silverman, R.H.; Barton, D.J. A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA 2008, 14, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Keel, A.Y.; Jha, B.K.; Kieft, J.S. Structural architecture of an RNA that competitively inhibits RNase L. RNA 2012, 18, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Mahy, B.W. Coxsackie B viruses: An introduction. Curr. Top. Microbiol. Immunol. 2008, 323, vii–xiii. [Google Scholar]
- Malathi, K.; Saito, T.; Crochet, N.; Barton, D.J.; Gale, M., Jr.; Silverman, R.H. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 2010, 16, 2108–2119. [Google Scholar] [CrossRef] [Green Version]
- Dutkiewicz, M.; Stachowiak, A.; Swiatkowska, A.; Ciesiolka, J. Structure and function of RNA elements present in enteroviral genomes. Acta Biochim. Pol. 2016, 63, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, J.M.; Tapprich, W.E. Structure of the 5′ nontranslated region of the coxsackievirus b3 genome: Chemical modification and comparative sequence analysis. J. Virol. 2007, 81, 650–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoll, J.; Heus, H.A.; van Kuppeveld, F.J.; Melchers, W.J. The structure-function relationship of the enterovirus 3′-UTR. Virus Res. 2009, 139, 209–216. [Google Scholar] [CrossRef]
- Glenet, M.; Heng, L.; Callon, D.; Lebreil, A.L.; Gretteau, P.A.; Nguyen, Y.; Berri, F.; Andreoletti, L. Structures and Functions of Viral 5′ Non-Coding Genomic RNA Domain-I in Group-B Enterovirus Infections. Viruses 2020, 12, 919. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef]
- Lindell, M.; Romby, P.; Wagner, E.G. Lead(II) as a probe for investigating RNA structure in vivo. RNA 2002, 8, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Gorska, A.; Blaszczyk, L.; Dutkiewicz, M.; Ciesiolka, J. Length variants of the 5′ untranslated region of p53 mRNA and their impact on the efficiency of translation initiation of p53 and its N-truncated isoform DeltaNp53. RNA Biol. 2013, 10, 1726–1740. [Google Scholar] [CrossRef] [Green Version]
- Dutkiewicz, M.; Ciesiolka, J. Structural characterization of the highly conserved 98-base sequence at the 3′ end of HCV RNA genome and the complementary sequence located at the 5′ end of the replicative viral strand. Nucleic Acids Res. 2005, 33, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [Green Version]
- Vasa, S.M.; Guex, N.; Wilkinson, K.A.; Weeks, K.M.; Giddings, M.C. ShapeFinder: A software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA 2008, 14, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloma, M.F.; Mathews, D.H. Improving RNA secondary structure prediction with structure mapping data. Methods Enzymol. 2015, 553, 91–114. [Google Scholar] [CrossRef]
- Ciesiolka, J.; Michalowski, D.; Wrzesinski, J.; Krajewski, J.; Krzyzosiak, W.J. Patterns of cleavages induced by lead ions in defined RNA secondary structure motifs. J. Mol. Biol. 1998, 275, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, M.; Ojdowska, A.; Gorska, A.; Swiatkowska, A.; Ciesiolka, J. The structural and phylogenetic profile of the 3′ terminus of coxsackievirus B3 negative strand. Virus Res. 2014, 188, 81–89. [Google Scholar] [CrossRef]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008, 9, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neubock, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofacker, I.L.; Fekete, M.; Stadler, P.F. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002, 319, 1059–1066. [Google Scholar] [CrossRef]
- Giudice, G.; Sánchez-Cabo, F.; Torroja, C.; Lara-Pezzi, E. ATtRACT-a database of RNA-binding proteins and associated motifs. Database 2016, 2016, baw035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiatkowska, A.; Dutkiewicz, M.; Machtel, P.; Janecki, D.M.; Kabacinska, M.; Zydowicz-Machtel, P.; Ciesiolka, J. Regulation of the p53 expression profile by hnRNP K under stress conditions. RNA Biol. 2020, 17, 1402–1415. [Google Scholar] [CrossRef]
- Michlewski, G.; Caceres, J.F. RNase-assisted RNA chromatography. RNA 2010, 16, 1673–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakun, M.; Karczmarski, J.; Poznanski, J.; Rubel, T.; Rozga, M.; Malinowska, A.; Sands, D.; Hennig, E.; Oledzki, J.; Ostrowski, J.; et al. An integrated LC-ESI-MS platform for quantitation of serum peptide ladders. Application for colon carcinoma study. Proteom. Clin. Appl. 2009, 3, 932–946. [Google Scholar] [CrossRef]
- Yang, D.; Wilson, J.E.; Anderson, D.R.; Bohunek, L.; Cordeiro, C.; Kandolf, R.; McManus, B.M. In vitro mutational and inhibitory analysis of the cis-acting translational elements within the 5′ untranslated region of coxsackievirus B3: Potential targets for antiviral action of antisense oligomers. Virology 1997, 228, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zydowicz-Machtel, P.; Swiatkowska, A.; Popenda, L.; Gorska, A.; Ciesiolka, J. Variants of the 5′-terminal region of p53 mRNA influence the ribosomal scanning and translation efficiency. Sci. Rep. 2018, 8, 1533. [Google Scholar] [CrossRef] [Green Version]
- Błaszczyk, L.; Ciesiołka, J. Secondary Structure and the Role in Translation Initiation of the 5′-Terminal Region of p53 mRNA. Biochemistry 2011, 50, 7080–7092. [Google Scholar] [CrossRef]
- Gorska, A.; Swiatkowska, A.; Dutkiewicz, M.; Ciesiolka, J. Modulation of p53 expression using antisense oligonucleotides complementary to the 5′-terminal region of p53 mRNA in vitro and in the living cells. PLoS ONE 2013, 8, e78863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutkiewicz, M.; Ojdowska, A.; Kuczynski, J.; Lindig, V.; Zeichhardt, H.; Kurreck, J.; Ciesiolka, J. Targeting Highly Structured RNA by Cooperative Action of siRNAs and Helper Antisense Oligomers in Living Cells. PLoS ONE 2015, 10, e0136395. [Google Scholar] [CrossRef]
- Han, Y.; Donovan, J.; Rath, S.; Whitney, G.; Chitrakar, A.; Korennykh, A. Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 2014, 343, 1244–1248. [Google Scholar] [CrossRef] [Green Version]
- Witteveldt, J.; Blundell, R.; Maarleveld, J.J.; McFadden, N.; Evans, D.J.; Simmonds, P. The influence of viral RNA secondary structure on interactions with innate host cell defences. Nucleic Acids Res. 2014, 42, 3314–3329. [Google Scholar] [CrossRef] [Green Version]
- Dubois, N.; Marquet, R.; Paillart, J.C.; Bernacchi, S. Retroviral RNA Dimerization: From Structure to Functions. Front. Microbiol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Bhattacharyya, D.; Mirihana Arachchilage, G.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front. Chem. 2016, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- van Domselaar, R.; de Poot, S.A.; Remmerswaal, E.B.; Lai, K.W.; ten Berge, I.J.; Bovenschen, N. Granzyme M targets host cell hnRNP K that is essential for human cytomegalovirus replication. Cell Death Differ. 2013, 20, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.S.; Seol, S.K.; Song, O.K.; Park, J.H.; Jang, S.K. An RNA-binding protein, hnRNP A1, and a scaffold protein, septin 6, facilitate hepatitis C virus replication. J. Virol. 2007, 81, 3852–3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.L.; Lin, J.Y.; Chen, B.S.; Weng, K.F.; Shih, S.R.; Calderon, J.D.; Tolbert, B.S.; Brewer, G. EV71 3C protease induces apoptosis by cleavage of hnRNP A1 to promote apaf-1 translation. PLoS ONE 2019, 14, e0221048. [Google Scholar] [CrossRef]
- Briata, P.; Chen, C.Y.; Ramos, A.; Gherzi, R. Functional and molecular insights into KSRP function in mRNA decay. Biochim. Biophys. Acta 2013, 1829, 689–694. [Google Scholar] [CrossRef] [PubMed]
- You, F.; Sun, H.; Zhou, X.; Sun, W.; Liang, S.; Zhai, Z.; Jiang, Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009, 10, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Chin, W.X.; Han, Q.; Ichiyama, K.; Lee, C.H.; Eyo, Z.W.; Ebina, H.; Takahashi, H.; Takahashi, C.; Tan, B.H.; et al. Characterization of RyDEN (C19orf66) as an Interferon-Stimulated Cellular Inhibitor against Dengue Virus Replication. PLoS Pathog. 2016, 12, e1005357. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Primer Name | Nucleotide Sequence |
|---|---|---|
| 1 | 1F | 5′-TAATACGACTCACTATAGGAAACAGCCTGTGGGTTGATCC-3′ |
| 2 | 1R | 5′- CTAGCATTCAGCCTGGTCTC-3′ |
| 3 | 1–2F | 5′-TAATACGACTCACTATAGGTCTAATACAGACATGGTGCGAAG-3′ |
| 4 | 1–2R | 5′-TCTGCCATTGCACAGAGTCAAG-3′ |
| 5 | 4F | 5′-TAATACGACTCACTATAGGTGTTGCTTCAGATGAGTATACCG-3′ |
| 6 | 4R | 5′-AAATTCAGACCATCCGTCATAG-3′ |
| 7 | 6–7F | 5′-TAATACGACTCACTATAGGCGAGTTCCTGAACAGACTTAAAC-3′ |
| 8 | 6–7R | 5′-TAGAGAGTATCTGACCTGTGTTC-3′ |
| 10 | 8–9F | 5′-TAATACGACTCACTATAGGCAATTAACACCAGCAAGTTTCC-3′ |
| 11 | 8–9R | 5′-TTCCTTTCGCTACCTTCTCTATG-3′ |
| 12 | 9F | 5′-TAATACGACTCACTATAGGGTTTCCAGTCATCAACACACC-3′ |
| 13 | 9R | 5′-GGAGTTGCACAAGTAGTCAATG-3′ |
| 14 | 9–10F | 5′-TAATACGACTCACTATAGGGAACCTACCAATGGTGACTTATG-3′ |
| 15 | 9–10R | 5′-GATTCGTGTATGTCTTTCATGG-3′ |
| 16 | 10F | 5′-TAATACGACTCACTATAGGCTTGGATACACGCACAAAGAG-3′ |
| 17 | 10R | 5′-GCACCGAATGCGGAGAATTTAC-3′ |
| 18 | 10+(R) | 5′-TTTTTTTTTTTCCGCACCGAATGC-3′ |
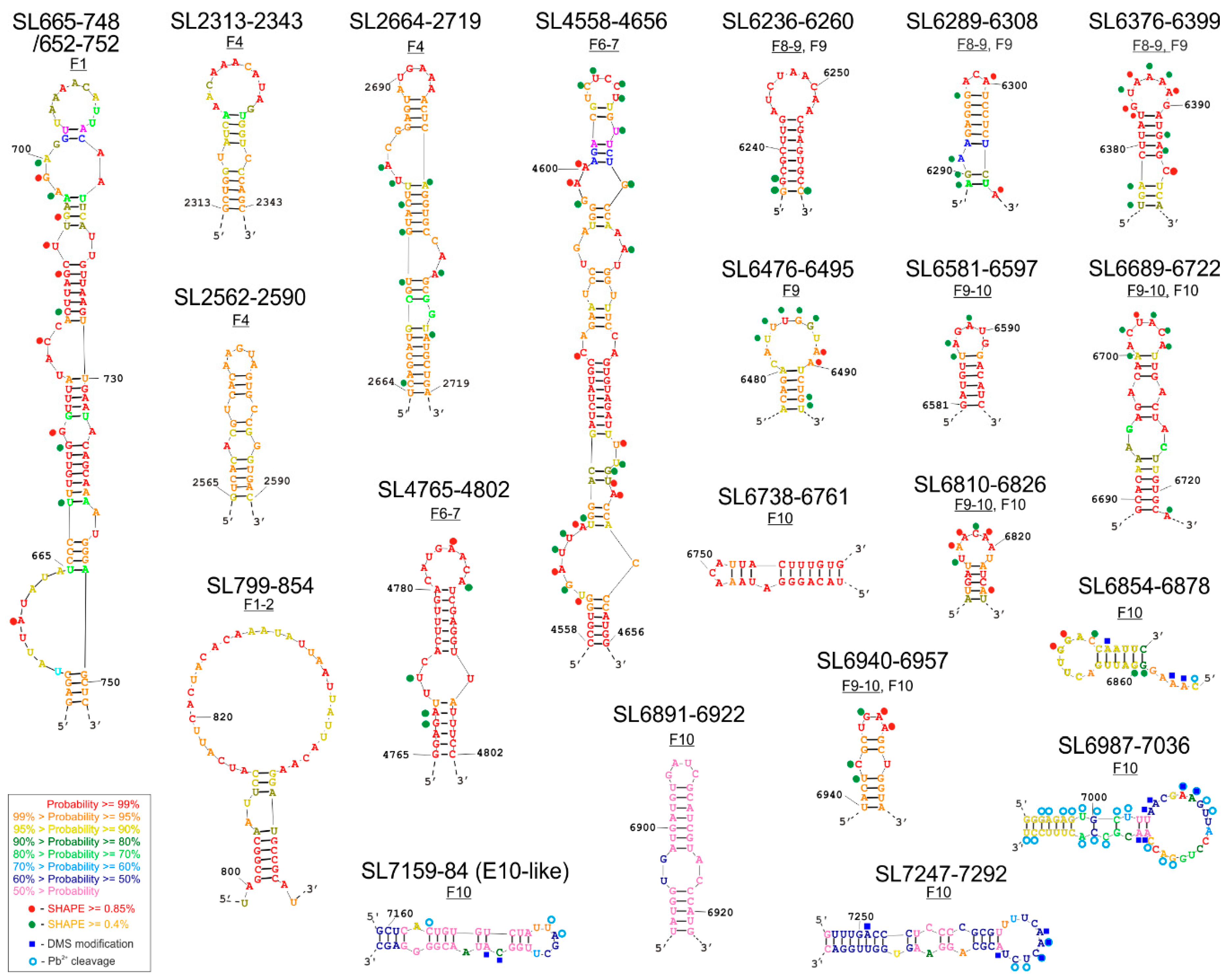
| RNA Structure Motif | Length (nt) | Genome Region | RNA Fragment in which the Motif was Experimentally Characterized | RNA Fragment in which the Motif was Thermodynamically Predicted | Motif Structure Conservation | Potential Interactions with Proteins According to ATtRACT Database and RNA-Centric Affinity Chromatography * |
|---|---|---|---|---|---|---|
| SL665-748 SL652-752 (with bulge) | 84 101 | 5′UTR /V4 | F1, F1–2 (SHAPE) | F1 (H), F1–2 (L) | (some CVB3) | CELF1, CELF 2, ELAVL2, ENOX1, GRSF1, HNRNP (A1, A2B1, F, H1, H2, H3, K, L), IGF2BP2, IGF2BP3, KHDRBS1, KHSRP, NOVA1, NOVA 2, OAS1, PCBP2, PIWIL1, PPIE, PTBP1, RBMS3, RBMX, RNASEL, SRP (19, 54, 68), SRSF (2, 5, 9, 10), TIA1, TIAL1, XPO5, YTHDC1, ZFP36, ZRANB2 |
| SL799-854 | 54 | V4 | - | F1–2 (H) | (CVB3) | A1CF, HNRNPL, IGF2BP3, MBNL1, NOVA1, NOVA2, PABPC1, PPIE, PTBP1, RBMY1A1, SRSF3, SRSF9, TIA1, TIAL1, XPO5, YBX1, YTHDC1, |
| SL2313-2343 | 31 | V1 | - | F4 (H) | (some CVB3) | HNRNPL, HNRNPLL, IGF2BP2, IGF2BP3, NOVA1, PABPC1, PTBP1, RBMX, SAMD4A, SRP14, ZNF346 |
| SL2562-2590 | 29 | V1 | - | F4 (H) | (some CVB3) | FUS, HNRNPL, KHSRP, MBNL1, NOVA1, NOVA2, RBMY1A1, SRSF (1, 2, 5, 6), YTHDC1, ZRANB2, RBMX, |
| SL2664-2719 | 55 | V1 | F4 (SHAPE) | F4 (H) | (CVB3) | CELF2, CPEB4, ELAVL2, FUS, IGF2BP2, IGF2BP3, KHDRBS1, LIN28A, MBNL1, NOVA1, NUDT21, PTBP1, RBFOX1, RNASEL, SRP54, SRP68, SRSF (1, 2, 5, 6, 9), SSB, TIA1, TIAL1, YTHDC1, ZRANB2, |
| SL4558-4656 | 99 | 2C | F6–7 (SHAPE) | F6–7 (H) | (E) | ACO1, CELF1, CELF2, CMTR1, ELAVL (1, 2, 4), ESRP2, FUS, GRSF1, HNRNP (F, H1, H2, H3, K, L), KHSRP, MBNL1, NOVA1, NOVA2, NUDT21, NXF1, PTBP1, RBM28, RC3H1, SRP14, SRSF (1, 2, 3, 6, 9), SSB, TIA1, TIAL1, TRA2B, XPO5, YBX1, ZFP36, ZNF346, |
| SL4765-4802 | 38 | 2C | F6–7 (SHAPE) | F6–7 (H) | (E) | CELF2, ELAVL (1, 2, 4), HNRNPL, NOVA1, NOVA2, OAS1, PHAX, PPIE, PTBP1, RC3H1, SRP54, SRSF (2, 3, 9), SSB, TIA1, TIAL1, YBX1, YBX2, ZRANB2 |
| SL6236-6260 | 25 | 3D | F8–9 (SHAPE) | F8–9 (H), F9 (H) | (CVB3) | ADAR, CMTR1, DDX58, DHX9, HNRNPL, MBNL1, OAS1, PIWIL1, PTBP1, QKI, SRSF3, YBX1, YBX2, YTHDC1 |
| SL6289-6308 | 20 | 3D | F8–9, F9 (SHAPE) | F8–9 (H), F9 (H) | (CVB3) (E) | F2, GRSF1, HNRNP (F, H1, H2, H3, L), KHSRP, NONO, PABPN1, PTBP1, PTBP2, SRP (19, 54, 68), SRSF (10, 2, 3, 5), TRA2B, YBX1, ZFP36, |
| SL6376-6399 | 24 | 3D | F8–9, F9 (SHAPE) | F8–9 (H), F9 (H) | (CVB3) | CELF2, NUDT21, PABPN1, PTBP1, QKI, RNASEL, SF1, SRP19 |
| SL6476-6495, | 20 | 3D | F9 (SHAPE) | F9 (H) | (CVB3) | ELAVL (1, 2, 4), HNRNPL, IGF2BP3, NXF1, PTBP1, SSB, ZRANB2 |
| SL6581-6597 | 17 | 3D | F9–10 (SHAPE) | F9–10 (H), F9 (L) | (CVB3) | CELF1, CELF2, HNRNPA1, HNRNPL, KHSRP, SRSF (1, 2, 3, 9), YBX1, |
| SL6689-6722 | 34 | 3D | F9–10 (SHAPE) | F9–10 (H), F10 (H) | (E) | CELF1, FXR2, HNRNP (H1*, H2, L), IGF2BP3, NXF1, OAS1, PTBP1, RBMY1A1, SRSF10, YTHDC1, ZFP36, |
| SL6738-6761 | 24 | 3D | - | F10 (H) | (E) | CELF1, ESRP1, GRSF1, HNRNP (A1*, F*, H1*, H2, H3, L), IGF2BP3, KHDRBS2, KHDRBS3, KHSRP*, NONO, PTBP1, RBM41, SRSF5, TIA1, QKI, SF1, TIAL1 |
| SL6810-6826 | 17 | 3D | F9–10, F10 (SHAPE, Pb2+, DMS) | F9–10 (H), F10 (H) | (E) | HNRNPL, NOVA1, NOVA2, QKI, SF1, TIAL1 |
| SL6854-6878 | 25 | 3D | F10 (SHAPE, Pb2+, DMS) | F10 (H) | (E) | EIF4B, ESRP1, GRSF1, HNRNP (F*, H1*, H2, H3), KHSRP*, NONO, OAS1, PTBP1, SRSF (10, 5, 9), TIA1, TIAL1 |
| SL6891–6922 | 32 | 3D | F10 (SHAPE, Pb2+, DMS) | F10 (L) | (E) | CELF2, CMTR1, FUS, HNRNPK*, NOVA1, PHAX, RBM46, SRSF (1, 3, 5, 6), YBX1, YTHDC1 |
| SL6940-6957 | 18 | 3D | F9–10, F10 (SHAPE, Pb2+, DMS) | F9–10 (H), F10 (H) | (E) | F2, MBNL1, PTBP1, SAMD4A, SRSF1 |
| SL6987-7036 | 50 | 3D | F10 (SHAPE, Pb2+, DMS) | F10 (L) | (E) | CPEB4, ELAVL2, F2, GRSF1, HNRNP (F*, H1*, H2, H3, L), KHSRP*, MBNL1, NONO, NOVA1, NXF1, PIWIL1, PTBP1, RC3H1, SRP68, SRSF (1, 2, 9), TIA1, TIAL1, YTHDC1, ZFP36, RBMX |
| E-10 like/ SL7159-7194 | 36 | 3D | F10 (SHAPE, Pb2+, DMS) | F10 (L) | (CVB3) (E) | CELF1, CELF2, GRSF1, HNRNP (A1*, F*, H1*, H2, H3), KHSRP*, NOVA1, NOVA2, OAS1, PIWIL1, PTBP1, SRP (19, 54, 68), SRSF2, SRSF5, TIAL1 |
| SL7247-7291 | 45 | 3D | F10 (SHAPE, Pb2+, DMS) | F10 (L) | (E) | AGO1, DHX9, ELAVL1, ELAVL2, F2, OAS1, PCBP1, PCBP2*, PTBP1, RC3H1, SFPQ, SRP (14, 19, 54, 68), SRSF (1, 5, 9), SSB, TIA1, TIAL1, TRA2B, XPO5, ZFP36, ZNF346, RBMX |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutkiewicz, M.; Kuczynski, J.; Jarzab, M.; Stachowiak, A.; Swiatkowska, A. New RNA Structural Elements Identified in the Coding Region of the Coxsackie B3 Virus Genome. Viruses 2020, 12, 1232. https://0-doi-org.brum.beds.ac.uk/10.3390/v12111232
Dutkiewicz M, Kuczynski J, Jarzab M, Stachowiak A, Swiatkowska A. New RNA Structural Elements Identified in the Coding Region of the Coxsackie B3 Virus Genome. Viruses. 2020; 12(11):1232. https://0-doi-org.brum.beds.ac.uk/10.3390/v12111232
Chicago/Turabian StyleDutkiewicz, Mariola, Jakub Kuczynski, Michal Jarzab, Aleksandra Stachowiak, and Agata Swiatkowska. 2020. "New RNA Structural Elements Identified in the Coding Region of the Coxsackie B3 Virus Genome" Viruses 12, no. 11: 1232. https://0-doi-org.brum.beds.ac.uk/10.3390/v12111232