Deciphering the Virome of Culex vishnui Subgroup Mosquitoes, the Major Vectors of Japanese Encephalitis, in Japan

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
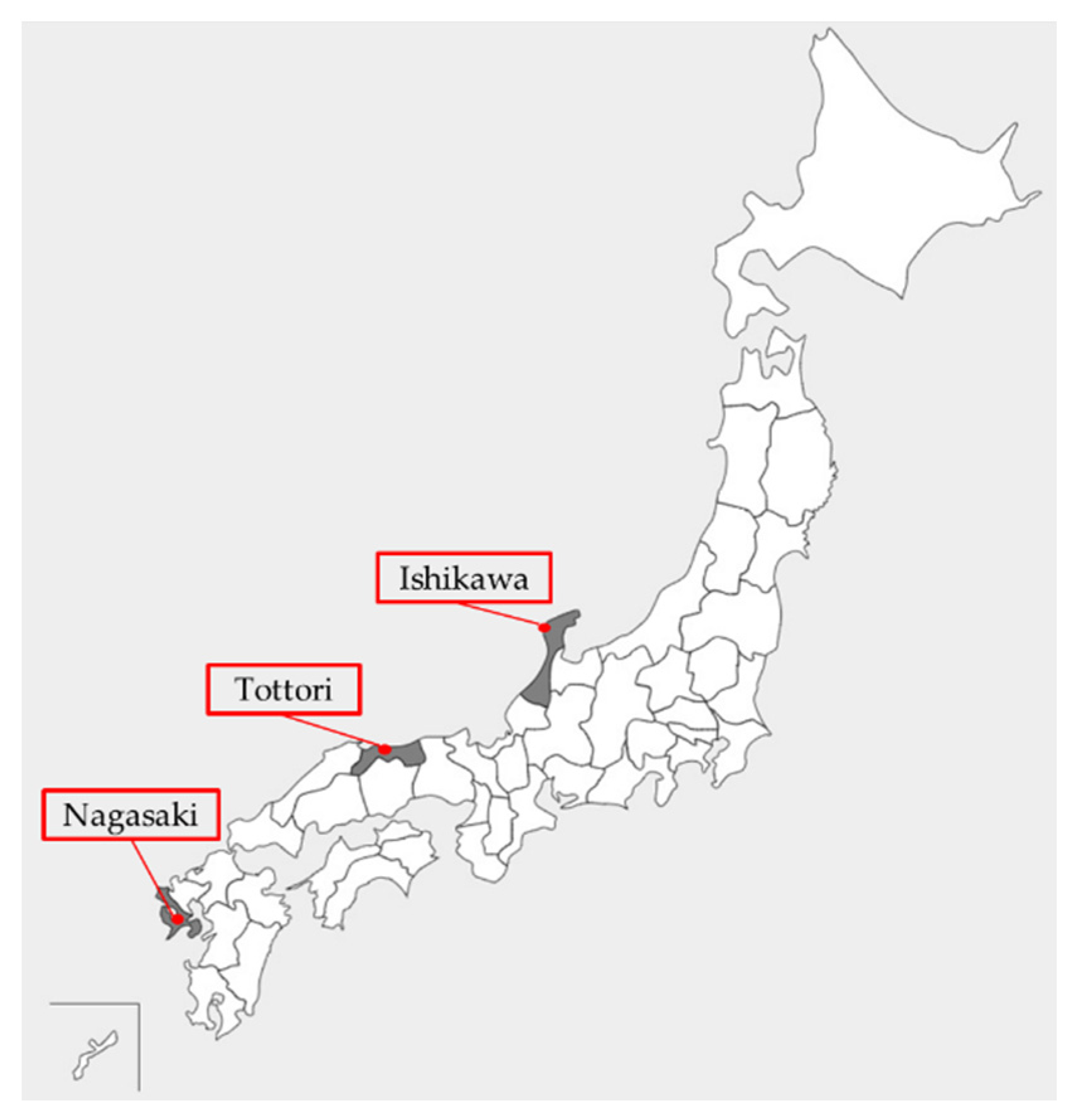
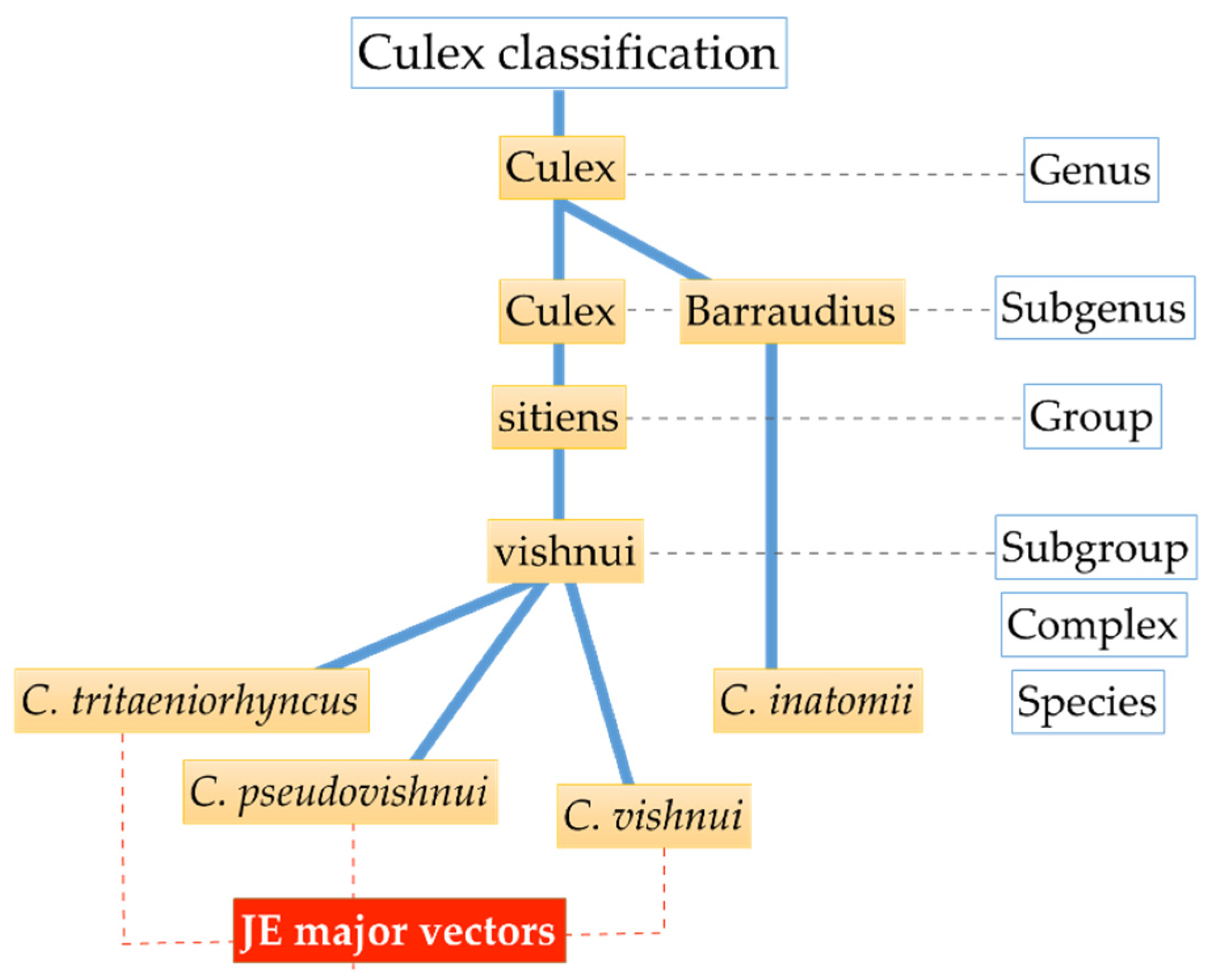
2.1. Mosquito Collection and Species Identification
2.2. Metagenomic Analysis of RNA Virome in Culex Mosquitoes
2.3. Virus Isolation
2.4. Pools Confirmation Using RT-PCR and Sequence Analysis of Viral RNA
2.5. Viral Genome Characterization and Phylogenetic Analyses
3. Results
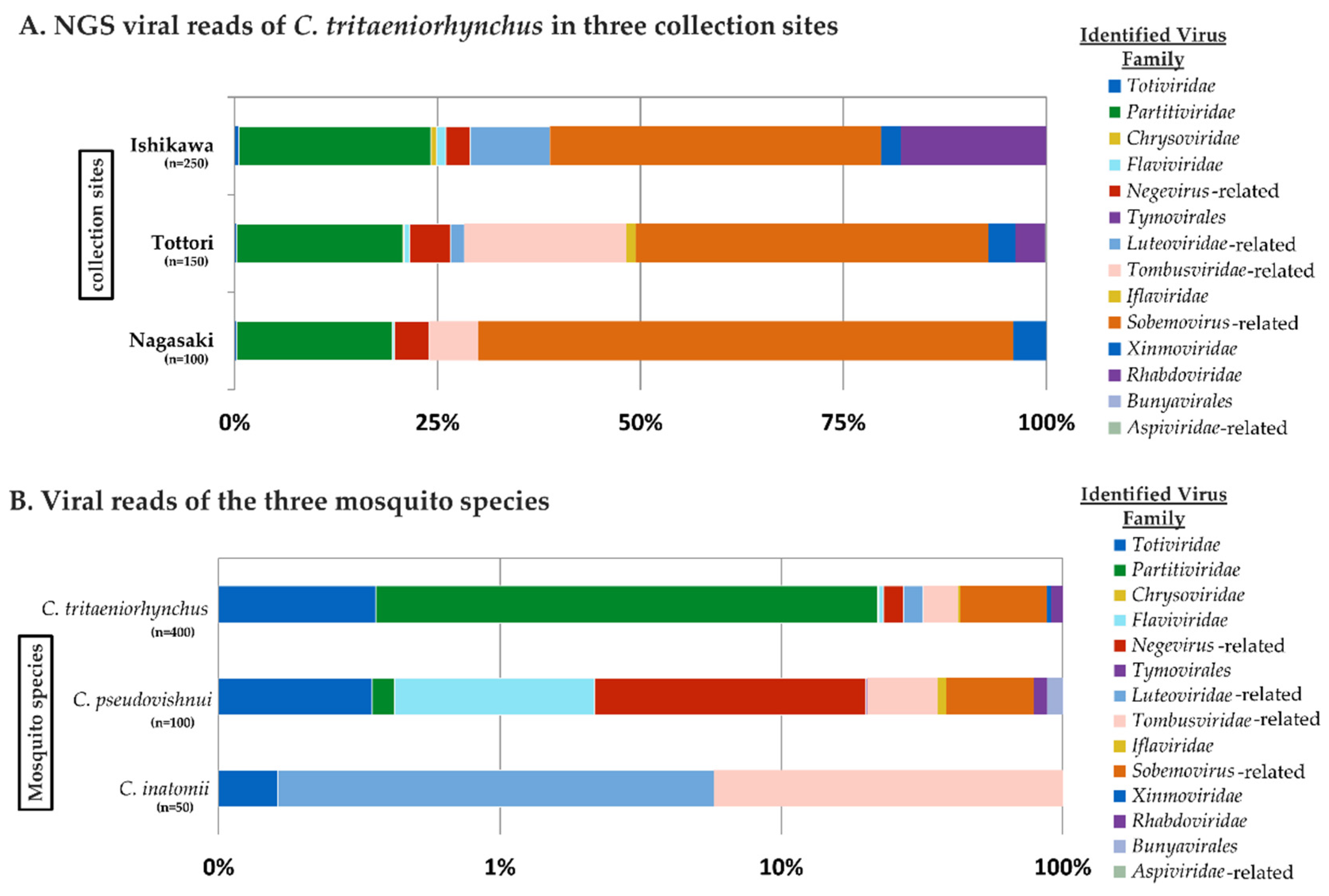
3.1. RNA Virome Analysis by Metagenomic Sequencing
3.2. Dataset Breakdown of Mosquito RNA Viruses
3.2.1. dsRNA Viruses
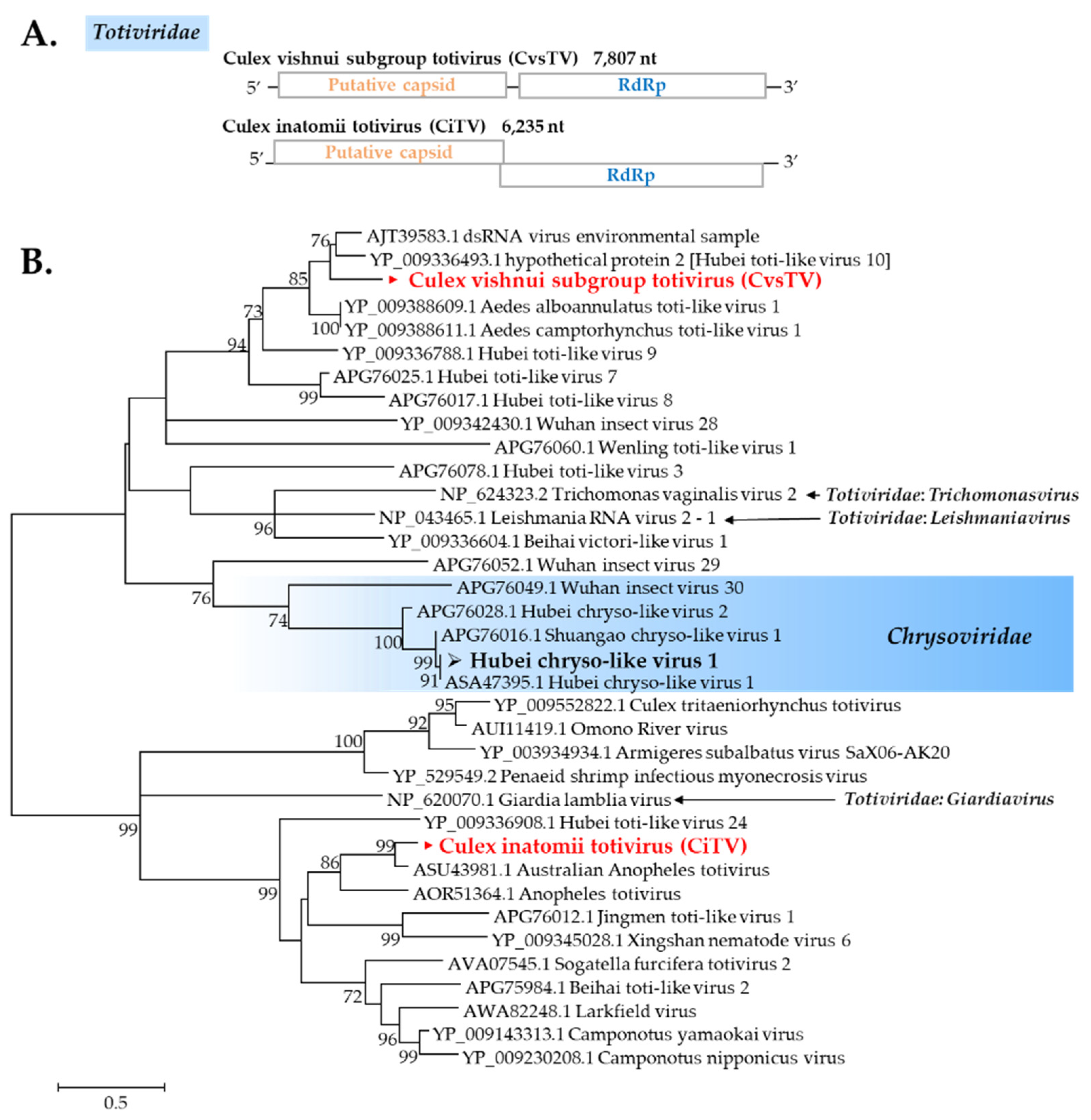
Totiviridae
Chrysoviridae
Partitiviridae
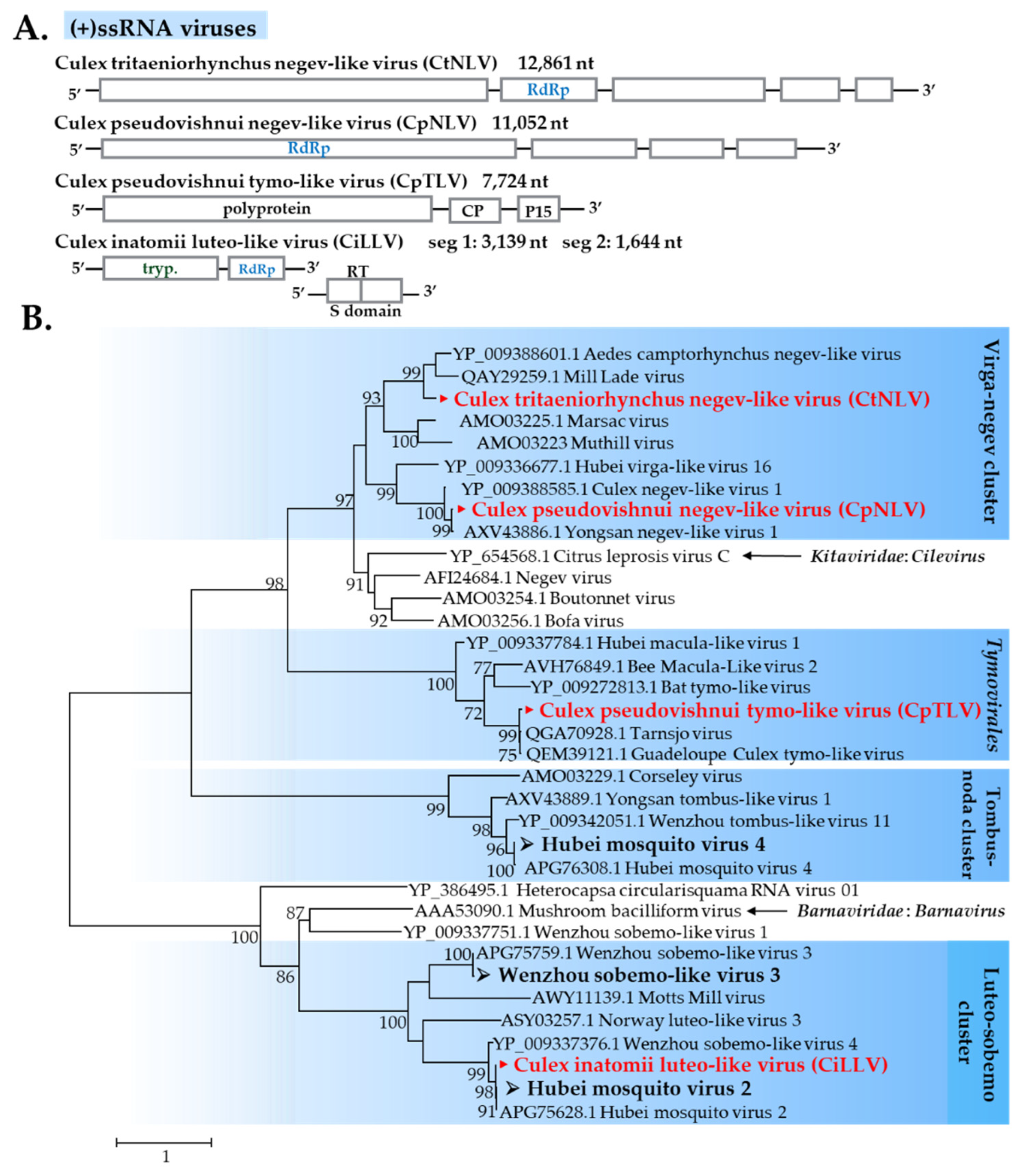
3.2.2. (+)ssRNA viruses
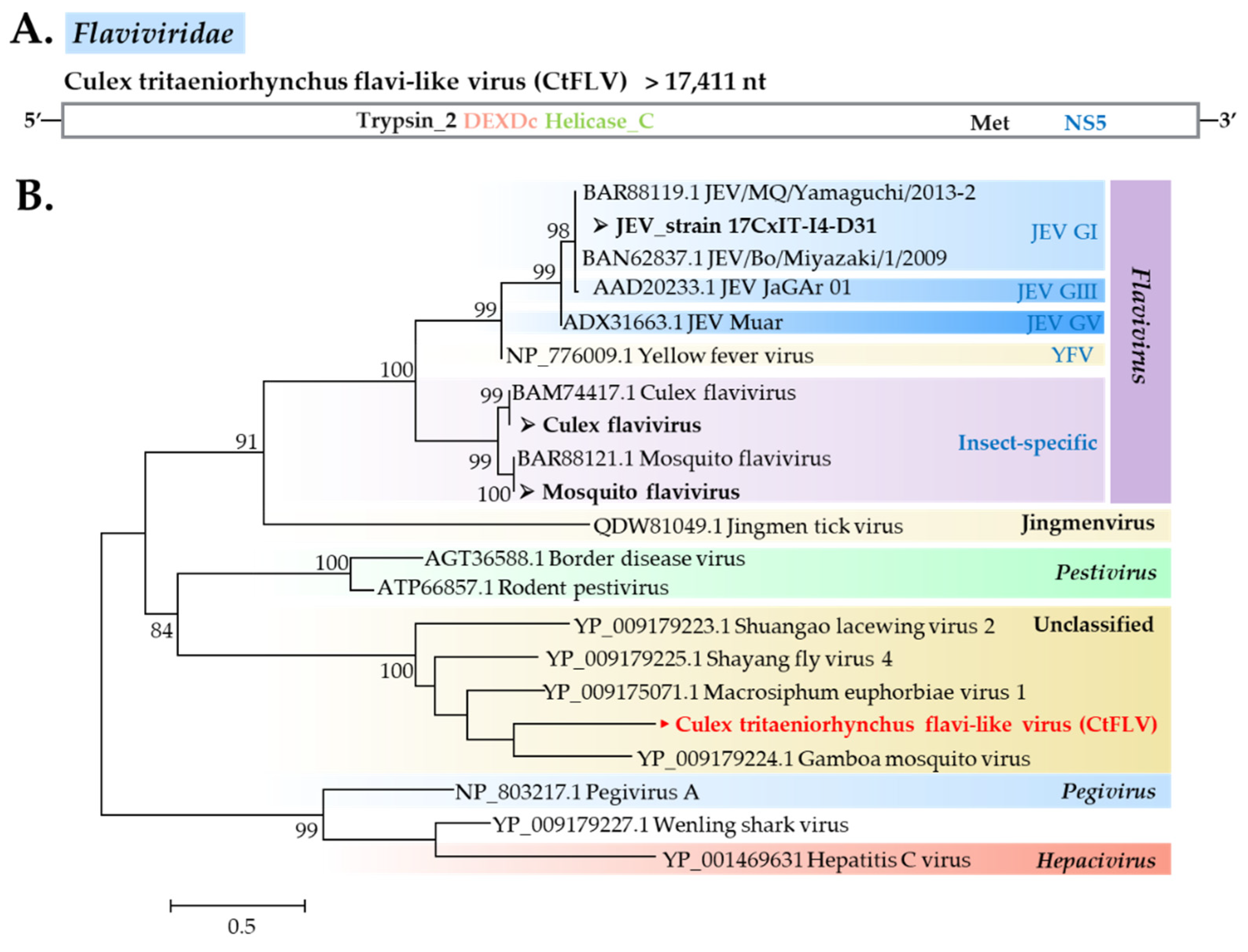
Flaviviridae
Negevirus-Related Viruses
Luteoviridae-Related Viruses
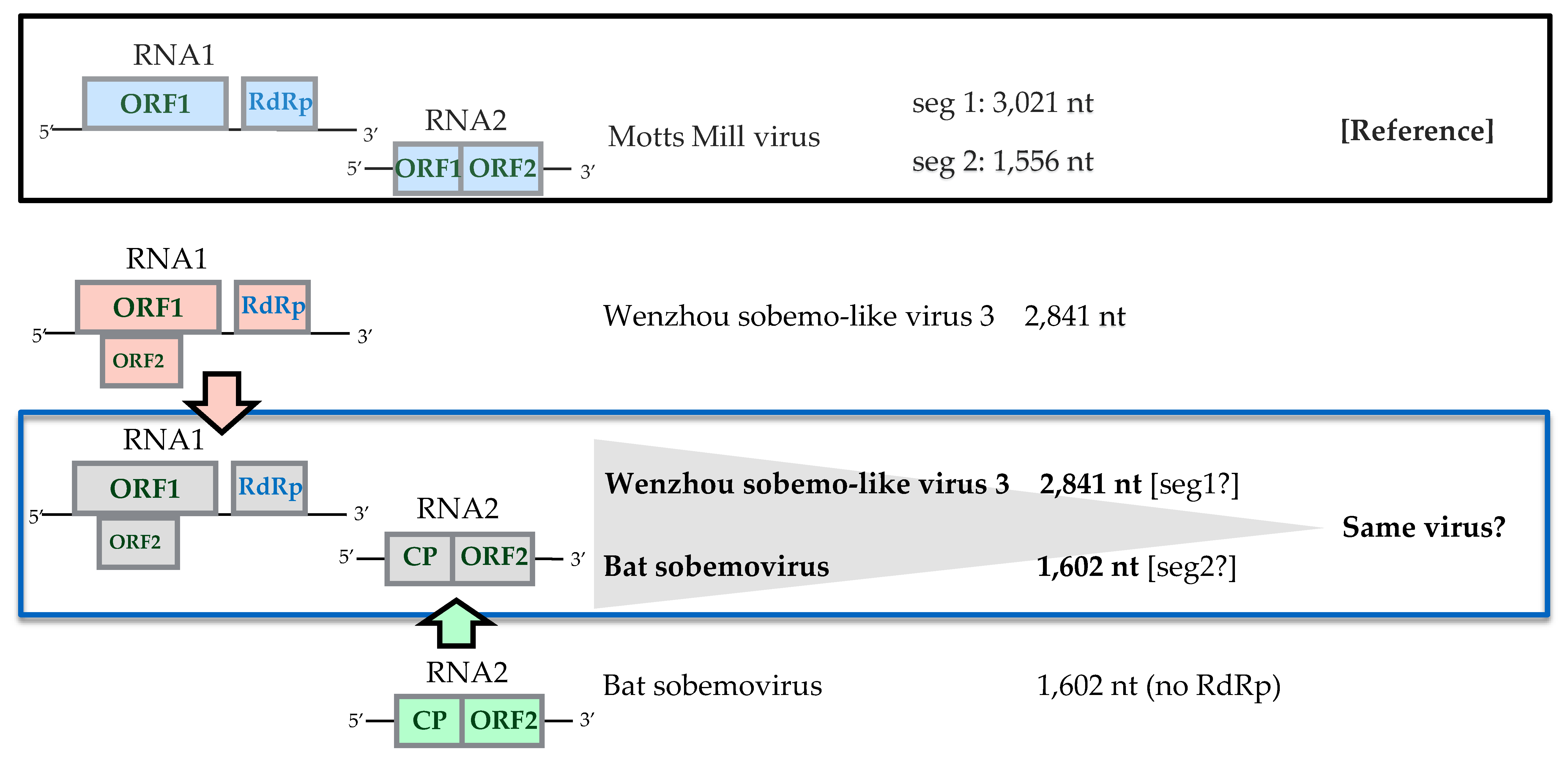
Sobemovirus-Related Viruses
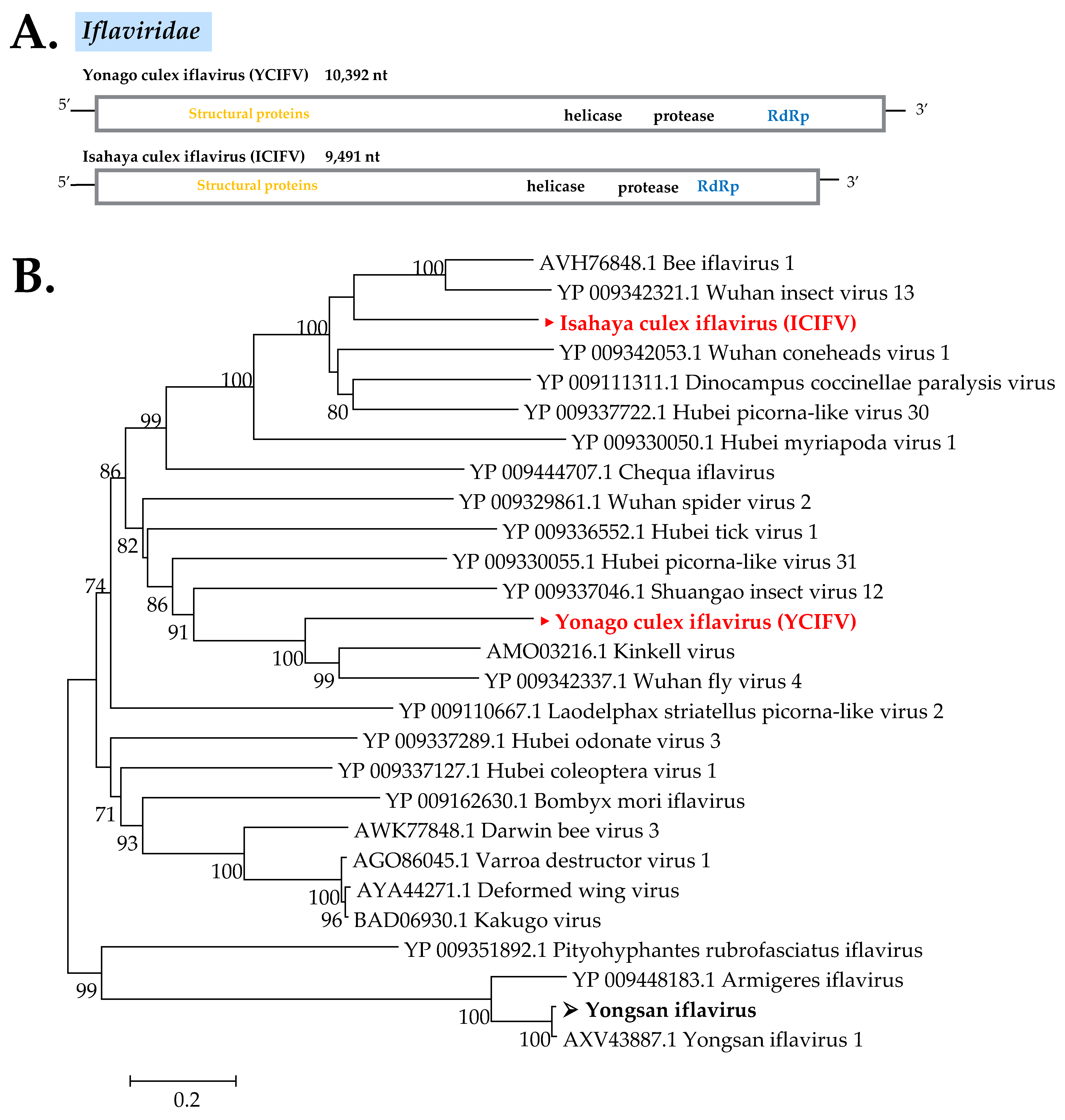
Iflaviridae
3.2.3. (−)ssRNA Viruses
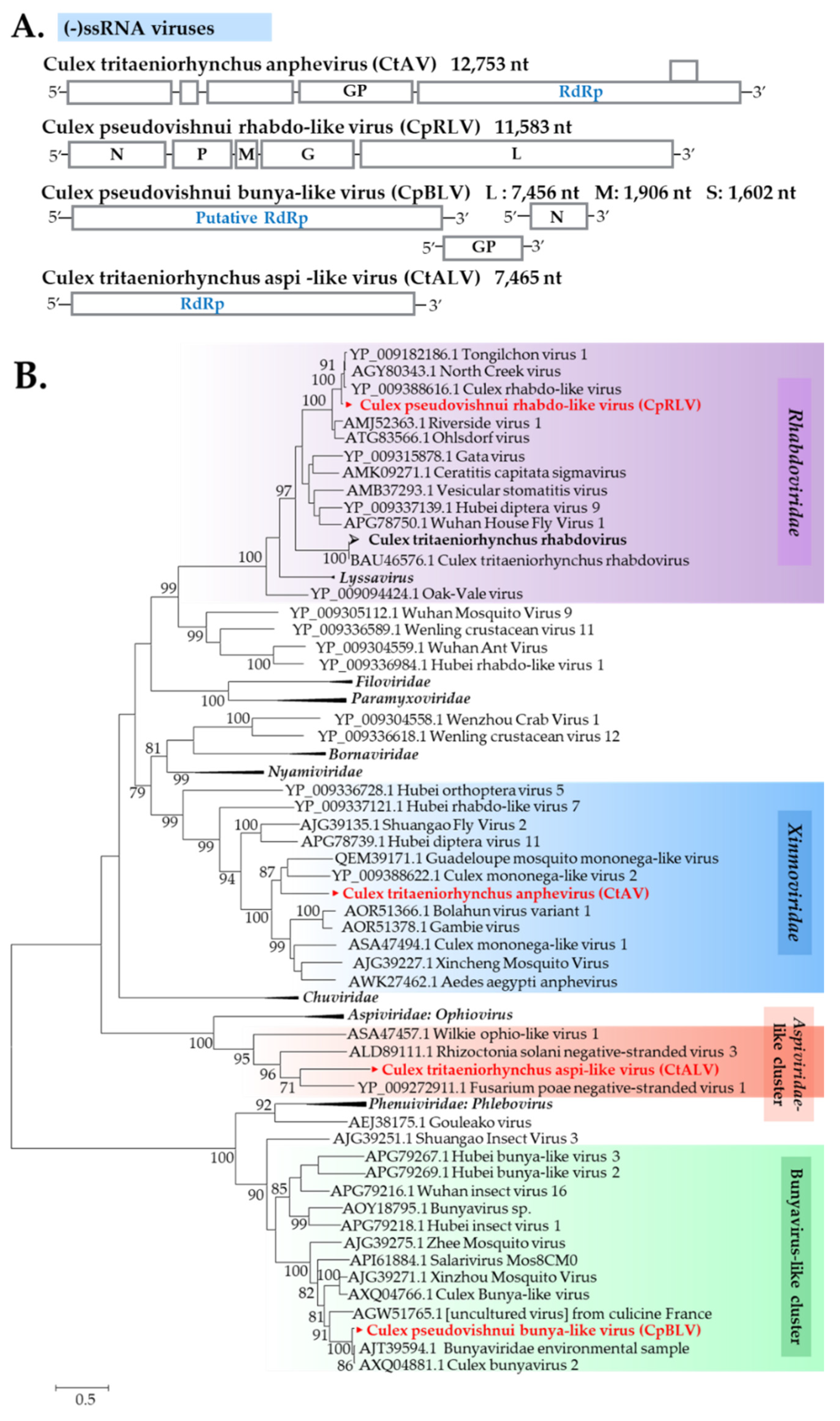
Xinmoviridae
Rhabdoviridae
Aspiviridae-Related Viruses
3.3. Attempt to Isolate Viruses Using a Mosquito Cell Line
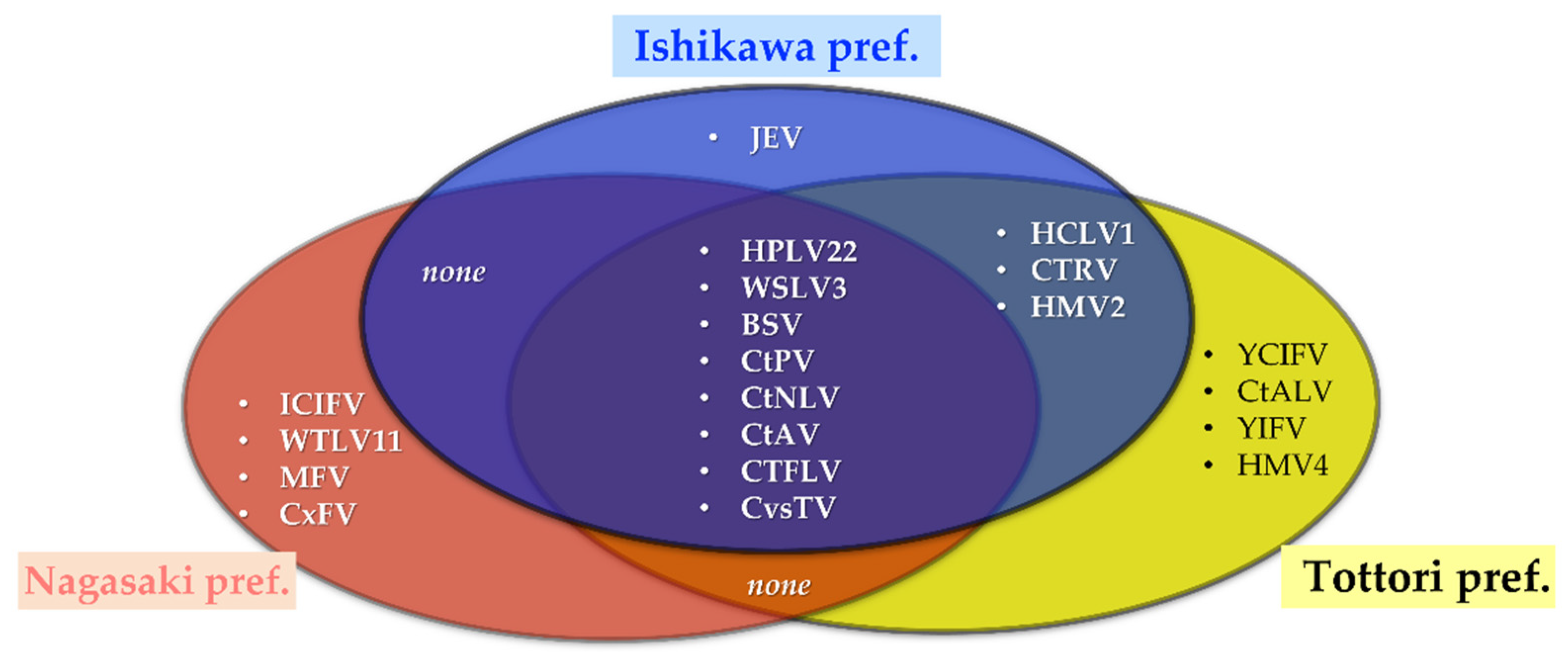
3.4. Pools Confirmation and Additional Individual Screening
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campbell, G.; Hills, S.; Fischer, M.; Jacobson, J.; Hoke, C.; Hombach, J.; Marfin, A.; Solomon, T.; Tsai, T.; Tsui, V.; et al. Estimated global incidence of Japanese encephalitis: A systematic review. Bull. World Health Organ. 2011, 89, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Erlanger, T.E.; Weiss, S.; Keiser, J.; Utzinger, J.; Wiedenmayer, K. Past, present, and future of Japanese encephalitis. Emerg. Infect. Dis. 2009, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Dungc, N.M.; Kneend, R.; Gainsboroughc, M.; Vaughnf, D.W.; Khanhc, V.T. Japanese encephalitis. J. Neurol. Neurosurg. Psychiatry 2000, 68, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Mourya, D.T.; Mishra, A.C.; Soman, R.S. Transmission of Japanese encephalitis virus in Culex pseudovishnui & C. tritaeniorhynchus mosquitoes. Indian J. Med. Res. 1991, 93, 250–252. [Google Scholar]
- Van den Hurk, A.F.; Ritchie, S.A.; Mackenzie, J.S. Ecology and geographical expansion of Japanese encephalitis virus. Annu. Rev. Entomol. 2009, 54, 17–35. [Google Scholar] [CrossRef] [Green Version]
- Buescher, E.L.; Scherer, W.F.; Rosenberg, M.Z.; Gresser, I.; Hardy, J.L.; Bullock, H.R. Ecologic studies of Japanese encephalitis virus in Japan. II. Mosquito infection. Am. J. Trop. Med. Hyg. 1959, 8, 651–664. [Google Scholar] [CrossRef]
- Atoni, E.; Wang, Y.; Karungu, S.; Waruhiu, C.; Zohaib, A.; Obanda, V.; Agwanda, B.; Mutua, M.; Xia, H.; Yuan, Z. Metagenomic virome analysis of Culex mosquitoes from Kenya and China. Viruses 2018, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.A.; Bielefeldt-Ohmann, H.; McLean, B.J.; O’Brien, C.A.; Colmant, A.M.; Piyasena, T.B.; Harrison, J.J.; Newton, N.D.; Barnard, R.T.; Prow, N.A.; et al. Commensal viruses of mosquitoes: Host restriction, transmission, and interaction with arboviral pathogens. Evol. Bioinform. Online 2017, 12, 35–44. [Google Scholar] [CrossRef]
- Öhlund, P.; Lundén, H.; Blomström, A.L. Insect-specific virus evolution and potential effects on vector competence. Virus Genes 2019, 55, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Hall-Mendelin, S.; McLean, B.J.; Bielefeldt-Ohmann, H.; Hobson-Peters, J.; Hall, R.A.; van den Hurk, A.F. The insect-specific Palm Creek virus modulates West Nile virus infection in and transmission by Australian mosquitoes. Parasit. Vectors 2016, 9, 414. [Google Scholar] [CrossRef] [Green Version]
- Vasilakis, N.; Tesh, R.B. Insect-specific viruses and their potential impact on arbovirus transmission. Curr. Opin. Virol. 2015, 15, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolling, B.G.; Weaver, S.C.; Tesh, R.B.; Vasilakis, N. Insect-specific virus discovery: Significance for the arbovirus community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jupatanakul, N.; Sim, S.; Dimopoulos, G. The insect microbiome modulates vector competence for arboviruses. Viruses 2014, 6, 4294–4313. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, M.; Rašić, G.; Darbro, J.; Krause, L.; Poo, Y.S.; Filipović, I.; Parry, R.; Asgari, S.; Devine, G.; Suhrbier, A. Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok. Sci. Rep. 2018, 8, 4690. [Google Scholar] [CrossRef] [Green Version]
- Xiao, P.; Li, C.; Zhang, Y.; Han, J.; Guo, X.; Xie, L.; Tian, M.; Li, Y.; Wang, M.; Liu, H.; et al. Metagenomic Sequencing from mosquitoes in China reveals a variety of insect and human viruses. Front. Cell. Infect. Microbiol. 2018, 8, 364. [Google Scholar] [CrossRef]
- Cholleti, H.; Hayer, J.; Fafetine, J.; Berg, M.; Blomström, A.L. Genetic characterization of a novel picorna-like virus in Culex spp. mosquitoes from Mozambique. Virol J. 2018, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Wang, Y.; Shi, C.; Atoni, E.; Zhao, L.; Yuan, Z. Comparative metagenomic profiling of viromes associated with four common mosquito species in China. Virol. Sin. 2018, 33, 59–66. [Google Scholar] [CrossRef]
- Shi, C.; Liu, Y.; Hu, X.; Xiong, J.; Zhang, B.; Yuan, Z. A metagenomic survey of viral abundance and diversity in mosquitoes from Hubei province. PLoS ONE 2015, 10, e0129845. [Google Scholar] [CrossRef]
- Tanaka, K.; Mizusawa, K.; Saugstad, E.S. A revision of the adult and larval mosquitoes of Japan (including the Ryukyu Archipelago and the Ogasawara Islands) and Korea (Diptera: Culicidae). Contrib. Am. Entomol. Inst. (Ann Arbor) 1979, 16, 1–987. [Google Scholar]
- Kobayashi, D.; Murota, K.; Itokawa, K.; Ejiri, H.; Amoa-Bosompem, M.; Faizah, A.N.; Watanabe, M.; Maekawa, Y.; Hayashi, T.; Noda, S.; et al. RNA virome analysis of questing ticks from Hokuriku District, Japan, and the evolutionary dynamics of tick-borne phleboviruses. Ticks Tick Borne Dis. 2020, 11. [Google Scholar] [CrossRef]
- Hoshino, K.; Isawa, H.; Tsuda, Y.; Sawabe, K.; Kobayashi, M. Isolation and characterization of a new insect flavivirus from Aedes albopictus and Aedes flavopictus mosquitoes in Japan. Virology 2009, 391, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghabrial, S.A.; Nibert, M.L. Victorivirus, a new genus of fungal viruses in the family Totiviridae. Arch. Virol. 2009, 154, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Ghabrial, S.A.; Castón, J.R.; Coutts, R.H.A.; Hillman, B.I.; Jiang, D.; Kim, D.H.; Moriyama, H.; Ictv report consortium. ICTV Virus Taxonomy Profile: Chrysoviridae. J. Gen. Virol. 2018, 99, 19–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Nibert, M.L.; Ghabrial, S.A.; Maiss, E.; Lesker, T.; Vainio, E.J.; Jiang, D.; Suzuki, N. Taxonomic reorganization of family Partitiviridae and other recent progress in partitivirus research. Virus Res. 2014, 188, 128–141. [Google Scholar] [CrossRef]
- Pettersson, J.H.; Shi, M.; Bohlin, J.; Eldholm, V.; Brynildsrud, O.B.; Paulsen, K.M.; Andreassen, Å.; Holmes, E.C. Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci. Rep. 2017, 7, 10870. [Google Scholar] [CrossRef] [Green Version]
- Solomon, T.; Ni, H.; Beasley, D.W.C.; Ekkelenkamp, M.; Cardosa, M.J.; Barrett, A.D.T. Origin and evolution of Japanese encephalitis virus in southeast asia. J. Virol. 2003, 77, 3091–3098. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.P.; Yoshida, Y.; Makino, Y.; Tadano, M.; Ono, T.; Ogawa, M. Short report: A major genotype of Japanese encephalitis virus currently circulating in Japan. Am. J. Trop. Med. Hyg. 2003, 69, 151–154. [Google Scholar] [CrossRef]
- Nunes, M.R.T.; Contreras-Gutierrez, M.A.; Guzman, H.; Martins, L.C.; Barbirato, M.F.; Savit, C.; Balta, V.; Uribe, S.; Vivero, R.; Suaza, J.D.; et al. Genetic characterization, molecular epidemiology, and phylogenetic relationships of insect-specific viruses in the taxon Negevirus. Virology 2017, 504, 152–167. [Google Scholar] [CrossRef]
- Adams, M.J.; Kreuze, J.F.; Martelli, G.P. Order Tymovirales. In Virus taxonomy, Ninth report of the International Committee on Taxonomy of Viruses; Andrew, M.Q.K., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: London, UK, 2012; pp. 901–903. [Google Scholar] [CrossRef]
- Ali, M.; Hameed, S.; Tahir, M. Luteovirus: Insights into pathogenicity. Arch. Virol. 2014, 159, 2853–2860. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.S.; Imrie, A.; Holmes, E.C. High-resolution metatranscriptomics reveals the ecological dynamics of mosquito-associated RNA viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dacheux, L.; Cervantes-Gonzalez, M.; Guigon, G.; Thiberge, J.M.; Vandenbogaert, M.; Maufrais, C.; Caro, V.; Bourhy, H. A preliminary study of viral metagenomics of French bat species in contact with humans: Identification of new mammalian viruses. PLoS ONE 2014, 9, e87194. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Nakashima, N.; Christian, P.D.; Bakonyi, T.; Bonning, B.C.; Valles, S.M.; Lightner, D. Family Iflaviridae. In Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; Andrew, M.Q.K., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: London, UK, 2012; pp. 846–849. [Google Scholar] [CrossRef]
- Maes, P.; Amarasinghe, G.K.; Ayllón, M.A.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Second update 2018. Arch. Virol. 2019, 164, 1233–1244. [Google Scholar] [CrossRef] [Green Version]
- Amarasinghe, G.K.; Aréchiga Ceballos, N.G.; Banyard, A.C.; Basler, C.F.; Bavari, S.; Bennett, A.J.; Blasdell, K.R.; Briese, T.; Bukreyev, A.; Caì, Y.; et al. Taxonomy of the order Mononegavirales: Update 2018. Arch. Virol. 2018, 163, 2283–2294. [Google Scholar] [CrossRef] [Green Version]
- Bourhy, H.; Cowley, J.A.; Larrous, F.; Holmes, E.C.; Walker, P.J. Phylogenetic relationships among rhabdoviruses inferred using the L polymerase gene. J. Gen. Virol. 2005, 86, 2849–2858. [Google Scholar] [CrossRef]
- Longdon, B.; Murray, G.G.; Palmer, W.J.; Day, J.P.; Parker, D.J.; Welch, J.J.; Obbard, D.J.; Jiggins, F.M. The evolution, diversity, and host associations of rhabdoviruses. Virus Evol. 2015, 1, vev014. [Google Scholar] [CrossRef] [Green Version]
- Walter, C.T.; Barr, J.N. Recent advances in the molecular and cellular biology of bunyaviruses. J. Gen. Virol. 2011, 92, 2467–2484. [Google Scholar] [CrossRef]
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the order Bunyavirales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [Green Version]
- Osaki, H.; Sasaki, A.; Nomiyama, K.; Tomioka, K. Multiple virus infection in a single strain of Fusarium poae shown by deep sequencing. Virus Genes 2016, 52, 835–847. [Google Scholar] [CrossRef]
- Walker, T.; Jeffries, C.L.; Mansfield, K.L.; Johnson, N. Mosquito cell lines: History, isolation, availability and application to assess the threat of arboviral transmission in the United Kingdom. Parasit. Vectors 2014, 7, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brackney, D.E.; Scott, J.C.; Sagawa, F.; Woodward, J.E.; Miller, N.A.; Schilkey, F.D.; Mudge, J.; Wilusz, J.; Olson, K.E.; Blair, C.D.; et al. C6/36 Aedes albopictus cells have a dysfunctional antiviral RNA interference response. PLoS Negl. Trop. Dis. 2010, 4, e856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infectious Disease Surveillance Center, National Institute of Infectious Diseases (NIID) Japan: Japanese encephalitis, Japan, 2003–2008. Infect. Agents Surveill. Rep. (IASR) 2009, 30, 147–148. Available online: https://idsc.niid.go.jp/iasr/30/352/tpc352.html (accessed on 14 January 2020).
- Infectious Disease Surveillance Center, NIID Japan: Japanese encephalitis, Japan, 2007–2016. Infectious Infect. Agents Surveill. Rep. (IASR) 2017, 38, 151–152. Available online: https://www.niid.go.jp/niid/en/iasr-vol38-e/865-iasr/7483-450te.html (accessed on 14 January 2020).
- Tadano, M.; Kanemura, K.; Hasegawa, H.; Makino, Y.; Fukunaga, T. Epidemiological and ecological studies of Japanese encephalitis in Okinawa, subtropical area in Japan. I. Investigations on antibody levels to Japanese encephalitis virus in swine sera and vector mosquito in Okinawa, Miyako and Ishigaki islands. Microbiol. Immunol. 1994, 38, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholleti, H.; Hayer, J.; Abilio, A.P.; Mulandane, F.C.; Verner-Carlsson, J.; Falk, K.I.; Fafetine, J.M.; Berg, M.; Blomström, A.L. Discovery of novel viruses in mosquitoes from the Zambezi valley of Mozambique. PLoS ONE 2016, 11, e0162751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Beller, L.; Deboutte, W.; Yinda, K.C.; Delang, L.; Vega-Rúa, A.; Failloux, A.B.; Matthijnssens, J. Stable distinct core eukaryotic viromes in different mosquito species from Guadeloupe, using single mosquito viral metagenomics. Microbiome 2019, 7, 121. [Google Scholar] [CrossRef]
- Ritchie, S.A.; Rochester, W. Wind-blown mosquitoes and introduction of Japanese encephalitis into Australia. Emerg. Infect. Dis. 2001, 7, 900–903. [Google Scholar] [CrossRef]
- Inglis, T.J.; O’Reilly, L.; Merritt, A.J.; Levy, A.; Heath, C.H. The aftermath of the Western Australian melioidosis outbreak. Am. J. Trop. Med. Hyg. 2011, 84, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Johansen, C.A.; van den Hurk, A.F.; Pyke, A.T.; Zborowski, P.; Phillips, D.A.; Mackenzie, J.S.; Ritchie, S.A. Entomological investigations of an outbreak of Japanese encephalitis virus in the Torres Strait, Australia, in 1998. J. Med. Entomol. 2001, 38, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Kuwata, R.; Isawa, H.; Hoshino, K.; Sasaki, T.; Kobayashi, M.; Maeda, K.; Sawabe, K. Analysis of mosquito-borne flavivirus superinfection in Culex tritaeniorhynchus (Diptera: Culicidae) cells persistently infected with Culex Flavivirus (Flaviviridae). J. Med. Entomol. 2015, 52, 222–229. [Google Scholar] [CrossRef] [PubMed]
- National Epidemiological Surveillance of Vaccine-Preventable Diseases: Seroprevalence of Japanese Encephalitis in Swine during Summer Season. Available online: https://www.niid.go.jp/niid/ja/y-graphs/1600-yosoku-index-e.html (accessed on 7 January 2020).
- Scherer, W.F.; Buescher, E.L. Ecologic studies of Japanese encephalitis virus in Japan: I. Introduction. Am. J. Trop. Med. Hyg. 1959, 8, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.; Isawa, H.; Ejiri, H.; Sasaki, T.; Sunahara, T.; Futami, K.; Tsuda, Y.; Katayama, Y.; Mizutani, T.; Minakawa, N.; et al. Complete genome sequencing and phylogenetic analysis of a Getah virus strain (Genus Alphavirus, Family Togaviridae) isolated from Culex tritaeniorhynchus mosquitoes in Nagasaki, Japan in 2012. Vector Borne Zoonotic Dis. 2016, 16, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.J.; Bushmaker, T.; Cameron, K.; Ondzie, A.; Niama, F.R.; Parra, H.J.; Mombouli, J.V.; Olson, S.H.; Munster, V.J.; Goldberg, T.L. Diverse RNA viruses of arthropod origin in the blood of fruit bats suggest a link between bat and arthropod viromes. Virology 2019, 528, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef] [Green Version]
- Kading, R.C.; Schountz, T. Flavivirus infections of bats: Potential role in Zika virus ecology. Am. J. Trop. Med. Hyg. 2016, 95, 993–996. [Google Scholar] [CrossRef] [Green Version]
- Saepulloh, M.; Dharmayanti, N.L.P.I.; Adjid, R.M.A.; Ratnawati, A.; Sendow, I. The presence of Japanese encephalitis virus infection in Pteropus sp. in West Kalimantan. Proc. Int. Sem. Livest. Prod. Vet. Technol. 2016, 549–553. [Google Scholar] [CrossRef]
- Wang, J.L.; Pan, X.L.; Zhang, H.L.; Fu, S.H.; Wang, H.Y.; Tang, Q.; Wang, L.F.; Liang, G.D. Japanese encephalitis viruses from bats in Yunnan, China. Emerg. Infect. Dis. 2009, 15, 939–942. [Google Scholar] [CrossRef]
- Hall-Mendelin, S.; Allcock, R.; Kresoje, N.; van den Hurk, A.F.; Warrilow, D. Detection of arboviruses and other micro-organisms in experimentally infected mosquitoes using massively parallel sequencing. PLoS ONE 2013, 8, e58026. [Google Scholar] [CrossRef] [Green Version]
- White, L.A. Susceptibility of Aedes albopictus C6/36 cells to viral infection. J. Clin. Microbiol. 1987, 25, 1221–1224. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mosquito Species | Mosquito Collection Site | GPS Coordinates | Collection Month | Methods | ||||
|---|---|---|---|---|---|---|---|---|
| Metagenomic Analysis | Virus Isolation | |||||||
| No. of Pools Tested | No. of Female Mosquitoes Tested | No. of Pools Tested | No. of Female Mosquitoes Tested | JEV-Positive Pools | ||||
| C. tritaeniorhynchus | Monzen-machi, Wajima city, Ishikawa | 37°29′ N, 136°74′ E | Jun–Oct | 5 | 250 | 12 | 300 | 1 |
| 2017 | ||||||||
| C. tritaeniorhynchus | Yonago waterbirds sanctuary, Yonago city, Tottori | 35°26′ N, 133°17′ E | Jun–Sep | 3 | 150 | 8 | 200 | 0 |
| 2017 | ||||||||
| C. tritaeniorhynchus | Teramine farm, Isahaya city, Nagasaki | 32°49′ N, 130°03′ E | Aug | 2 | 100 | 54 | 1,350 | 0 |
| 2017 | ||||||||
| C. pseudovishnui | Teramine farm, Isahaya city, Nagasaki | 32°49′ N, 130°03′ E | Aug | 2 | 100 | 17 | 421 | 0 |
| 2017 | ||||||||
| C. inatomii | Yonago waterbirds sanctuary, Yonago city, Totttori | 35°26′ N, 133°17′ E | Jun | 1 | 52 | 2 | 52 | 0 |
| 2017 | ||||||||
| Total | 13 | 652 | 93 | 2323 | 1 | |||
| Virus Category | VIRUS TAXON | Virus Name | Closely Related Viruses (Accession no.) | Blastn | Blastx | Location †/Species Origin ‡ | Segment | Accession no. [This Study] | Complete CDs ¶ | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identity (%) * | Query Cover (%) | e-Value | Identity (%) * | Query Cover (%) | e-Value | ||||||||
| dsRNA | Totiviridae | Culex vishnui subgroup totivirus (CvsTV) | dsRNA virus environmental sample (AJT39583) | - | - | - | 46 | 39 | 0.0 | Japan/CtrCps | - | LC514295 | Y |
| Totiviridae | Culex inatomii totivirus (CiTV) | Australian Anopheles Totivirus (ASU43981) | - | - | - | 61 | 37 | 0.0 | Tottori/Cnt | - | LC514398 | Y | |
| Partitiviridae | Culex pseudovishnui partitivirus (CpPV) | Hubei partiti-like virus 56 (APG78242) | - | - | - | 56 | 85 | 0.0 | Nagasaki/Cps | 1 | LC514399 | Y | |
| Partitiviridae | Hubei partiti-like virus 22 (HPLV22) | (APG78283) | 99 | 98 | 0.0 | 99 | 98 | 0.0 | Tottori/Ctr Nagasaki/Ctr | 1 | LC514400 LC514401 | Y Y | |
| Partitiviridae | Culex tritaeniorhynchus partitivirus (CtPV) | Hubei partiti-like virus 19 (APG78260) | - | - | - | 49 | 84 | 1 × 10−170 | Japan/Ctr | 1 | LC514402 | Y | |
| Chrysoviridae | Hubei chryso-like virus 1 (HCLV1) | (ASA47395) | 84 | 99 | 0.0 | - | - | - | Ishikawa/Ctr | 1 | LC514396 | Y | |
| (+)ssRNA | Flaviviridae | Culex tritaeniorhynchus flavi-like virus (CtFLV) | Shayang fly virus 4 (YP_009179225) | - | - | - | 41 | 71 | 0.0 | Japan/Ctr | - | LC514290 | N |
| Flaviviridae | Mosquito flavivirus (MFV) | (BAR88121) | 98 | 99 | 0.0 | 99 | 93 | 0.0 | Nagasaki/Ctr Nagasaki/Cps | - | LC513840 LC513841 | Y Y | |
| Flaviviridae | Japanese Encephalitis Virus (JEV) | (AB981184) | 99 | 100 | 0.0 | 99 | 97 | 0.0 | Ishikawa/Ctr | - | LC513838 | Y | |
| Flaviviridae | Culex Flavivirus (CxFV) | (BAM74417) | 97 | 100 | 0.0 | 99 | 93 | 0.0 | Nagasaki/Ctr | - | LC513839 | Y | |
| Negevirus-related | Culex tritaeniorhynchus negev-like virus (CtNLV) | Mill Lade virus (QAY29259) | - | - | - | 47 | 22 | 0.0 | Japan/Ctr | - | LC507097 | Y | |
| Negevirus-related | Culex pseudovishnui negev-like virus (CpNLV) | Yongsan negev-like virus 1 (AXV43886) | - | - | - | 87 | 73 | 0.0 | Nagasaki/Cps | - | LC512731 | Y | |
| (+)ssRNA | Tymovirales | Culex pseudovishnui tymo-like virus (CpTLV) | Tarnsjo virus (QGA70928) | 79 | 76 | 0.0 | 74 | 84 | 0.0 | Nagasaki/Cps | - | LC512732 | Y |
| Luteoviridae-related | Hubei mosquito virus 2 (HMV2) | APG75628 | 99 | 99 | 0.0 | 100 | 34 | 0.0 | Ishikawa/Ctr Tottori/Ctr | 1 1 | LC513829 LC513830 | Y Y | |
| Luteoviridae-related | Culex inatomii luteo-like virus (CiLLV) | Hubei mosquito virus 2 (APG75628) | 83 | 99 | 0.0 | - | - | - | Tottori/Cnt | 1 | LC513833 | Y | |
| Tombusviridae-related | Hubei mosquito virus 4 (HMV4) | (APG76308) | 91 | 82 | 0.0 | - | - | - | Tottori/Ctr Nagasaki/Cnt | - | LC512733 LC512734 | Y N | |
| 90 | 98 | 0.0 | - | - | - | Tottori/Ctr Nagasaki/Cnt | - | LC512735 LC512736 | N N | ||||
| Tombusviridae-related | Wenzhou tombus-like virus 11 (WTLV11) | (YP_009342051) | 99 | 99 | 0.0 | 100 | 28 | 0.0 | Nagasaki/Ctr Nagasaki/Cps | - | LC512737 LC512738 | Y Y | |
| Iflaviridae | Isahaya Culex Iflavirus (ICIFV) | Wuhan fly virus 4 (YP 009342337) | - | - | - | 43 | 71 | 0.0 | Nagasaki/Cps | - | LC513835 | Y | |
| Iflaviridae | Yongsan Iflavirus 1 (YIFV1) | (AXV43887) | 96 | 100 | 0.0 | 97 | 96 | 0.0 | Tottori/Ctr | - | LC513837 | N | |
| Iflaviridae | Yonago Culex Iflavirus (YCIFV) | Wuhan insect virus 13 (YP 009342321) | - | - | - | 38 | 82 | 0.0 | Tottori/Ctr | - | LC513836 | Y | |
| Sobemovirus-related | Wenzhou sobemo-like virus 3 (WSLV3) | (APG75759) | 91 | 92 | 0.0 | 96 | 47 | 0.0 | Ishikawa/Ctr Tottori/Ctr Nagasaki/Ctr Nagasaki/Cps | - | LC512854 LC512855 LC512856 LC512857 | N N N Y | |
| Sobemovirus-related | Bat sobemovirus (BSV) | (AGN73380) | 90 | 42 | 0.0 | 94 | 42 | 4×10−133 | Ishikawa/Ctr Tottori/Ctr Nagasaki/Ctr Nagasaki/Cps | - | LC512858 LC512859 LC512860 LC512861 | N N Y Y | |
| (−)ssRNA | Xinmoviridae | Culex tritaeniorhynchus anphevirus (CtAV) | Guadeloupe mosquito mononega-like virus (QEM39171.1) | - | - | - | 38 | 47 | 0.0 | Ishikawa/Ctr Tottori/Ctr | - | LC514054 LC514055 | Y Y |
| Rhabdoviridae | Culex tritaeniorhynchus rhabdovirus (CTRV) | (BAU46576) | 99 | 100 | 0.0 | 99 | 56 | 0.0 | Japan/Ctr | - | LC514403 | Y | |
| Rhabdoviridae | Culex pseudovishnui rhabdo-like virus (CpRLV) | Tongilchon virus 1 (YP_009182186) | 76 | 55 | 0.0 | 88 | 54 | 0.0 | Nagasaki/Cps Nagasaki/Cps | - | LC514056 LC514057 | Y Y | |
| Bunyavirales | Culex pseudovishnui bunya-like virus (CpBLV) | Bunyaviridae environmental sample (AJT39594) | 80 | 98 | 0.0 | - | - | - | Nagasaki/Cps Nagasaki/Cps | L | LC514291 LC514293 | N Y | |
| Aspiviridae-related | Culex tritaeniorhynchus Aspi-like virus (CtALV) | Fusarium poae negative-stranded virus (YP_009272911) | - | - | - | 30 | 80 | 0.0 | Tottori/Ctr | - | LC514058 | Y | |
| Virus Name | Mosquito Groups * | ||||
|---|---|---|---|---|---|
| Ctr-Ishikawa | Ctr-Tottori | Ctr-Nagasaki | Cps-Nagasaki | Cnt-Tottori | |
| WSLV3 | + | + | + | + | − |
| BSV | + | + | + | + | − |
| CvsTV | + | + | + | + | − |
| HPLV22 | + | + | + | − | − |
| CtPV | + | + | + | − | − |
| CtNLV | + | + | + | − | − |
| CtAV | + | + | + | − | − |
| CtFLV | + | + | + | − | − |
| HCLV1 | + | + | − | − | − |
| CTRV | + | + | − | − | − |
| HMV2 | + | + | − | − | − |
| HMV4 | − | + | − | − | + |
| ICIFV | − | − | + | + | − |
| WTLV11 | − | − | + | + | − |
| MFV | − | − | + | + | − |
| CxFV | − | − | + | − | − |
| JEV | + | − | − | − | − |
| YCIFV | − | + | − | − | − |
| CtALV | − | + | − | − | − |
| YIFV | − | + | − | − | − |
| CpPV | − | − | − | + | − |
| CpNLV | − | − | − | + | − |
| CpBLV | − | − | − | + | − |
| CpRLV | − | − | − | + | − |
| CpTLV | − | − | − | + | − |
| CiTV | − | − | − | − | + |
| CiLLV | − | − | − | − | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faizah, A.N.; Kobayashi, D.; Isawa, H.; Amoa-Bosompem, M.; Murota, K.; Higa, Y.; Futami, K.; Shimada, S.; Kim, K.S.; Itokawa, K.; et al. Deciphering the Virome of Culex vishnui Subgroup Mosquitoes, the Major Vectors of Japanese Encephalitis, in Japan. Viruses 2020, 12, 264. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030264
Faizah AN, Kobayashi D, Isawa H, Amoa-Bosompem M, Murota K, Higa Y, Futami K, Shimada S, Kim KS, Itokawa K, et al. Deciphering the Virome of Culex vishnui Subgroup Mosquitoes, the Major Vectors of Japanese Encephalitis, in Japan. Viruses. 2020; 12(3):264. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030264
Chicago/Turabian StyleFaizah, Astri Nur, Daisuke Kobayashi, Haruhiko Isawa, Michael Amoa-Bosompem, Katsunori Murota, Yukiko Higa, Kyoko Futami, Satoshi Shimada, Kyeong Soon Kim, Kentaro Itokawa, and et al. 2020. "Deciphering the Virome of Culex vishnui Subgroup Mosquitoes, the Major Vectors of Japanese Encephalitis, in Japan" Viruses 12, no. 3: 264. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030264