Characterization of Clinical and Carrier Streptococcus agalactiae and Prophage Contribution to the Strain Variability

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Condition
2.2. DNA Isolation and PCR
2.3. Multilocus Sequence Typing
2.4. Whole Genome Sequencing, Sequence Analysis, and Prophage Identification
2.5. Detection of Prophages within S. agalactiae Strain Collection
2.6. Analysis of Prophage Induction by WGS
3. Results
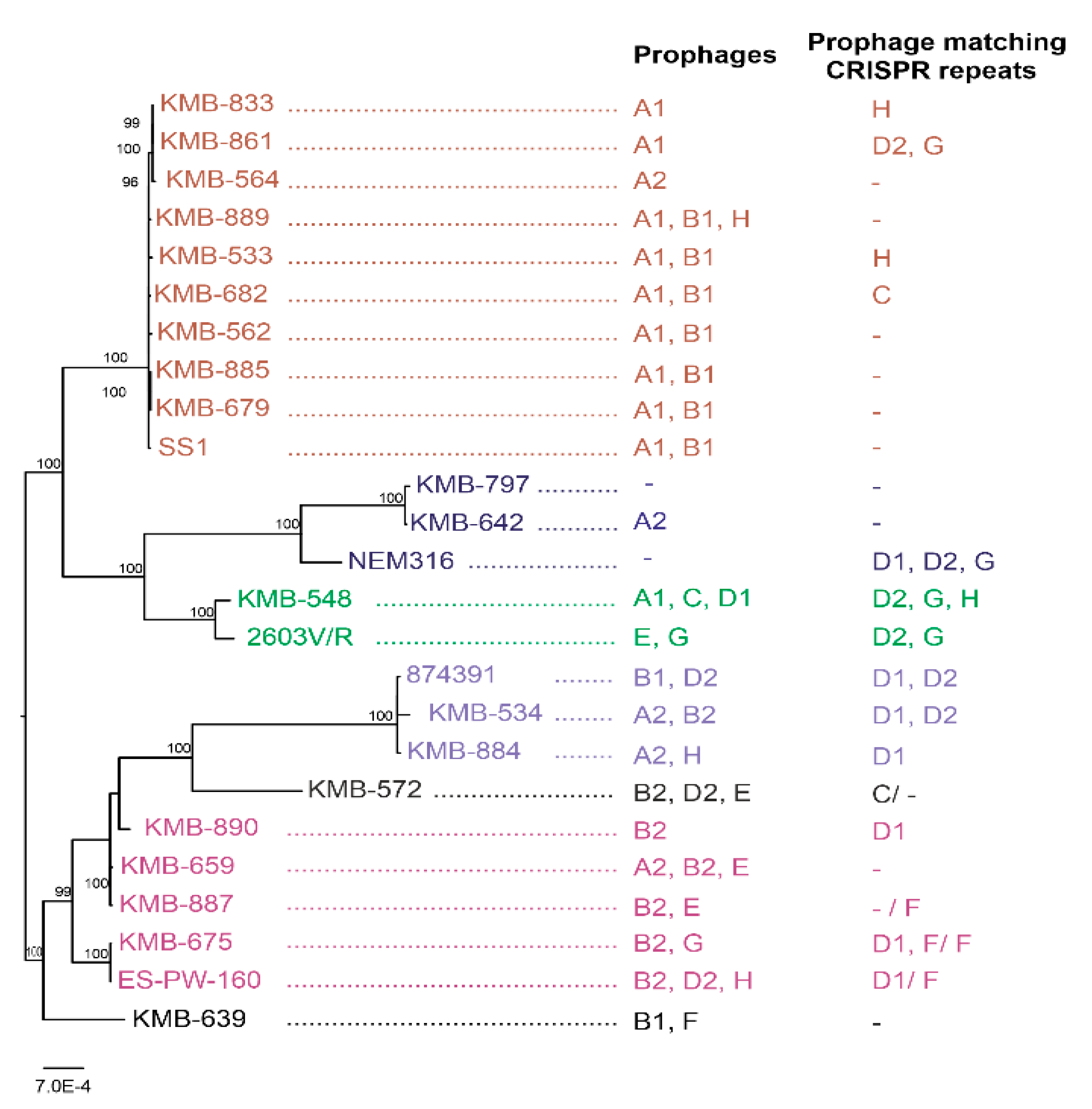
3.1. Analysis of GBS Strains
3.2. Whole Genome Sequencing
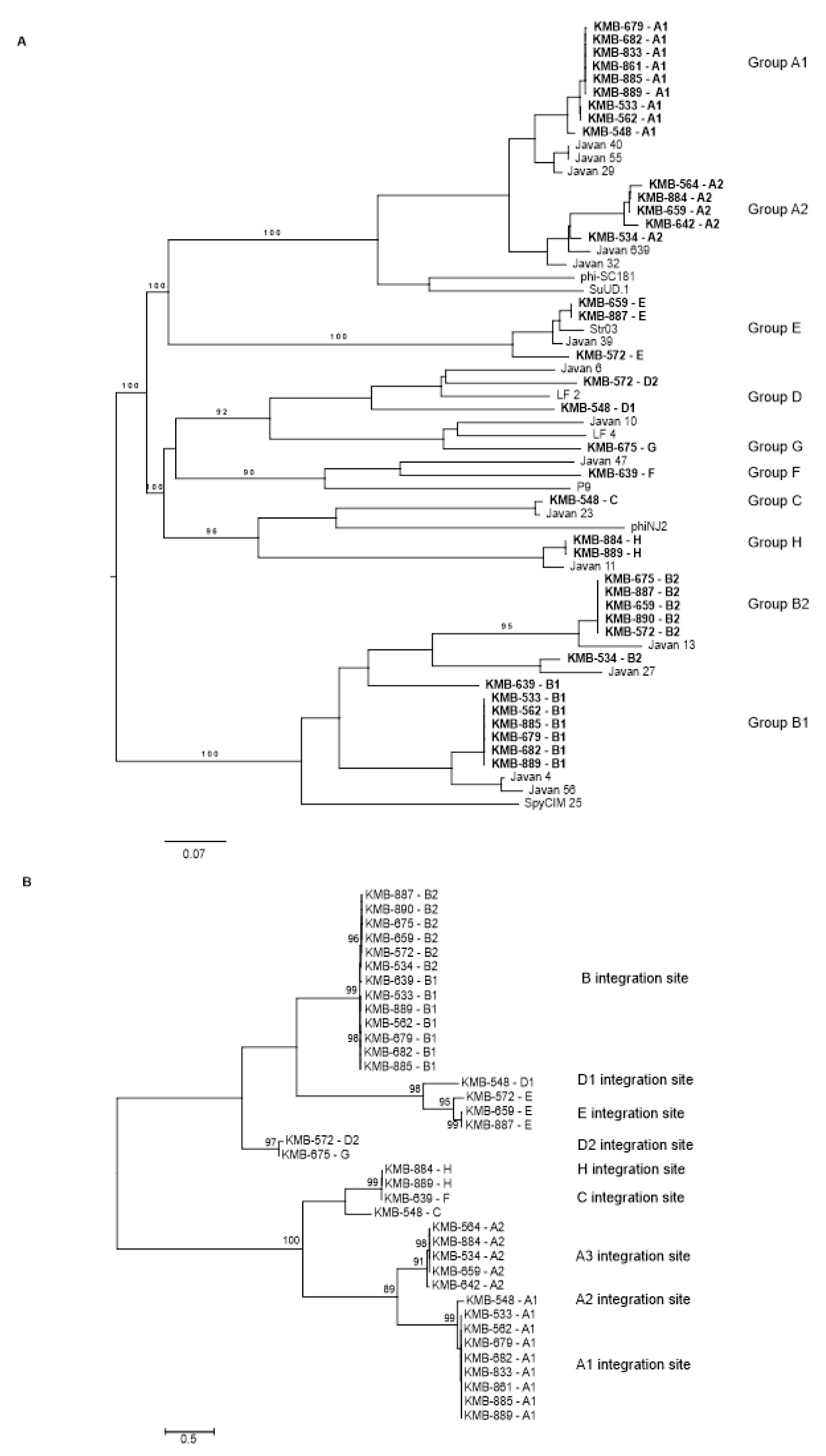
3.3. Prophage Detection in S. agalactiae
3.4. CRISPR-Cas Detection in S. agalactiae Genomes
3.5. PCR-Based Prophage Identification
3.6. Prophage Induction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Edmond, K.M.; Kortsalioudaki, C.; Scott, S.; Schrag, S.J.; Zaidi, A.K.; Cousens, S.; Heath, P.T. Group B streptococcal disease in infants aged younger than 3 months: Systematic review and meta-analysis. Lancet 2012, 379, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Puopolo, K.M.; Lynfield, R.; Cummings, J.J. Management of Infants at Risk for Group B Streptococcal Disease. Pediatrics 2019, 144, e20191881. [Google Scholar] [CrossRef] [Green Version]
- Watkins, L.K.F.; McGee, L.; Schrag, S.J.; Beall, B.; Jain, J.H.; Pondo, T.; Farley, M.M.; Harrison, L.H.; Zansky, S.M.; Baumbach, J.; et al. Epidemiology of Invasive Group B Streptococcal Infections among Nonpregnant Adults in the United States, 2008-2016. JAMA Intern. Med. 2019, 179, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Le Doare, K.; Heath, P.T. An overview of global GBS epidemiology. Vaccine 2013, 31, D7–D12. [Google Scholar] [CrossRef] [PubMed]
- Nizet, V.; Doran, K.S. Group B Streptococcus meningitis. Meningitis Cell. Mol. Basis 2013, 26, 118–132. [Google Scholar] [CrossRef] [Green Version]
- Tien, N.; Ho, C.M.; Lin, H.J.; Shih, M.C.; Ho, M.W.; Lin, H.C.; Lin, H.S.; Chang, C.C.; Lu, J.J. Multilocus sequence typing of invasive group B Streptococcus in central area of Taiwan. J. Microbiol. Immunol. Infect. 2011, 44, 430–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGee, L.; Chochua, S.; Li, Z.; Mathis, S.; Rivers, J.; Metcalf, B.; Ryan, A.; Alden, N.; Farley, M.M.; Harrison, L.H.; et al. Multistate, Population-Based Distributions of Candidate Vaccine Targets, Clonal Complexes, and Resistance Features of Invasive Group B Streptococci Within the United States, 2015–2017. Clin. Infect. Dis. 2020, 30329, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Davies, E.V.; Winstanley, C.; Fothergill, J.L.; James, C.E. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol. Lett. 2016, 363, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Brueggemann, A.B.; Harrold, C.L.; Javan, R.R.; van Tonder, A.J.; McDonnell, A.J.; Edwards, B.A. Pneumococcal prophages are diverse, but not without structure or history. Sci. Rep. 2017, 7, 42976. [Google Scholar] [CrossRef] [Green Version]
- Javan, R.R.; Ramos-Sevillano, E.; Akter, A.; Brown, J.; Brueggemann, A. Prophages and satellite prophages are widespread among Streptococcus species and may play a role in pneumococcal pathogenesis. bioRxiv 2018, 502740. [Google Scholar] [CrossRef]
- Beres, S.B.; Sylva, G.L.; Barbian, K.D.; Lei, B.; Hoff, J.S.; Mammarella, N.D.; Liu, M.Y.; Smoot, J.C.; Porcella, S.F.; Parkins, L.D.; et al. Genome sequence of a serotype M3 strain of group A Streptococcus: Phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc. Natl. Acad. Sci. USA 2002, 99, 10078–10083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, M.; Severina, E.; Tomasz, A. A high incidence of prophage carriage among natural isolates of Streptococcus pneumoniae. J. Bacteriol. 1999, 181, 3618–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russel, H.; Norcross, N.L.; Kahn, D.E. Isolation and Characterization of Streptococcus agalactiae Bacteriophage. J. Gen. Virol. 1969, 5, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [Green Version]
- Salloum, M.; van der Mee-Marquet, N.; Valentin-Domelier, A.S.; Quentin, R. Diversity of prophage DNA regions of streptococcus agalactiae clonal lineages from adults and neonates with invasive infectious disease. PLoS ONE 2011, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Van Der Mee-Marquet, N.; Domelier, A.S.; Mereghetti, L.; Lanotte, P.; Rosenau, A.; Van Leeuwen, W.; Quentin, R. Prophagic DNA fragments in Streptococcus agalactiae strains and association with neonatal meningitis. J. Clin. Microbiol. 2006, 44, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Van der Mee-Marquet, N.; Diene, S.M.; Barbera, L.; Courtier-Martinez, L.; Lafont, L.; Ouachée, A.; Valentin, A.S.; Dos Santos, S.; Quentin, R.; François, P. Analysis of the prophages carried by human infecting isolates provides new insight into the evolution of Group B Streptococcus species. Clin. Microbiol. Infect. 2018, 24, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Domelier, A.S.; Van Der Mee-Marquet, N.; Sizaret, P.Y.; Héry-Arnaud, G.; Lartigue, M.F.; Mereghetti, L.; Quentin, R. Molecular characterization and lytic activities of Streptococcus agalactiae bacteriophages and determination of lysogenic-strain features. J. Bacteriol. 2009, 191, 4776–4785. [Google Scholar] [CrossRef] [Green Version]
- Bai, Q.; Zhang, W.; Yang, Y.; Tang, F.; Nguyen, X.; Liu, G.; Lu, C. Characterization and genome sequencing of a novel bacteriophage infecting Streptococcus agalactiae with high similarity to a phage from Streptococcus pyogenes. Arch. Virol. 2013, 158, 1733–1741. [Google Scholar] [CrossRef]
- Furfaro, L.L.; Payne, M.S.; Chang, B.J. Host range, morphological and genomic characterisation of bacteriophages with activity against clinical Streptococcus agalactiae isolates. PLoS ONE 2020, 15, e0235002. [Google Scholar] [CrossRef]
- Chotár, M.; Vidová, B.; Godány, A. Development of specific and rapid detection of bacterial pathogens in dairy products by PCR. Folia Microbiol. 2006, 51, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Poyart, C.; Tazi, A.; Réglier-Poupet, H.; Billoët, A.; Tavares, N.; Raymond, J.; Trieu-Cuot, P. Multiplex PCR assay for rapid and accurate capsular typing of group B streptococci. J. Clin. Microbiol. 2007, 45, 1985–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creti, R.; Fabretti, F.; Orefici, G.; Von Hunolstein, C. Multiplex PCR Assay for Direct Identification of Group B Streptococcal Alpha-Protein-Like Protein Genes. J. Clin. Microbiol. 2004, 42, 1326–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.; Bohnsack, J.F.; Takahashi, S.; Oliver, K.A.; Chan, M.; Kunst, F.; Glaser, P.; Rusniok, C.; Crook, D.W.M.; Rosalind, M.; et al. Multilocus Sequence Typing System for Group B Streptococcus. J. Clin. Microbiol. 2003, 41, 2530–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, A.P.; Vaz, C.; Monteiro, P.T.; Melo-Cristino, J.; Ramirez, M.; Carriço, J.A. PHYLOViZ: Phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinform. 2012, 13, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolley, K.A.; Maiden, M.C.J. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; Li, D.; Hao, W.; Schwarz, S.; Shan, X.; Liu, B.; Zhang, S.M.; Li, X.S.; Du, X.D. A prophage and two ICESa2603-family integrative and conjugative elements (ICEs) carrying optrA in Streptococcus suis. J. Antimicrob. Chemother. 2019, 74, 2876–2879. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, C.; Princivalli, M.S.; Brenciani, A.; Varaldo, P.E.; Facinelli, B. Different genetic elements carrying the tet(W) gene in two human clinical isolates of Streptococcus suis. Antimicrob. Agents Chemother. 2011, 55, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Brenciani, A.; Bacciaglia, A.; Vignaroli, C.; Pugnaloni, A.; Varaldo, P.E.; Giovanetti, E. Φm46.1, the main Streptococcus pyogenes element carrying mef(A) and tet(O) genes. Antimicrob. Agents Chemother. 2010, 54, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Bossers, A.; Harders, F.; Lu, C.; Smith, H. Comparative genomic analysis of twelve Streptococcus suis (pro)phages. Genomics 2013, 101, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, S.V.; McShan, W.M. Chromosomal islands of Streptococcus pyogenes and related streptococci: Molecular switches for survival and virulence. Front. Cell. Infect. Microbiol. 2014, 4, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, R.; Artiushin, S.; Timoney, J.F. P9, a temperate bacteriophage of Streptococcus equi. Int. Congr. Ser. 2006, 1289, 165–168. [Google Scholar] [CrossRef]
- Harhala, M.; Barylski, J.; Humińska-Lisowska, K.; Lecion, D.; Wojciechowicz, J.; Lahutta, K.; Kus, M.; Kropinski, A.M.; Nowak, S.; Nowicki, G.; et al. Two novel temperate bacteriophages infecting Streptococcus pyogenes: Their genomes, morphology and stability. PLoS ONE 2018, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Russell, N.J.; Seale, A.C.; O’Driscoll, M.; O’Sullivan, C.; Bianchi-Jassir, F.; Gonzalez-Guarin, J.; Lawn, J.E.; Baker, C.J.; Bartlett, L.; Cutland, C.; et al. Maternal Colonization with Group B Streptococcus and Serotype Distribution Worldwide: Systematic Review and Meta-analyses. Clin. Infect. Dis. 2017, 65, S100–S111. [Google Scholar] [CrossRef]
- Rojo-Bezares, B.; Azcona-Gutiérrez, J.M.; Martin, C.; Jareño, M.S.; Torres, C.; Sáenz, Y. Streptococcus agalactiae from pregnant women: Antibiotic and heavy-metal resistance mechanisms and molecular typing. Epidemiol. Infect. 2016, 144, 3205–3214. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Tong, J.J.; Ma, X.H.; Song, F.L.; Fan, L.; Guo, C.M.; Shi, W.; Yu, S.J.; Yao, K.H.; Yang, Y.H. Serotypes, antibiotic susceptibilities, and multi-locus sequence type profiles of Streptococcus agalactiae isolates circulating in Beijing, China. PLoS ONE 2015, 10, 1–13. [Google Scholar] [CrossRef]
- Madrid, L.; Seale, A.C.; Kohli-Lynch, M.; Edmond, K.M.; Lawn, J.E.; Heath, P.T.; Madhi, S.A.; Baker, C.J.; Bartlett, L.; Cutland, C.; et al. Infant Group B Streptococcal Disease Incidence and Serotypes Worldwide: Systematic Review and Meta-analyses. Clin. Infect. Dis. 2017, 65, S160–S172. [Google Scholar] [CrossRef] [Green Version]
- Flores, A.R.; Galloway-Peña, J.; Sahasrabhojane, P.; Saldaña, M.; Yao, H.; Su, X.; Ajami, N.J.; Holder, M.E.; Petrosino, J.F.; Thompson, E.; et al. Sequence type 1 group B Streptococcus, an emerging cause of invasive disease in adults, evolves by small genetic changes. Proc. Natl. Acad. Sci. USA 2015, 112, 6431–6436. [Google Scholar] [CrossRef] [Green Version]
- Ulett, K.B.; Benjamin, W.H.; Zhuo, F.; Xiao, M.; Kong, F.; Gilbert, G.L.; Schembri, M.A.; Ulett, G.C. Diversity of group B streptococcus serotypes causing urinary tract infection in adults. J. Clin. Microbiol. 2009, 47, 2055–2060. [Google Scholar] [CrossRef] [Green Version]
- Piccinelli, G.; Biscaro, V.; Gargiulo, F.; Caruso, A.; De Francesco, M.A. Characterization and antibiotic susceptibility of Streptococcus agalactiae isolates causing urinary tract infections. Infect. Genet. Evol. 2015, 34, 1–6. [Google Scholar] [CrossRef]
- Björnsdóttir, E.S.; Martins, E.R.; Erlendsdóttir, H.; Haraldsson, G.; Melo-Cristino, J.; Kristinsson, K.G.; Ramirez, M. Changing epidemiology of group B streptococcal infections among adults in Iceland: 1975–2014. Clin. Microbiol. Infect. 2016, 22, 379.e9–379.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teatero, S.; Ferrieri, P.; Martin, I.; Demczuk, W.; McGeer, A.; Fittipaldi, N. Serotype Distribution, Population Structure, and Antimicrobial Resistance of Group B Streptococcus Strains Recovered from Colonized Pregnant Women. J. Clin. Microbiol. 2017, 55, 412–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renard, A.; Barbera, L.; Courtier-Martinez, L.; Dos Santos, S.; Valentin, A.S.; Mereghetti, L.; Quentin, R.; Van Der Mee-Marquet, N.L. PhiD12-like livestock-associated prophages are associated with novel subpopulations of streptococcus agalactiae infecting neonates. Front. Cell. Infect. Microbiol. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croucher, N.J.; Mostowy, R.; Wymant, C.; Turner, P.; Bentley, S.D.; Fraser, C. Horizontal DNA Transfer Mechanisms of Bacteria as Weapons of Intragenomic Conflict. PLoS Biol. 2016, 14, 1–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovich, L.; Sigal, N.; Borovok, I.; Nir-Paz, R.; Herskovits, A.A. Prophage excision activates listeria competence genes that promote phagosomal escape and virulence. Cell 2012, 150, 792–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crestani, C.; Forde, T.L.; Zadoks, R.N. Development and Application of a Prophage Integrase Typing Scheme for Group B Streptococcus. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Fléchard, M.; Lucchetti-Miganeh, C.; Hallet, B.; Hols, P.; Gilot, P. Intensive targeting of regulatory competence genes by transposable elements in streptococci. Mol. Genet. Genom. 2019, 294, 531–548. [Google Scholar] [CrossRef]
- Campbell, A.M. Preferential Orientation Preferential Orientation of Natural Lambdoid Prophages and Bacterial Chromosome Organization. Theor. Popul. Biol. 2002, 61, 503–507. [Google Scholar] [CrossRef]
- Wen, Y.; Behiels, E.; Devreese, B. Toxin-Antitoxin systems: Their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 2014, 70, 240–249. [Google Scholar] [CrossRef]
- Wang, X.; Wood, T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef] [Green Version]
- Hoskisson, P.A.; Smith, M.C. Hypervariation and phase variation in the bacteriophage “resistome”. Curr. Opin. Microbiol. 2007, 10, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Salloum, M.; Van Der Mee-Marquet, N.; Domelier, A.S.; Arnault, L.; Quentin, R. Molecular Characterization and Prophage DNA Contents of Streptococcus agalactiae Strains Isolated from Adult Skin and Osteoarticular Infections. J. Clin. Microbiol. 2010, 48, 1261–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lier, C.; Baticle, E.; Horvath, P.; Haguenoer, E.; Valentin, A.S.; Glaser, P.; Mereghetti, L.; Lanotte, P. Analysis of the type II-A CRISPR-Cas system of Streptococcus agalactiae reveals distinctive features according to genetic lineages. Front. Genet. 2015, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauruelle, C.; Pastuszka, A.; Horvath, P.; Perrotin, F.; Mereghetti, L.; Lanotte, P. CRISPR: A Useful Genetic Feature to Follow Vaginal Carriage of Group B Streptococcus. Front. Microbiol. 2017, 8, 1981. [Google Scholar] [CrossRef]
- Beauruelle, C.; Pastuszka, A.; Mereghetti, L.; Lanotte, P. Group b streptococcus vaginal carriage in pregnant women as deciphered by clustered regularly interspaced short palindromic repeat analysis. J. Clin. Microbiol. 2018, 56, 1–9. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prophage Group | Sub-Groups | No. of Prophages in Sequenced Genomes (%) | Genome Size (kbp) | GC Content (%) | No. of Genes | Phage Status | Integration Site | Induction | No. of Prophages in All Strains (%) |
|---|---|---|---|---|---|---|---|---|---|
| A | A1 | 9 | 39.9–40.6 | 42.6 | 40–42 | complete | A1, A2 | no | 87 (70.7) |
| A2 | 5 | 38.7–45.7 | 43 | 42–53 | complete | A3 | yes, no | ||
| B | B1 | 7 | 16.2–17.7 | 35.7 | 24–29 | satellite | B | no | 76 (61.8) |
| B2 | 6 | 17.4–17.9 | 34.5 | 27–29 | satellite | B | no | ||
| CFH | C | 1 | 33.2 | 35.4 | 45 | complete | C | yes | 3 (2.4) |
| F | 1 | 34 | 37 | 49 | complete | C | yes | 2 (1.6) 2 | |
| H | 2 | 36.5 | 39.7 | 53 | complete | C, H | NT 1 | 3 (2.4) | |
| DG | D1 | 1 | 42.2 | 38.7 | 49 + 1tRNA | complete | D1 | no | 1 (0.8) 3 |
| D2 | 1 | 44.2 | 36.3 | 65 + 1tRNA | complete | D2 | yes | 3 (2.4) 3 | |
| G | 1 | 38.6 | 37.1 | 60 | complete | D2 | NT | 3 (2.4) | |
| E | E | 3 | 32.4 | 40 | 50 | complete | E | yes, no | 7 (5.7) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lichvariková, A.; Soltys, K.; Szemes, T.; Slobodnikova, L.; Bukovska, G.; Turna, J.; Drahovska, H. Characterization of Clinical and Carrier Streptococcus agalactiae and Prophage Contribution to the Strain Variability. Viruses 2020, 12, 1323. https://0-doi-org.brum.beds.ac.uk/10.3390/v12111323
Lichvariková A, Soltys K, Szemes T, Slobodnikova L, Bukovska G, Turna J, Drahovska H. Characterization of Clinical and Carrier Streptococcus agalactiae and Prophage Contribution to the Strain Variability. Viruses. 2020; 12(11):1323. https://0-doi-org.brum.beds.ac.uk/10.3390/v12111323
Chicago/Turabian StyleLichvariková, Aneta, Katarina Soltys, Tomas Szemes, Livia Slobodnikova, Gabriela Bukovska, Jan Turna, and Hana Drahovska. 2020. "Characterization of Clinical and Carrier Streptococcus agalactiae and Prophage Contribution to the Strain Variability" Viruses 12, no. 11: 1323. https://0-doi-org.brum.beds.ac.uk/10.3390/v12111323