RNA Viruses of Amblyomma variegatum and Rhipicephalus microplus and Cattle Susceptibility in the French Antilles

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ticks and Cattle Sera Collected in Guadeloupe and Martinique
2.2. Nucleic Acid Extraction
2.3. High-Throughput Sequencing, Bioinformatic and Phylogenetic Analyses
2.4. Tick-Borne Virus Screening in Ticks from the French Antilles
2.5. Endogenous Viral Element Analysis
2.6. Serological Screening of Cattle Exposed to Tick Bites
3. Results
3.1. Virome Composition of Caribbean Cattle-Associated Ticks
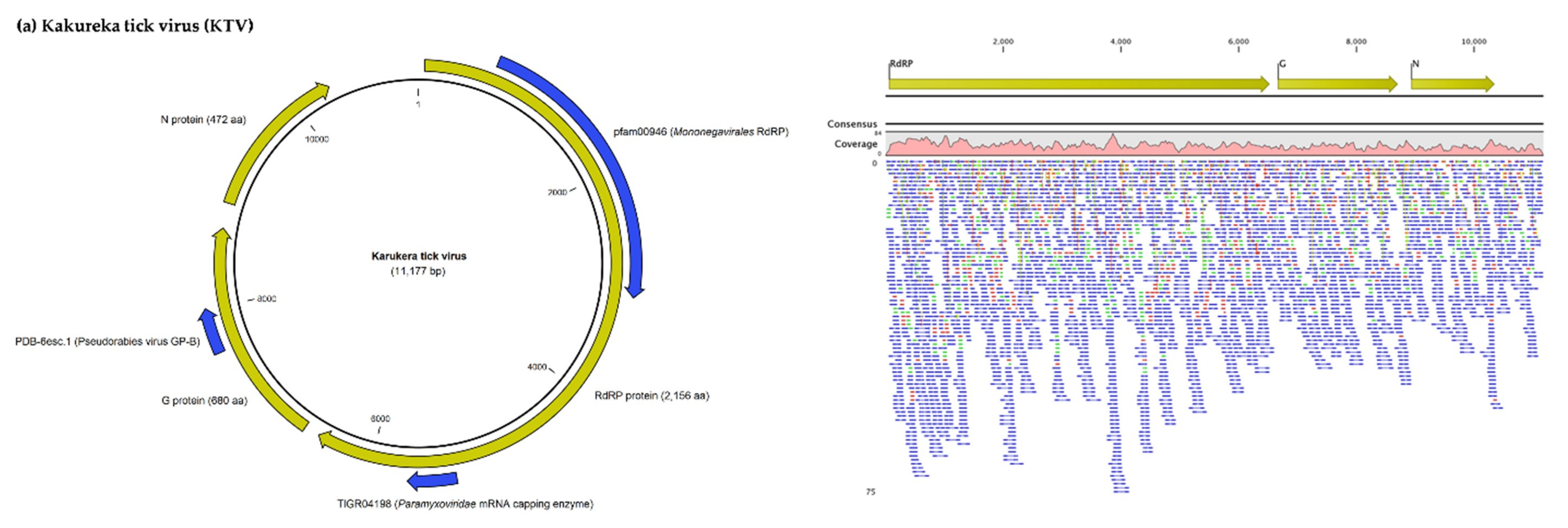
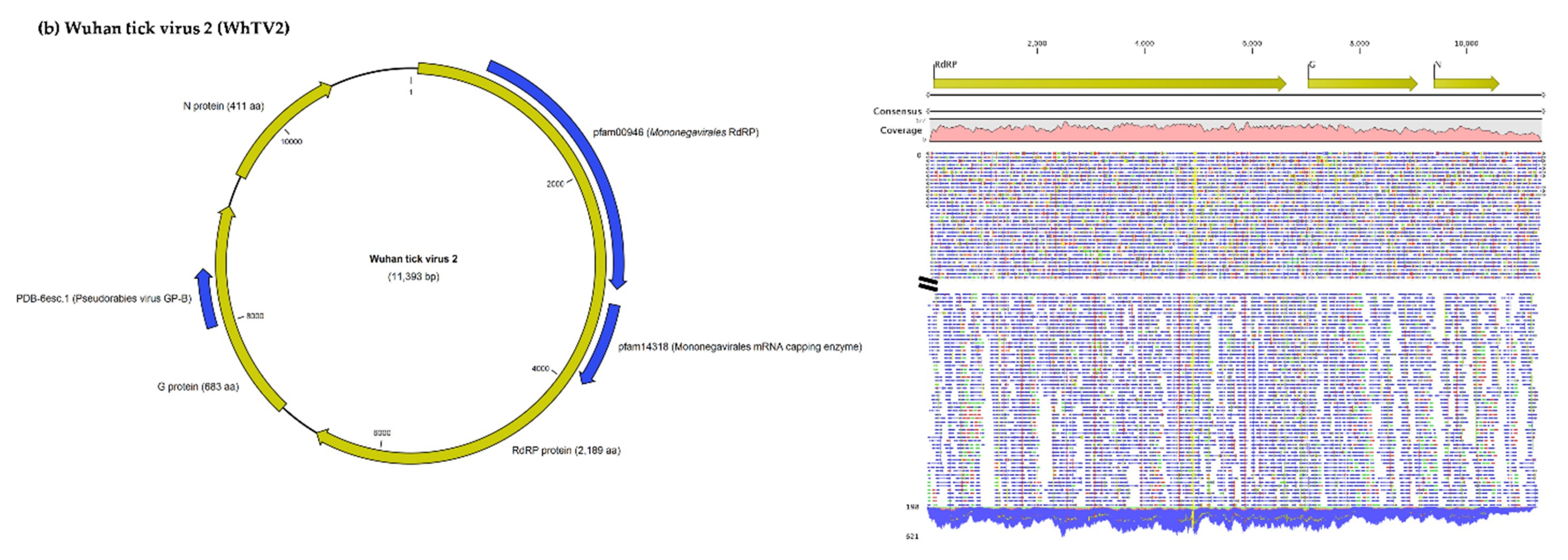
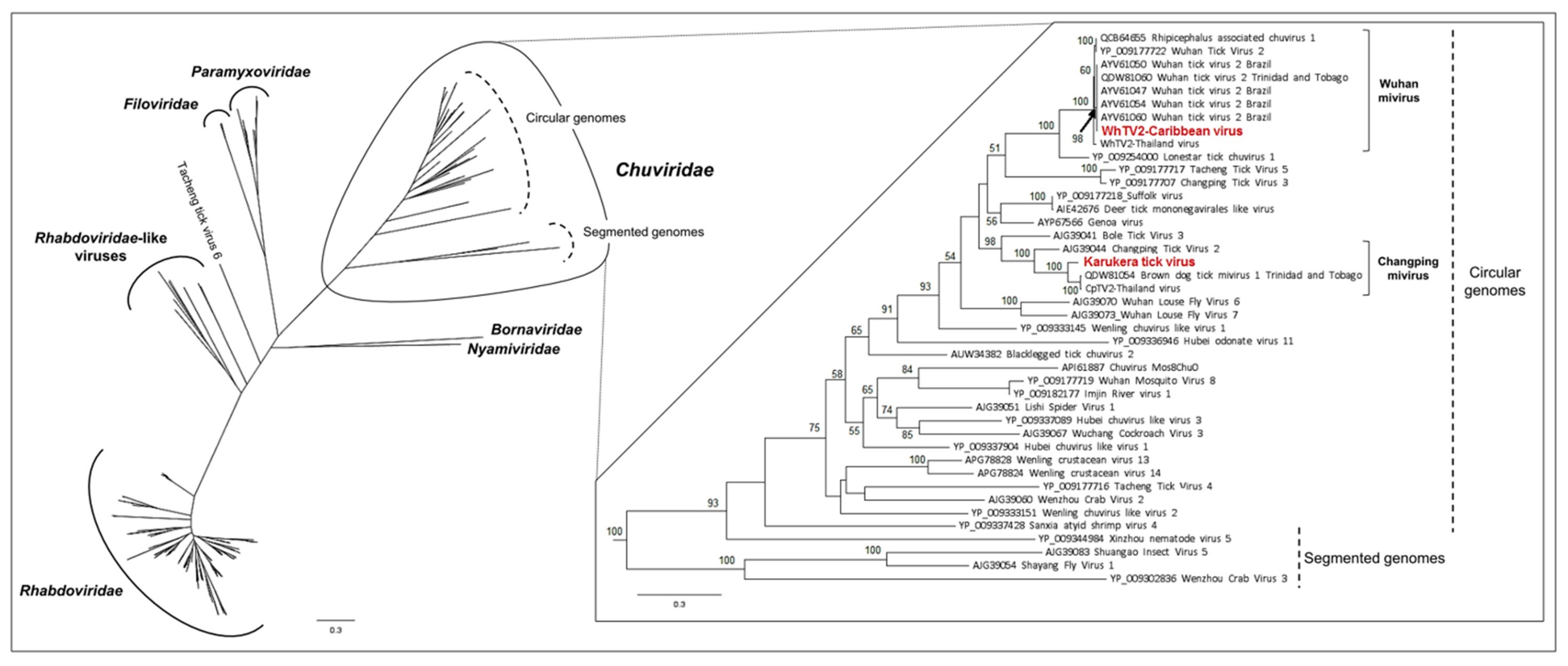
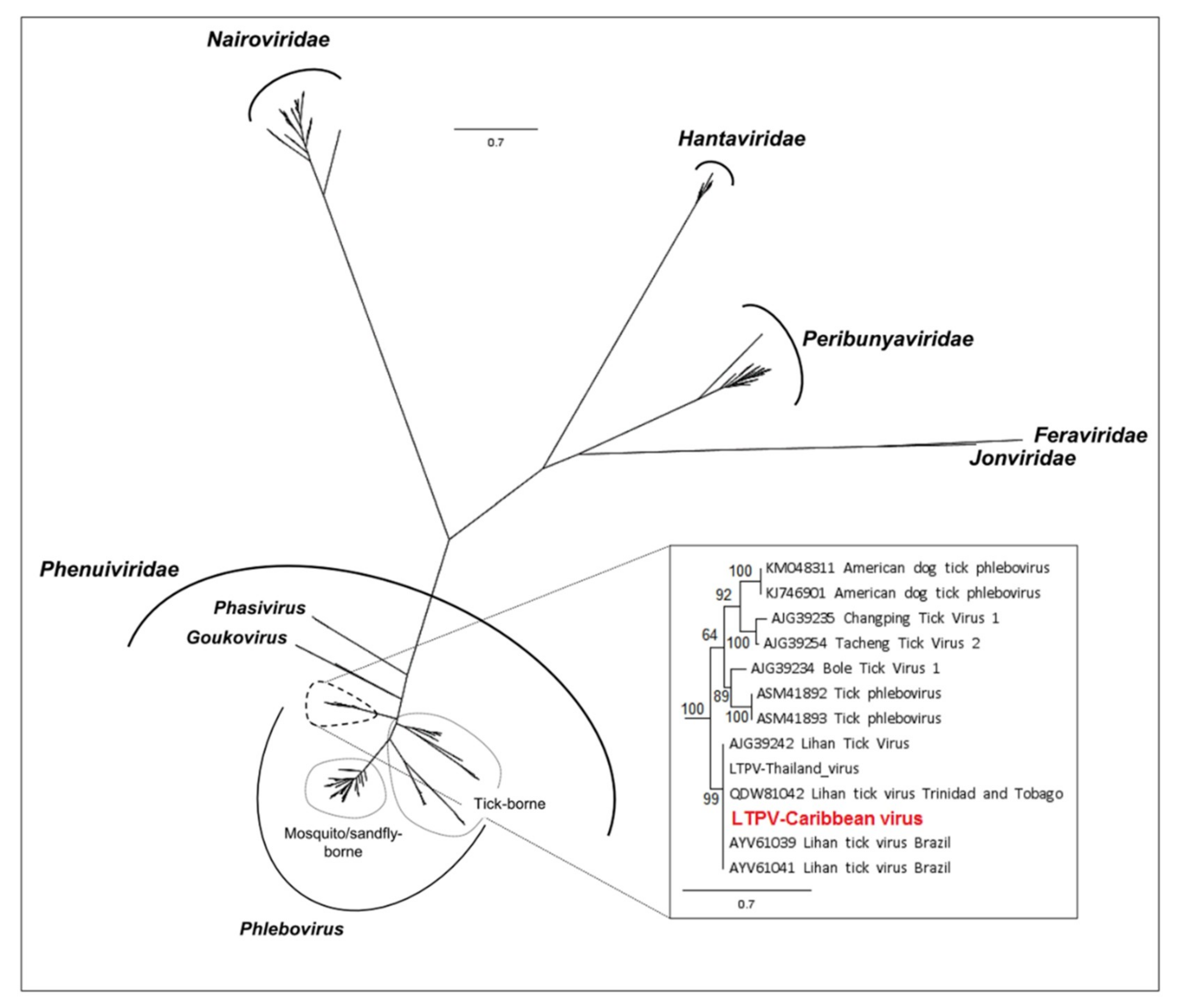
3.1.1. Viruses Belonging to the Chuviridae Family
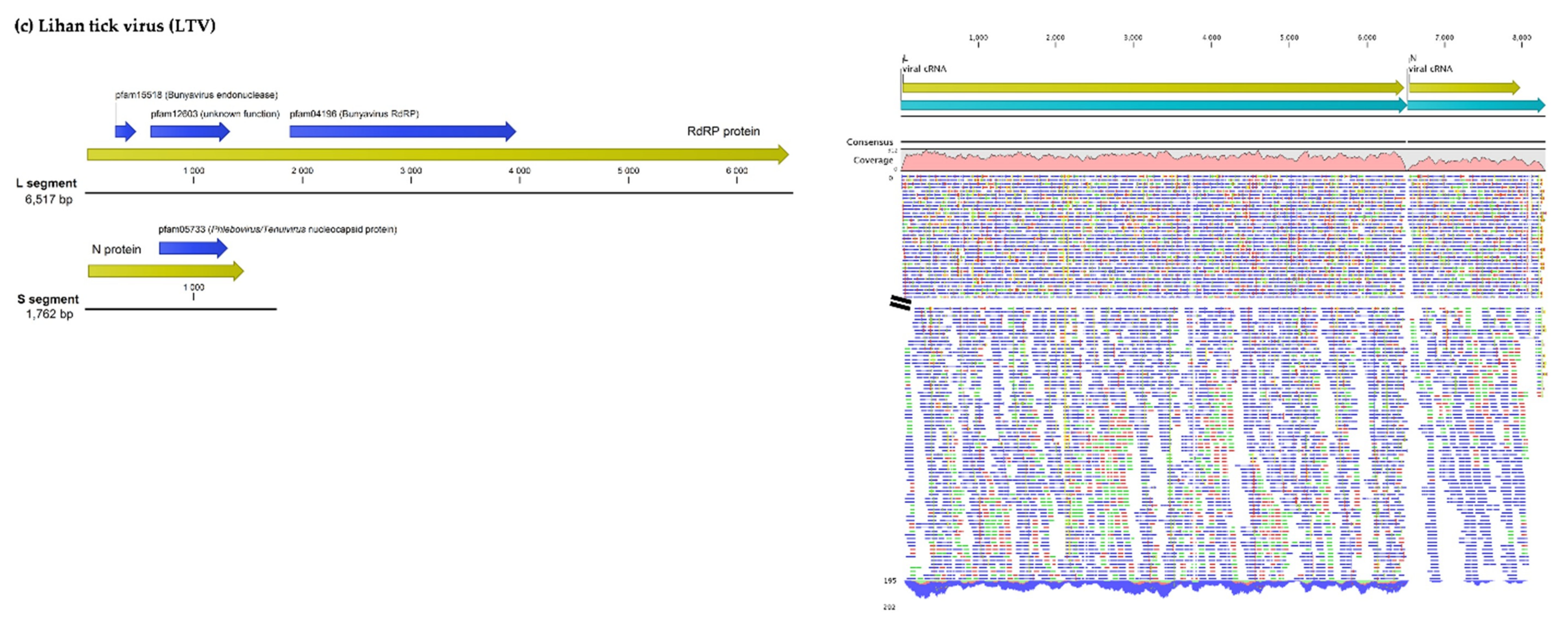
3.1.2. Viruses Belonging to the Phenuiviridae Family
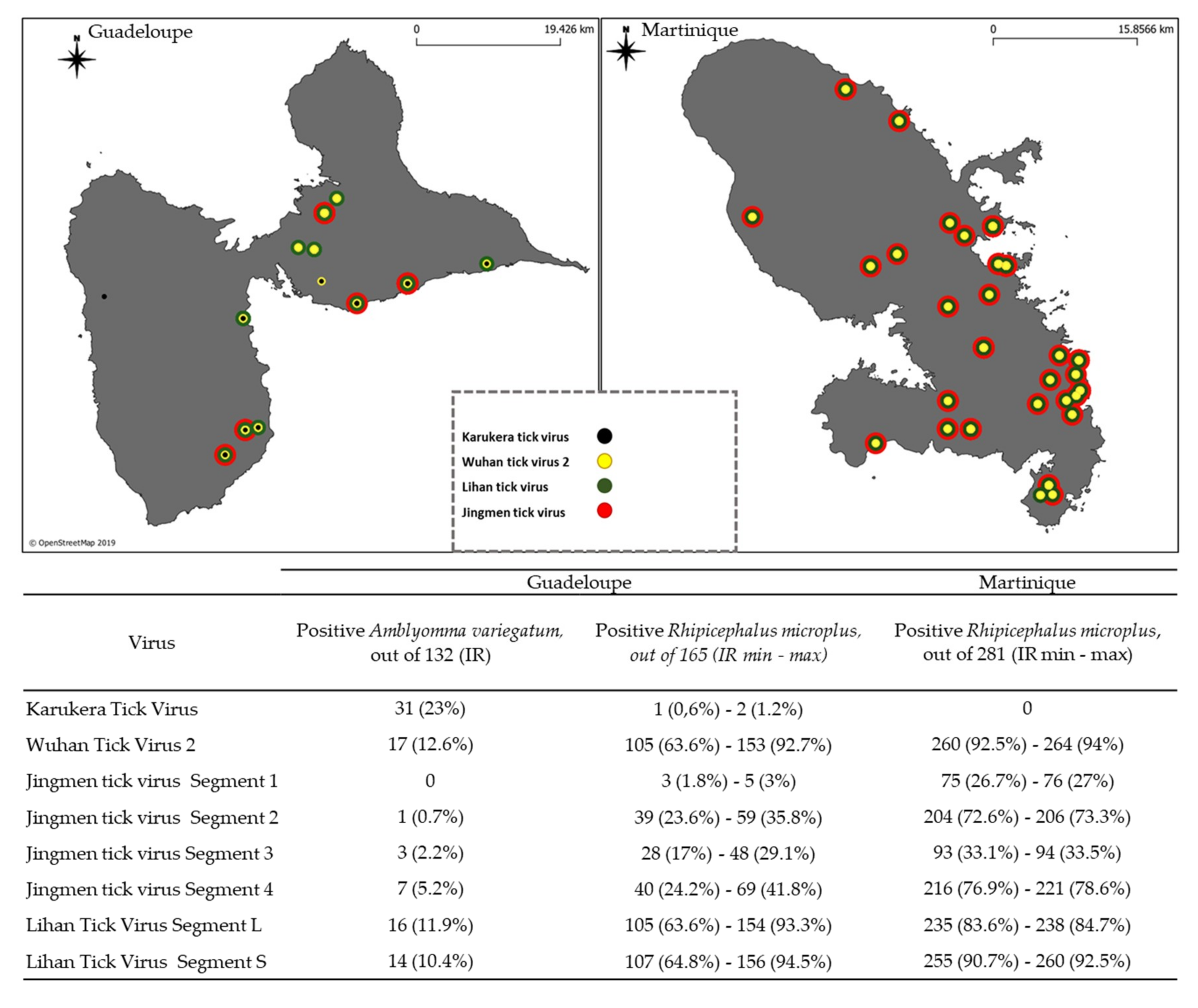
3.2. Screening of Tick-Borne Viruses in Individual Tick Samples from Guadeloupe and Martinique
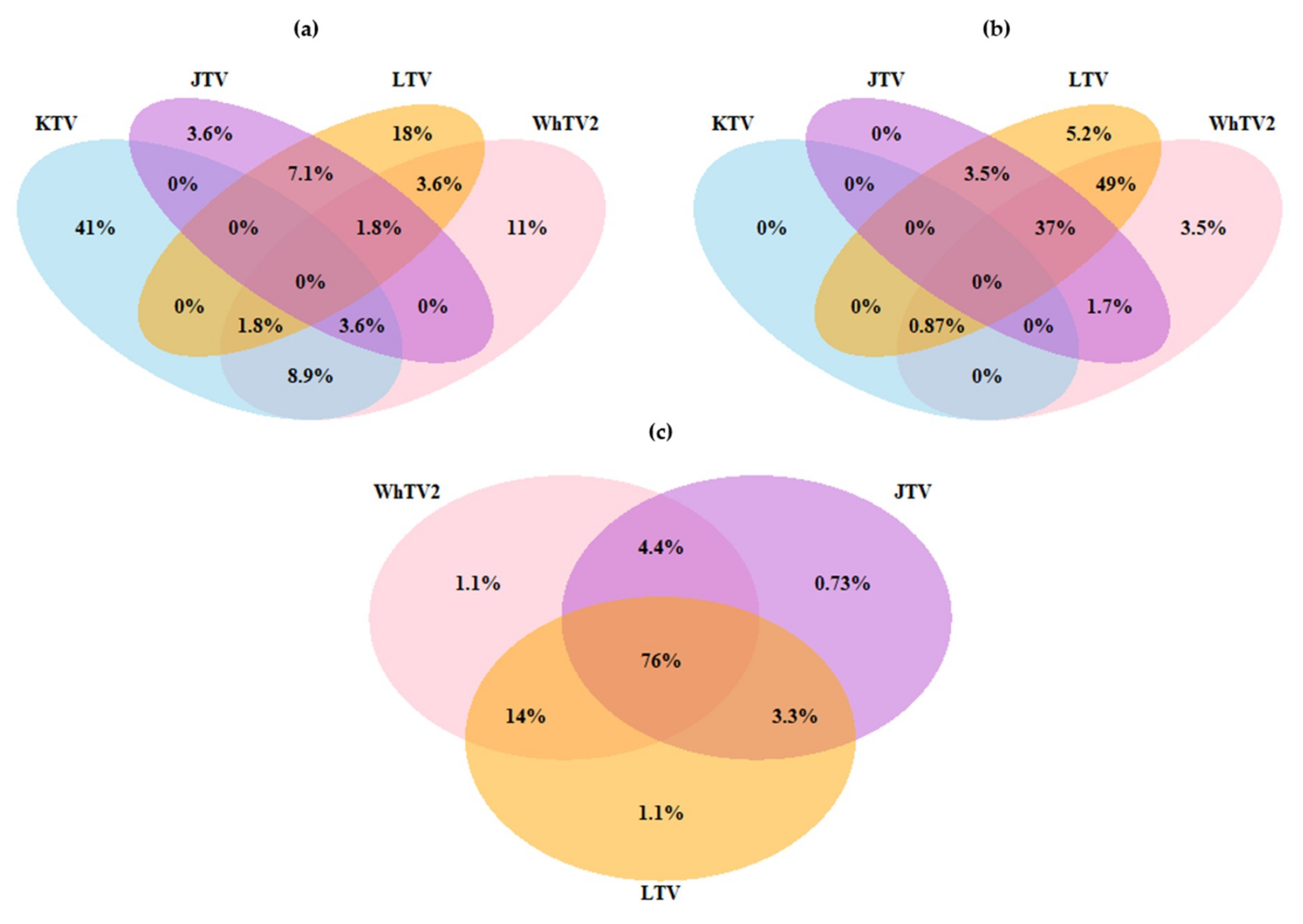
3.3. Viral Co-Infections
3.4. Search for Endogenous Viral Elements
3.5. Serological Screening of Guadeloupean Cattle Exposed to Tick Bites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jongejan, F.; Uilenberg, G. The global importance of ticks. Parasitology 2004, 129. [Google Scholar] [CrossRef]
- De la Fuente, J.; Antunes, S.; Bonnet, S.; Cabezas-Cruz, A.; Domingos, A.G.; Estrada-Peña, A.; Johnson, N.; Kocan, K.M.; Mansfield, K.L.; Nijhof, A.M.; et al. Tick-Pathogen Interactions and Vector Competence: Identification of Molecular Drivers for Tick-Borne Diseases. Front. Cell Infect. Microbiol. 2017, 7, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazimírová, M.; Thangamani, S.; Bartíková, P.; Hermance, M.; Holíková, V.; Štibrániová, I.; Nuttall, P.A. Tick-Borne Viruses and Biological Processes at the Tick-Host-Virus Interface. Front. Cell Infect. Microbiol. 2017, 7, 339. [Google Scholar] [CrossRef] [Green Version]
- Labuda, M.; Nuttall, P.A. Tick-borne viruses. Parasitology 2004, 129, S221–S245. [Google Scholar] [CrossRef] [PubMed]
- Bichaud, L.; de Lamballerie, X.; Alkan, C.; Izri, A.; Gould, E.A.; Charrel, R.N. Arthropods as a source of new RNA viruses. Microb. Pathog. 2014, 77, 136–141. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Vasilakis, N.; Tian, J.-H.; Li, C.-X.; Chen, L.-J.; Eastwood, G.; Diao, X.-N.; Chen, M.-H.; Chen, X.; et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J. Virol. 2015, 90, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Moutailler, S.; Popovici, I.; Devillers, E.; Vayssier-Taussat, M.; Eloit, M. Diversity of viruses in Ixodes ricinus, and characterization of a neurotropic strain of Eyach virus. New Microbes New Infect. 2016, 11, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, J.H.-O.; Shi, M.; Bohlin, J.; Eldholm, V.; Brynildsrud, O.B.; Paulsen, K.M.; Andreassen, Å.; Holmes, E.C. Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Junglen, S.; Drosten, C. Virus discovery and recent insights into virus diversity in arthropods. Curr. Opin. Microbiol. 2013, 16, 507–513. [Google Scholar] [CrossRef]
- Bolling, B.G.; Weaver, S.C.; Tesh, R.B.; Vasilakis, N. Insect-Specific Virus Discovery: Significance for the Arbovirus Community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, R.A.; Bielefeldt-Ohmann, H.; McLean, B.J.; O’Brien, C.A.; Colmant, A.M.G.; Piyasena, T.B.H.; Harrison, J.J.; Newton, N.D.; Barnard, R.T.; Prow, N.A.; et al. Commensal Viruses of Mosquitoes: Host Restriction, Transmission, and Interaction with Arboviral Pathogens. Evol. Bioinform. Online 2017, 12, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Hu, C.; Zhang, D.; Tang, S.; Zhang, Z.; Kou, Z.; Fan, Z.; Bente, D.; Zeng, C.; Li, T. Metagenomic Profile of the Viral Communities in Rhipicephalus spp. Ticks from Yunnan, China. PLoS ONE 2015, 10, e0121609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Temmam, S.; Chrétien, D.; Bigot, T.; Dufour, E.; Petres, S.; Desquesnes, M.; Devillers, E.; Dumarest, M.; Yousfi, L.; Jittapalapong, S.; et al. Monitoring Silent Spillovers Before Emergence: A Pilot Study at the Tick/Human Interface in Thailand. Front. Microbiol. 2019, 10, 2315. [Google Scholar] [CrossRef]
- Tokarz, R.; Williams, S.H.; Sameroff, S.; Sanchez Leon, M.; Jain, K.; Lipkin, W.I. Virome Analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis Ticks Reveals Novel Highly Divergent Vertebrate and Invertebrate Viruses. J. Virol. 2014, 88, 11480–11492. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, R.; Sameroff, S.; Tagliafierro, T.; Jain, K.; Williams, S.H.; Cucura, D.M.; Rochlin, I.; Monzon, J.; Carpi, G.; Tufts, D.; et al. Identification of Novel Viruses in Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis Ticks. mSphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, J.M.; Ng, T.F.F.; Suzuki, Y.; Tsujimoto, H.; Deng, X.; Delwart, E.; Rasgon, J.L. Bunyaviruses are common in male and female Ixodes scapularis ticks in central Pennsylvania. PeerJ 2016, 4, e2324. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, S.R.; Castro-Jorge, L.A.; Ribeiro, J.M.C.; Gardinassi, L.G.; Garcia, G.R.; Brandão, L.G.; Rodrigues, A.R.; Okada, M.I.; Abrão, E.P.; Ferreira, B.R.; et al. Characterisation of divergent flavivirus NS3 and NS5 protein sequences detected in Rhipicephalus microplus ticks from Brazil. Mem. Inst. Oswaldo Cruz 2014, 109, 38–50. [Google Scholar] [CrossRef]
- De Souza, W.M.; Fumagalli, M.J.; de O Torres Carrasco, A.; Romeiro, M.F.; Modha, S.; Seki, M.C.; Gheller, J.M.; Daffre, S.; Nunes, M.R.T.; Murcia, P.R.; et al. Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil. Sci. Rep. 2018, 8, 16315. [Google Scholar] [CrossRef]
- Cholleti, H.; Hayer, J.; Mulandane, F.C.; Falk, K.; Fafetine, J.; Berg, M.; Blomström, A.-L. Viral metagenomics reveals the presence of highly divergent quaranjavirus in Rhipicephalus ticks from Mozambique. Infect. Ecol. Epidemiol. 2018, 8, 1478585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, E.; Rose, K.; Eden, J.-S.; Lo, N.; Abeyasuriya, T.; Shi, M.; Doggett, S.L.; Holmes, E.C. Extensive Diversity of RNA Viruses in Australian Ticks. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sameroff, S.; Tokarz, R.; Charles, R.A.; Jain, K.; Oleynik, A.; Che, X.; Georges, K.; Carrington, C.V.; Lipkin, W.I.; Oura, C. Viral Diversity of Tick Species Parasitizing Cattle and Dogs in Trinidad and Tobago. Sci. Rep. 2019, 9, 10421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greay, T.L.; Gofton, A.W.; Paparini, A.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Recent insights into the tick microbiome gained through next-generation sequencing. Parasites Vectors 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.I.; Binetruy, F.; Hernández-Jarguín, A.M.; Duron, O. The Tick Microbiome: Why Non-pathogenic Microorganisms Matter in Tick Biology and Pathogen Transmission. Front. Cell Infect. Microbiol. 2017, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.A.; Montgomery, B.L.; Popovici, J.; Iturbe-Ormaetxe, I.; Johnson, P.H.; Muzzi, F.; Greenfield, M.; Durkan, M.; Leong, Y.S.; Dong, Y.; et al. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 2011, 476, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, P.; Lundén, H.; Blomström, A.-L. Insect-specific virus evolution and potential effects on vector competence. Virus Genes 2019, 55, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Gondard, M.; Cabezas-Cruz, A.; Charles, R.A.; Vayssier-Taussat, M.; Albina, E.; Moutailler, S. Ticks and Tick-Borne Pathogens of the Caribbean: Current Understanding and Future Directions for More Comprehensive Surveillance. Front. Cell Infect. Microbiol. 2017, 7, 490. [Google Scholar] [CrossRef]
- Aitken, T.H.G.; Jonkers, A.H.; Tikasingh, E.S.; Worth, C.B. Hughes Virus from Trinidadian Ticks And Terns. J. Med. Entomol. 1968, 5, 501–503. [Google Scholar] [CrossRef]
- Jonkers, A.H.; Casals, J.; Aitken, T.H.G.; Spence, L. Soldado Virus, a New Agent from Trinidadian Ornithodoros Ticks. J. Med. Entomol. 1973, 10, 517–519. [Google Scholar] [CrossRef]
- Danielová, V.; Marhoul, Z.; Dusbábek, F.; Ryba, J.; Fernández, A.; de la Cruz, J.; Abreu, R.; Herrera, M.; Rodriquez, P.; Cantelar, N. Isolation of Hughes virus from ticks in Cuba. Acta Virol. 1982, 26, 186–189. [Google Scholar] [PubMed]
- Butler, J.F.; Gibbs, E.P.J. Distribution of potential soft tick vectors of African swine fever in the Caribbean region (Acari: Argasidae). Prev. Vet. Med. 1984, 2, 63–70. [Google Scholar] [CrossRef]
- Málková, D.; Holubová, J.; Cerný, V.; Daniel, M.; Fernández, A.; de la Cruz, J.; Herrera, M.; Calisher, C.H. Estero real virus: A new virus isolated from argasid ticks Ornithodoros tadaridae in Cuba. Acta Virol. 1985, 29, 247–250. [Google Scholar]
- Penrith, M.-L. African swine fever. Onderstepoort J. Vet. Res. 2009, 76, 91–95. [Google Scholar] [CrossRef]
- Gondard, M.; Delannoy, S.; Pinarello, V.; Aprelon, R.; Devillers, E.; Galon, C.; Pradel, J.; Vayssier-Taussat, M.; Albina, E.; Moutailler, S. Upscaling surveillance of tick-borne pathogens in the French Caribbean islands. bioRxiv 2019. [Google Scholar] [CrossRef]
- Walker, A.R.; Bouattour, A.; Camicas, J.-L.; Estrada-Pena, A.; Horak, I.G.; Latif, A.A.; Pegram, R.G.; Preston, P.M. Ticks of Domestic Animals in Africa: A Guide to Identification of Species; Bioscience Reports; Wisconsin University-Madison: Madison, WI, USA, 2003; ISBN 0-9545173-0-X. [Google Scholar]
- Bigot, T.; Temmam, S.; Pérot, P.; Eloit, M. RVDB-prot, a reference viral protein database and its HMM profiles. F1000Research 2019, 8, 530. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, F.; Correia, D.; Lefort, V.; Doppelt-Azeroual, O.; Mareuil, F.; Cohen-Boulakia, S.; Gascuel, O. NGPhylogeny.fr: New generation phylogenetic services for non-specialists. Nucl. Acids Res. 2019, 47, W260–W265. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gondard, M.; Michelet, L.; Nisavanh, A.; Devillers, E.; Delannoy, S.; Fach, P.; Aspan, A.; Ullman, K.; Chirico, J.; Hoffmann, B.; et al. Prevalence of tick-borne viruses in Ixodes ricinus assessed by high-throughput real-time PCR. Pathog. Dis. 2018, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbelo, P.D.; Ching, K.H.; Bush, E.R.; Han, B.L.; Iadarola, M.J. Antibody-profiling technologies for studying humoral responses to infectious agents. Expert Rev. Vaccines 2010, 9, 567–578. [Google Scholar] [CrossRef]
- Villa, E.C.; Maruyama, S.R.; de Miranda-Santos, I.K.F.; Palacios, G.; Ladner, J.T. Complete Coding Genome Sequence for Mogiana Tick Virus, a Jingmenvirus Isolated from Ticks in Brazil. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temmam, S.; Bigot, T.; Chrétien, D.; Gondard, M.; Pérot, P.; Pommelet, V.; Dufour, E.; Petres, S.; Devillers, E.; Hoem, T.; et al. Insights into the Host Range, Genetic Diversity, and Geographical Distribution of Jingmenviruses. mSphere 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauliard, N.; Billecocq, A.; Flick, R.; Bouloy, M. Rift Valley fever virus noncoding regions of L, M and S segments regulate RNA synthesis. Virology 2006, 351, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feschotte, C.; Gilbert, C. Endogenous viruses: insights into viral evolution and impact on host biology. Nat. Rev. Genet. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell-Sakyi, L.; Attoui, H. Endogenous tick viruses and modulation of tick-borne pathogen growth. Front. Cell Infect. Microbiol. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Katzourakis, A.; Gifford, R.J. Endogenous Viral Elements in Animal Genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef]
- Qin, X.-C.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Gao, D.-Y.; He, J.-R.; Wang, J.-B.; Li, C.-X.; Kang, Y.-J.; Yu, B.; et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef] [Green Version]
- Costard, S.; Wieland, B.; de Glanville, W.; Jori, F.; Rowlands, R.; Vosloo, W.; Roger, F.; Pfeiffer, D.U.; Dixon, L.K. African swine fever: how can global spread be prevented? Philos. Trans. R. Soc. Lond. B 2009, 364, 2683–2696. [Google Scholar] [CrossRef]
- Brinkmann, A.; Dinçer, E.; Polat, C.; Hekimoğlu, O.; Hacıoğlu, S.; Földes, K.; Özkul, A.; Öktem, İ.M.A.; Nitsche, A.; Ergünay, K. A metagenomic survey identifies Tamdy orthonairovirus as well as divergent phlebo-, rhabdo-, chu- and flavi-like viruses in Anatolia, Turkey. Ticks Tick Borne Dis. 2018, 9, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Barré, N.; Uilenberg, G. Spread of parasites transported with their hosts: case study of two species of cattle tick. Rev. Off. Int. Epizoot. 2010, 29, 135–147, 149–160 . [Google Scholar]
- Low, V.L.; Tay, S.T.; Kho, K.L.; Koh, F.X.; Tan, T.K.; Lim, Y.A.L.; Ong, B.L.; Panchadcharam, C.; Norma-Rashid, Y.; Sofian-Azirun, M. Molecular characterisation of the tick Rhipicephalus microplus in Malaysia: new insights into the cryptic diversity and distinct genetic assemblages throughout the world. Parasites Vectors 2015, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinçer, E.; Brinkmann, A.; Hekimoğlu, O.; Hacıoğlu, S.; Földes, K.; Karapınar, Z.; Polat, P.F.; Oğuz, B.; Orunç Kılınç, Ö.; Hagedorn, P.; et al. Generic amplification and next generation sequencing reveal Crimean-Congo hemorrhagic fever virus AP92-like strain and distinct tick phleboviruses in Anatolia, Turkey. Parasites Vectors 2017, 10, 335. [Google Scholar] [CrossRef]
- Jones, L.D.; Davies, C.R.; Williams, T.; Cory, J.; Nuttall, P.A. Non-viraemic transmission of Thogoto virus: Vector efficiency of Rhipicephalus appendiculatus and Amblyomma variegatum. Trans. R. Soc. Trop. Med. Hyg. 1990, 84, 846–848. [Google Scholar] [CrossRef]
- Labuda, M.; Nuttall, P.A.; Kozuch, O.; Elecková, E.; Williams, T.; Zuffová, E.; Sabó, A. Non-viraemic transmission of tick-borne encephalitis virus: A mechanism for arbovirus survival in nature. Experientia 1993, 49, 802–805. [Google Scholar] [CrossRef]
- Havlíková, S.; Ličková, M.; Klempa, B. Non-viraemic transmission of tick-borne viruses. Acta Virol. 2013, 57, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Hermance, M.E.; Thangamani, S. Tick–Virus–Host Interactions at the Cutaneous Interface: The Nidus of Flavivirus Transmission. Viruses 2018, 10, 362. [Google Scholar] [CrossRef] [Green Version]
- Varela-Stokes, A.S.; Park, S.H.; Kim, S.A.; Ricke, S.C. Microbial Communities in North American Ixodid Ticks of Veterinary and Medical Importance. Front. Vet. Sci. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Swei, A.; Kwan, J.Y. Tick microbiome and pathogen acquisition altered by host blood meal. ISME J. 2017, 11, 813–816. [Google Scholar] [CrossRef]
- Chicana, B.; Couper, L.I.; Kwan, J.Y.; Tahiraj, E.; Swei, A. Comparative Microbiome Profiles of Sympatric Tick Species from the Far-Western United States. Insects 2019, 10, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couper, L.I.; Kwan, J.Y.; Ma, J.; Swei, A. Drivers and patterns of microbial community assembly in a Lyme disease vector. Ecol. Evol. 2019, 9, 7768–7779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimhan, S.; Fikrig, E. Tick microbiome: the force within. Trends Parasitol. 2015, 31, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, N.M.; Liu, L.; Jutras, B.L.; Yadav, A.K.; Narasimhan, S.; Gopalakrishnan, V.; Ansari, J.M.; Jefferson, K.K.; Cava, F.; Jacobs-Wagner, C.; et al. Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc. Natl. Acad. Sci. USA 2017, 114, E781–E790. [Google Scholar] [CrossRef] [Green Version]
- Duron, O.; Binetruy, F.; Noël, V.; Cremaschi, J.; McCoy, K.D.; Arnathau, C.; Plantard, O.; Goolsby, J.; Pérez de León, A.A.; Heylen, D.J.A.; et al. Evolutionary changes in symbiont community structure in ticks. Mol. Ecol. 2017, 26, 2905–2921. [Google Scholar] [CrossRef] [Green Version]
- Macaluso, K.R.; Sonenshine, D.E.; Ceraul, S.M.; Azad, A.F. Rickettsial infection in Dermacentor variabilis (Acari: Ixodidae) inhibits transovarial transmission of a second Rickettsia. J. Med. Entomol. 2002, 39, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Nováková, M.; Šmajs, D. Rickettsial Endosymbionts of Ticks. Ticks Tick Borne Pathog. 2018. [Google Scholar] [CrossRef] [Green Version]
- Jia, N.; Liu, H.-B.; Ni, X.-B.; Bell-Sakyi, L.; Zheng, Y.-C.; Song, J.-L.; Li, J.; Jiang, B.-G.; Wang, Q.; Sun, Y.; et al. Emergence of human infection with Jingmen tick virus in China: A retrospective study. eBioMedicine 2019, 43, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Emmerich, P.; Jakupi, X.; von Possel, R.; Berisha, L.; Halili, B.; Günther, S.; Cadar, D.; Ahmeti, S.; Schmidt-Chanasit, J. Viral metagenomics, genetic and evolutionary characteristics of Crimean-Congo hemorrhagic fever orthonairovirus in humans, Kosovo. Infect. Genet. Evol. 2018, 65, 6–11. [Google Scholar] [CrossRef]
- Wang, Z.-D.; Wang, B.; Wei, F.; Han, S.-Z.; Zhang, L.; Yang, Z.-T.; Yan, Y.; Lv, X.-L.; Li, L.; Wang, S.-C.; et al. A New Segmented Virus Associated with Human Febrile Illness in China. N. Engl. J. Med. 2019, 380, 2116–2125. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Target | Design Name | Sequence (5′–3′) | Amplicon Size (bp) |
|---|---|---|---|---|
| Karukera Tick Virus | Putative RdRP 1 gene | KTVL_Poly_F | CACATGTCTCGGAGCGAGG | 136 |
| KTVL_Poly_R | TTCCTGAACGTCTGAGGCTG | |||
| KTVL_Poly_S | AAAGCTATTCGGGCACGTCATTAAAGTGG | |||
| Wuhan Tick Virus | Putative RdRP 1 gene | WTV_Poly_F | GACCCAGGGAGAGTTAGATG | 119 |
| WTV_Poly_R | ACCTGCTGTTCCATGAGCTC | |||
| WTV_Poly_S | TAGCCCGTAAACTCTTGGGATTTCGTATGC | |||
| Jingmen Tick Virus Segment 1 | Putative NS5-like gene | JTV_Seg1_F | ACGTGAAGGAAATATCATTCTGC | 100 |
| JTV_Seg1_R | GCGAATATCTCTCCCACGTC | |||
| JTV_Seg1_P | TCCCACAGGTACTGGCCGGTAAAGTA | |||
| Jingmen Tick Virus Segment 2 | Putative Glycoprotein | JTV_Seg2_F | ATCTTCAGCGCTATCACCGC | 95 |
| JTV_Seg2_R | CGGTTTTGTCGGCGAATGATG | |||
| JTV_Seg2_P | ATTGCAGCGATGAGTGGGACGAGCG | |||
| Jingmen Tick Virus Segment 3 | Putative NS3-like gene | JTV_Seg3_F | CGTGGGGAAGGACAAAAGC | 102 |
| JTV_Seg3_R | CCTTATCTCTCCGCTAGTGG | |||
| JTV_Seg3_P | AAGGCAGCTTGCATAGAGATGACCGC | |||
| Jingmen Tick Virus Segment 4 | Putative membrane protein gene | JTV_Seg4_F | ACAGCGTGCTAGTCTTCGC | 79 |
| JTV_Seg4_R | GGGAGTTGAAAGTGTATGCCA | |||
| JTV_Seg4_P | AGGCACGTTTGTGATGGTTCAGGACAG | |||
| Lihan Tick Virus Segment L | Putative RdRP 1 gene | LTV_SegL_F | ACATGGGTGTATCCAACACAC | 127 |
| LTV_SegL_R | ACCGACATAGCCCATCGAG | |||
| LTV_SegL_P | ACAGGAGTCTAAACAAGGACGGGTGCAT | |||
| Lihan Tick Virus Segment S | Putative nucleopasid protein (N) gene | LTV_SegS_F | TTGACGTTCTACTCGGCCAC | 123 |
| LTV_SegS_R | TACTGCCTGCGTCATGAGTG | |||
| LTV_SegS_P | AATTCTAGCCGCTCACCATTCTGCCCA |
| Family | Genus | Closest Viral Sequence (GenBank Accession Number) | % Identity (aa) | Abundance (nt) | |
|---|---|---|---|---|---|
| ssRNA+ | Flaviviridae | unclassified | Jingmen tick virus (MH133317-20) | 72%–100% | 6,003,829 |
| Tymoviridae | Marafivirus | Peach virus D (NC_033828) | 85%–86% | 1,087,064 | |
| Maize rayado fino virus (NC_002786) | 96%–100% | 20,750 | |||
| Oat blue dwarf virus (NC_001793) | 94% | 10,556 | |||
| Citrus sudden death-associated virus (DQ185573) | 93%–100% | 3390 | |||
| Olive latent virus 3 (NC_013920) | 95%–96% | 687 | |||
| Maculavirus | Bee Macula-Like virus 2 (MF998084) | 96% | 6615 | ||
| Grapevine Red Globe virus (KX109927) | 92%–100% | 1824 | |||
| Grapevine fleck virus (NC_003347) | 79%–100% | 639 | |||
| Tymovirus | Erysimum latent virus (NC_001977) | 95%–100% | 2709 | ||
| unclassified | Bee Macula-like virus (KT162925) | 59%–100% | 551,038 | ||
| Varroa Tymo-like virus (NC_027619) | 87%–100% | 502,827 | |||
| unclassified Tymovirales | Peach virus T (KY348615) | 98% | 213 | ||
| Fusarium graminearum mycotymovirus 1 (KT360947) | 100% | 72 | |||
| ssRNA− | Chuviridae | Mivirus | Wuhan tick virus 2 (NC_028266) | 82%–100% | 184,236 |
| Changping tick virus 2 (NC_028260) | 58%–95% | 1596 | |||
| unclassified | Lonestar tick chuvirus 1 (NC_030204) | 100% | 144 | ||
| Phenuiviridae | Phlebovirus | Lihan tick virus (KM817672 - KM817736) | 76%–100% | 277,395 | |
| unclassified RNA viruses | Hubei sobemo-like virus 15 (NC_032208) | 50%–95% | 96,054 | ||
| Hubei partiti-like virus 7 (KX884117) | 80%–83% | 147 | |||
| Wuhan fly virus 5 (NC_033485) | 76% | 75 | |||
| Wenling chuvirus-like virus 1 (NC_032409) | 87% | 72 | |||
| dsRNA | Partitiviridae | unclassified | Maize associated partiti-like virus (MF372918) | 53%–96% | 16,857 |
| Virus | Sequence | Closest Homology | C% | E-value | I% | Accession Number |
|---|---|---|---|---|---|---|
| Karukera Tick Virus | Complete genome | Brown dog tick mivirus 1 (Trinidad and Tobago) | 89 | 0 | 71.7 | MN025520.1 |
| L protein (RNA polymerase) | Polymerase (Mivirus sp.) | 100 | 0 | 82.4 | QDW81054.1 | |
| G protein (Glycoprotein) | Glycoprotein (Mivirus sp.) | 99 | 0 | 77.1 | QDW81055.1 | |
| N protein (Nucleoprotein) | Nucleoprotein (Mivirus sp.) | 94 | 0 | 61.7 | QDW81056.1 | |
| Wuhan Tick Virus 2 | Complete genome | Wuhan tick virus 2 isolate WTV2_100 (Brazil) | 98 | 0 | 99.4 | MH155927.1 |
| L protein (RNA polymerase) | Polymerase (Wuhan tick virus 2) * | 100 | 0 | 98.1 | YP_009177722.1 | |
| G protein (Glycoprotein) | Glycoprotein (Wuhan tick virus 2) | 100 | 0 | 99.1 | QDW81058.1 | |
| N protein (Nucleoprotein) | Nucleoprotein (Wuhan tick virus 2) | 100 | 0 | 99.8 | AYV61049.1 | |
| Lihan Tick Virus | Complete Segment L | Lihan Tick Virus isolate LTV_L_100 (Brazil) | 99 | 0 | 99.3 | MH155914.1 |
| L protein (RNA polymerase) | RNA-dependent RNA polymerase (Lihan Tick Virus) | 100 | 0 | 99.8 | AYV61041.1 | |
| Complete Segment S | Lihan Tick Virus strain LH-1 (China) * | 100 | 0 | 97.8 | KM817736.1 | |
| N protein (Nucleoprotein) | Nucleoprotein (Lihan Tick Virus) | 99 | 0 | 100 | AYV61046.1 |
| Virus | Guadeloupe | Martinique | |
|---|---|---|---|
| Positive Amblyomma variegatum, out of 132 (IR) | Positive Rhipicephalus microplus, out of 165 (IR min–max) | Positive Rhipicephalus microplus, out of 281 (IR min–max) | |
| Karukera Tick Virus | 0 | 1 (0.6%)–2(1.2%) | 0 |
| Wuhan Tick Virus 2 | 0 | 41 (24.8%)–59 (35.8%) | 87 (31%)–90 (32%) |
| Jingmenvirus Segment 1 | 0 | 0 | 3 (1.1%) |
| Jingmenvirus Segment 2 | 0 | 0 | 23 (8.2%) |
| Jingmenvirus Segment 3 | 0 | 4 (2.4%)–7 (4.2%) | 8 (2.8%) |
| Jingmenvirus Segment 4 | 0 | 1 (0.6%)–2(1.2%) | 14 (5%) |
| Lihan Tick Virus Segment L | 0 | 1 (0.6%)–2(1.2%) | 16 (5.7%) |
| Lihan Tick Virus Segment S | 0 | 0 | 1 (0.4%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gondard, M.; Temmam, S.; Devillers, E.; Pinarello, V.; Bigot, T.; Chrétien, D.; Aprelon, R.; Vayssier-Taussat, M.; Albina, E.; Eloit, M.; et al. RNA Viruses of Amblyomma variegatum and Rhipicephalus microplus and Cattle Susceptibility in the French Antilles. Viruses 2020, 12, 144. https://0-doi-org.brum.beds.ac.uk/10.3390/v12020144
Gondard M, Temmam S, Devillers E, Pinarello V, Bigot T, Chrétien D, Aprelon R, Vayssier-Taussat M, Albina E, Eloit M, et al. RNA Viruses of Amblyomma variegatum and Rhipicephalus microplus and Cattle Susceptibility in the French Antilles. Viruses. 2020; 12(2):144. https://0-doi-org.brum.beds.ac.uk/10.3390/v12020144
Chicago/Turabian StyleGondard, Mathilde, Sarah Temmam, Elodie Devillers, Valérie Pinarello, Thomas Bigot, Delphine Chrétien, Rosalie Aprelon, Muriel Vayssier-Taussat, Emmanuel Albina, Marc Eloit, and et al. 2020. "RNA Viruses of Amblyomma variegatum and Rhipicephalus microplus and Cattle Susceptibility in the French Antilles" Viruses 12, no. 2: 144. https://0-doi-org.brum.beds.ac.uk/10.3390/v12020144