Suramin Inhibits Chikungunya Virus Replication by Interacting with Virions and Blocking the Early Steps of Infection

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Compounds, and Viruses
2.2. Preparation of 35S-labeled Viruses and Purification of Virus Stocks
2.3. Virus Attachment Assay
2.4. Bulk Fusion Assay
2.5. 3H-Suramin-Virus Binding Assay
2.6. Reverse Genetics
2.7. RNA Isolation and RT-qPCR
2.8. Cytopathic Effect (CPE) Reduction Assay
2.9. Plaque Number Reduction Assay
2.10. Molecular Modelling
2.11. Statistics
3. Results and Discussion
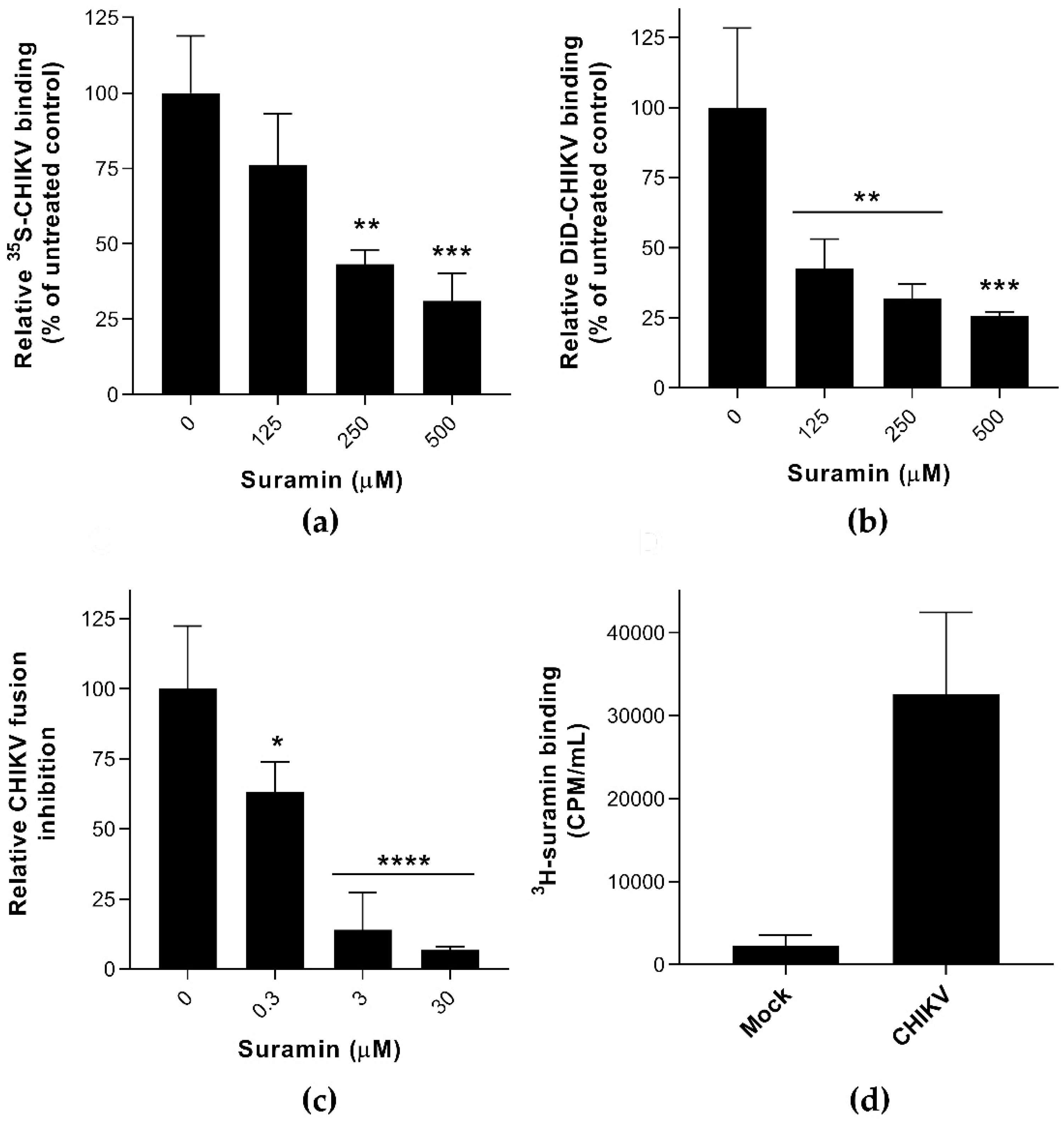
3.1. Suramin Inhibits Viral Attachment and Fusion by Interacting with the Chikungunya Virion
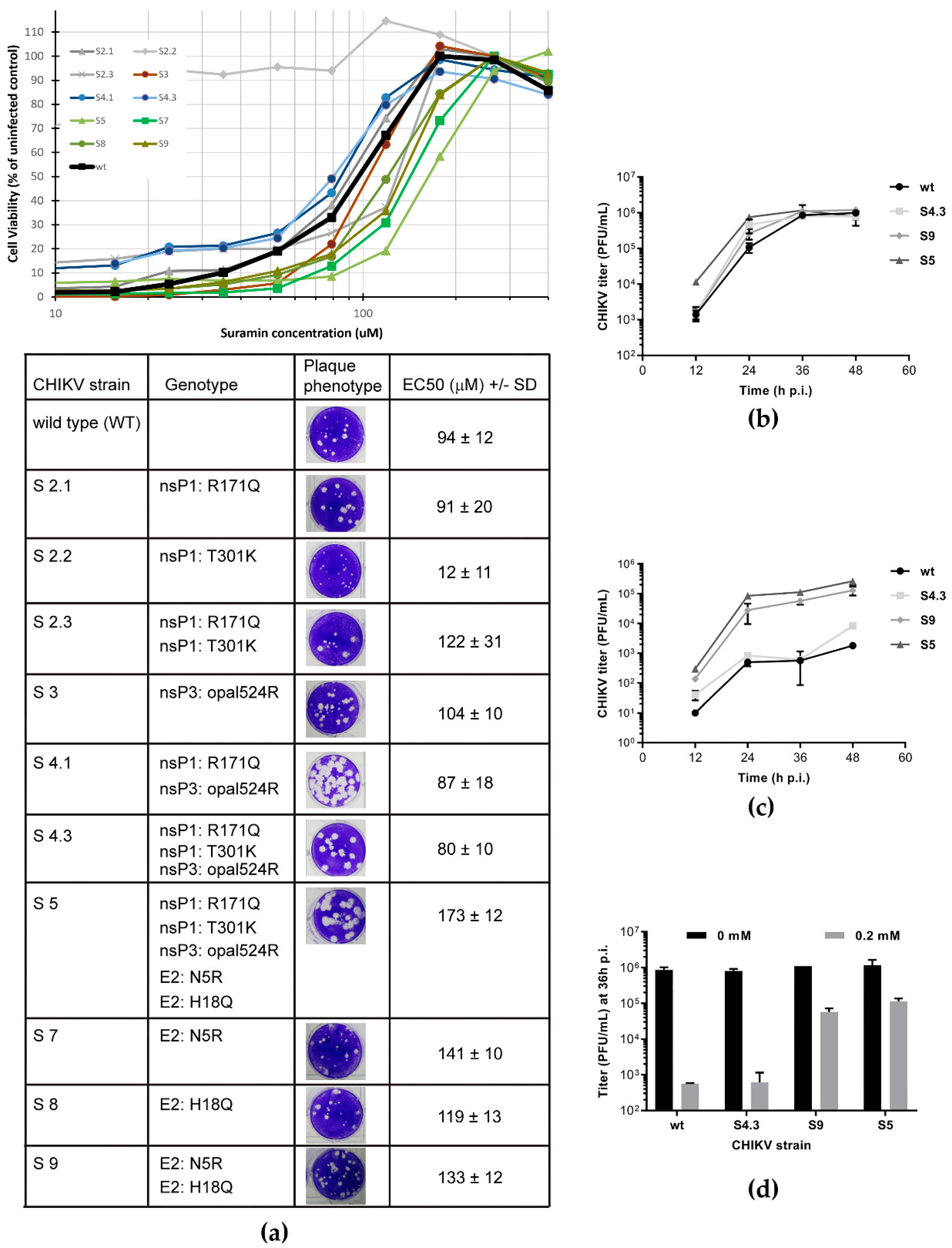
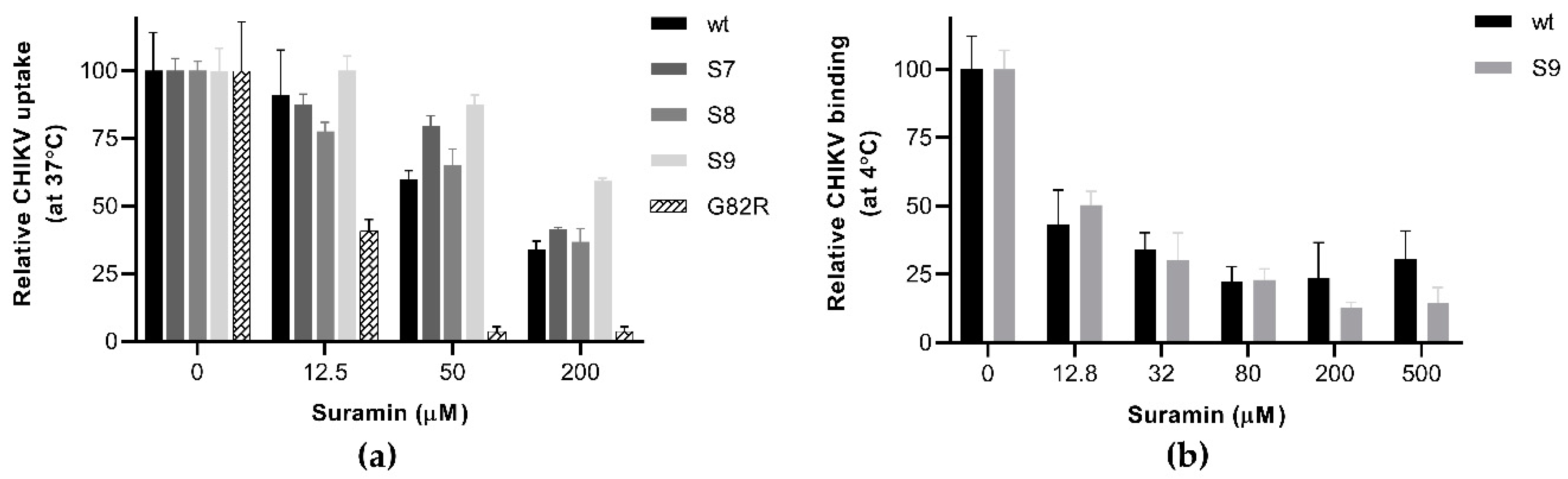
3.2. Suramin-Resistant CHIKV Variants Acquired Mutations in the Envelope Protein E2
3.3. The N5R and H18Q Mutations in E2 Enhance CHIKV Entry
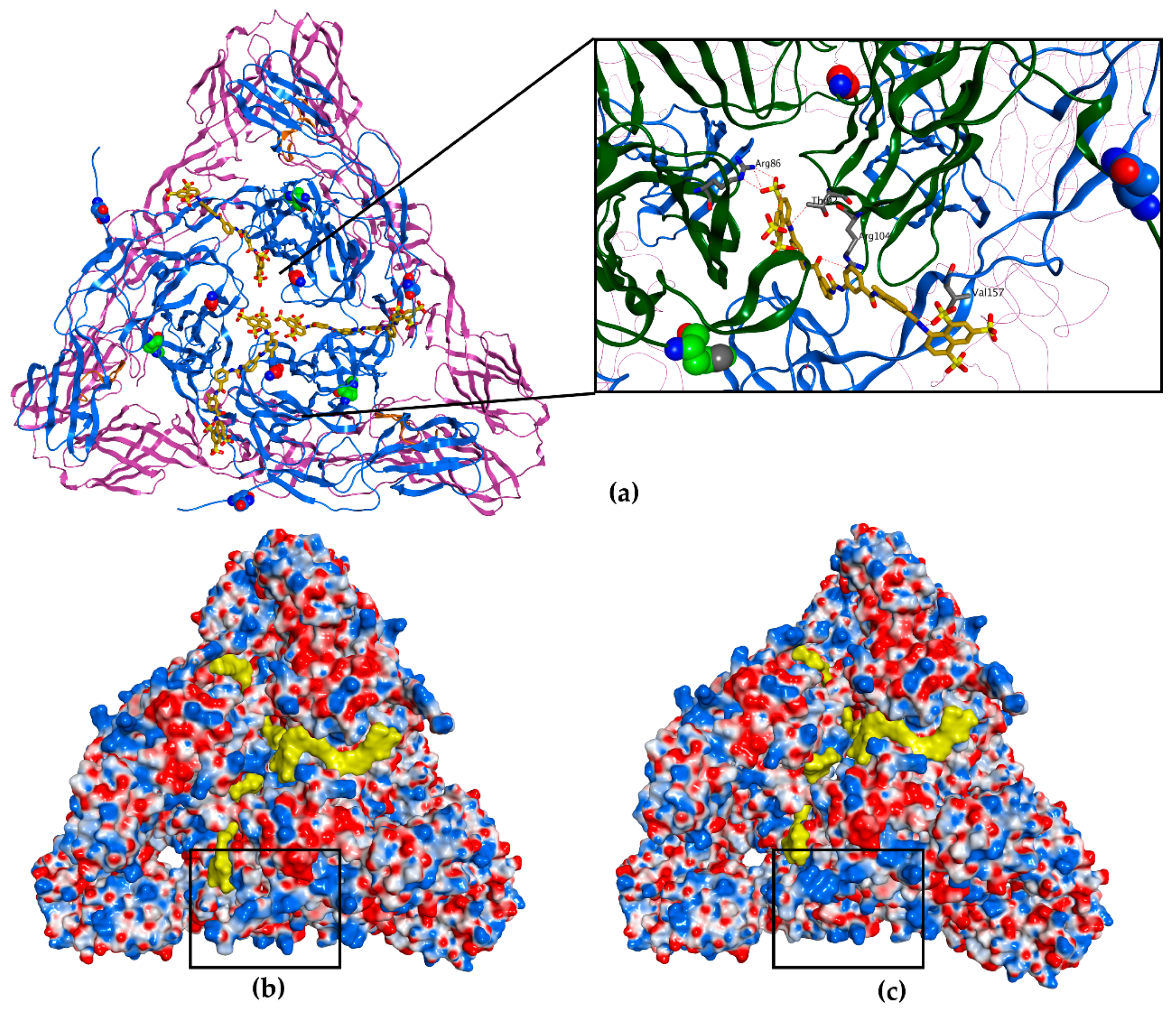
3.4. Molecular Modelling Predicts Suramin to Bind between Two Adjacent E2 Proteins in a Mature Spike
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Molecular Modeling—In Depth Description of Molecular Modelling

References
- Weaver, S.C.; Lecuit, M. Chikungunya virus and the global spread of a mosquito-borne disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.C.; Forrester, N.L. Chikungunya: Evolutionary history and recent epidemic spread. Antivir. Res. 2015, 120, 32–39. [Google Scholar] [CrossRef]
- Silva, L.A.; Dermody, T.S. Chikungunya virus: Epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Investig. 2017, 127, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Von Seidlein, L.; Kekulé, A.S.; Strickman, D. Novel Vector Control Approaches: The Future for Prevention of Zika Virus Transmission? PLoS Med. 2017, 14, e1002219. [Google Scholar] [CrossRef] [Green Version]
- Tharmarajah, K.; Mahalingam, S.; Zaid, A. Chikungunya: Vaccines and therapeutics. F1000Research 2017, 6, 2114. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.V.; Lopes, T.R.; De Oliveira-Filho, E.F.; Oliveira, R.A.; Durães-Carvalho, R.; Gil, L.H.V.G. Current status, challenges and perspectives in the development of vaccines against yellow fever, dengue, Zika and chikungunya viruses. Acta Trop. 2018, 182, 257–263. [Google Scholar] [CrossRef]
- Ching, K.-C.; Ng, L.F.P.; Chai, C.L. A compendium of small molecule direct-acting and host-targeting inhibitors as therapies against alphaviruses. J. Antimicrob. Chemother. 2017, 72, 2973–2989. [Google Scholar] [CrossRef]
- Albulescu, I.C.; Van Hoolwerff, M.; Wolters, L.A.; Bottaro, E.; Nastruzzi, C.; Yang, S.C.; Tsay, S.-C.; Hwu, J.R.; Snijder, E.J.; Van Hemert, M. Suramin inhibits chikungunya virus replication through multiple mechanisms. Antivir. Res. 2015, 121, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-J.; Wang, Y.-M.; Lu, J.-W.; Wu, T.-Y.; Lin, L.-I.; Kuo, S.-C.; Lin, C.-C. Suramin Inhibits Chikungunya Virus Entry and Transmission. PLoS ONE 2015, 10, e0133511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henss, L.; Beck, S.; Weidner, T.; Biedenkopf, N.; Sliva, K.; Weber, C.; Becker, S.; Schnierle, B.S. Suramin is a potent inhibitor of Chikungunya and Ebola virus cell entry. Virol. J. 2016, 13, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, S.-C.; Wang, Y.-M.; Ho, Y.-J.; Chang, T.-Y.; Lai, Z.-Z.; Tsui, P.-Y.; Wu, T.-Y.; Lin, C.-C. Suramin treatment reduces chikungunya pathogenesis in mice. Antivir. Res. 2016, 134, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, J.E.; Vaney, M.-C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F.A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.; Khomandiak, S.; Ashbrook, A.W.; Weller, R.; Heise, M.T.; Morrison, T.E.; Dermody, T.S. A Single-Amino-Acid Polymorphism in Chikungunya Virus E2 Glycoprotein Influences Glycosaminoglycan Utilization. J. Virol. 2013, 88, 2385–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weger-Lucarelli, J.; Aliota, M.T.; Wlodarchak, N.; Kamlangdee, A.; Swanson, R.; Osorio, J.E. Dissecting the Role of E2 Protein Domains in Alphavirus Pathogenicity. J. Virol. 2016, 90, 2418–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.M.; Long, F.; Edeling, M.A.; Lin, H.; Van Duijl-Richter, M.K.; Fong, R.H.; Kahle, K.M.; Smit, J.M.; Jin, J.; Simmons, G.; et al. Broadly Neutralizing Alphavirus Antibodies Bind an Epitope on E2 and Inhibit Entry and Egress. Cell 2015, 163, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Liss, N.M.; Chen, N.-H.; Liao, M.; Fox, J.M.; Shimak, R.M.; Fong, R.H.; Chafets, D.; Bakkour, S.; Keating, S.; et al. Neutralizing Monoclonal Antibodies Block Chikungunya Virus Entry and Release by Targeting an Epitope Critical to Viral Pathogenesis. Cell Rep. 2015, 13, 2553–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Tumkosit, U.; Nakamura, S.; Motooka, D.; Kishishita, N.; Priengprom, T.; Sa-Ngasang, A.; Kinoshita, T.; Takeda, N.; Maeda, Y. Genome-Wide Screening Uncovers the Significance of N-Sulfation of Heparan Sulfate as a Host Cell Factor for Chikungunya Virus Infection. J. Virol. 2017, 91, e00432-17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Van Duijl-Richter, M.K.S.; Hoornweg, T.E.; Rodenhuis-Zybert, I.; Smit, J.M. Early Events in Chikungunya Virus Infection—From Virus CellBinding to Membrane Fusion. Viruses 2015, 7, 3647–3674. [Google Scholar] [CrossRef]
- Li, L.; Jose, J.; Xiang, Y.; Kuhn, R.J.; Rossmann, M.G. Structural changes of envelope proteins during alphavirus fusion. Nature 2010, 468, 705–708. [Google Scholar] [CrossRef]
- Kuhn, R.J. Fields’ Virology (Chapter 22—Togaviride). In Fields’ Virology; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 629–650. [Google Scholar]
- Hoornweg, T.E.; Van Duijl-Richter, M.K.S.; Nuñez, N.V.A.; Albulescu, I.C.; Van Hemert, M.; Smit, J.M. Dynamics of Chikungunya Virus Cell Entry Unraveled by Single-Virus Tracking in Living Cells. J. Virol. 2016, 90, 4745–4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albulescu, I.C.; Kovacikova, K.; Tas, A.; Snijder, E.J.; Van Hemert, M. Suramin inhibits Zika virus replication by interfering with virus attachment and release of infectious particles. Antivir. Res. 2017, 143, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholte, F.E.M.; Taş, A.; Martina, B.E.E.; Cordioli, P.; Narayanan, K.; Makino, S.; Snijder, E.J.; Van Hemert, M. Characterization of Synthetic Chikungunya Viruses Based on the Consensus Sequence of Recent E1-226V Isolates. PLoS ONE 2013, 8, e71047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemical Computing Group. Molecular Operating Environment (MOE 2018.10). 2018. Available online: http://www.chemcomp.com (accessed on 22 January 2020).
- Schrödinger. Schrödinger Release 2017-1. 2017. Available online: https://www.schrodinger.com/citations/#Maestro (accessed on 22 January 2020).
- Tomoda, H.; Kishimoto, Y.; Lee, Y.C. Temperature effect on endocytosis and exocytosis by rabbit alveolar macrophages. J. Biol. Chem. 1989, 264, 15445–15450. [Google Scholar] [PubMed]
- Hawking, F. Suramin: With Special Reference to Onchocerciasis. Adv. Pharmacol. 1978, 15, 289–322. [Google Scholar]
- Ren, P.; Zheng, Y.; Wang, W.; Hong, L.; Delpeyroux, F.; Arenzana-Seisdedos, F.; Altmeyer, R. Suramin interacts with the positively charged region surrounding the 5-fold axis of the EV-A71 capsid and inhibits multiple enterovirus A. Sci. Rep. 2017, 7, 42902. [Google Scholar] [CrossRef] [Green Version]
- Yahi, N.; Sabatier, J.M.; Nickel, P.; Mabrouk, K.; Gonzalez-Scarano, F.; Fantini, J. Suramin inhibits binding of the V3 region of HIV-1 envelope glycoprotein gp120 to galactosylceramide, the receptor for HIV-1 gp120 on human colon epithelial cells. J. Biol. Chem. 1994, 269, 24349–24353. [Google Scholar]
- Coffey, L.L.; Vignuzzi, M. Host alternation of chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J. Virol. 2010, 85, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Ashbrook, A.W.; Burrack, K.S.; Silva, L.A.; Montgomery, S.A.; Heise, M.T.; Morrison, T.E.; Dermody, T.S. Residue 82 of the Chikungunya Virus E2 Attachment Protein Modulates Viral Dissemination and Arthritis in Mice. J. Virol. 2014, 88, 12180–12192. [Google Scholar] [CrossRef] [Green Version]
- Moyen, N.; Thiberville, S.-D.; Pastorino, B.; Nougairède, A.; Thirion, L.; Mombouli, J.-V.; Dimi, Y.; Leparc-Goffart, I.; Capobianchi, M.R.; Lepfoundzou, A.D.; et al. First Reported Chikungunya Fever Outbreak in the Republic of Congo, 2011. PLoS ONE 2014, 9, e115938. [Google Scholar] [CrossRef] [Green Version]
- Mounce, B.C.; Cesaro, T.; Vlajnić, L.; Vidiņa, A.; Vallet, T.; Weger-Lucarelli, J.; Passoni, G.; Stapleford, K.A.; Levraud, J.-P.; Vignuzzi, M. Chikungunya Virus Overcomes Polyamine Depletion by Mutation of nsP1 and the Opal Stop Codon To Confer Enhanced Replication and Fitness. J. Virol. 2017, 91, e00344-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef] [PubMed]
- Myles, K.M.; Kelly, C.L.H.; Ledermann, J.P.; Powers, A.M. Effects of an Opal Termination Codon Preceding the nsP4 Gene Sequence in the O’Nyong-Nyong Virus Genome on Anopheles gambiae Infectivity. J. Virol. 2006, 80, 4992–4997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.E.; Long, K.M.; Whitmore, A.C.; Sanders, W.; Thurlow, L.R.; Brown, J.A.; Morrison, C.R.; Vincent, H.; Peck, K.; Browning, C.; et al. Disruption of the Opal Stop Codon Attenuates Chikungunya Virus-Induced Arthritis and Pathology. MBio 2017, 8, e01456-17. [Google Scholar] [CrossRef] [Green Version]
- Kovacikova, K.; Morren, B.M.; Tas, A.; Albulescu, I.C.; van Rijswijk, R.; Jarhad, D.B.; Jeong, L.S. 6′-beta-Fluoro-homoaristeromycin and 6′-fluoro-homoneplanocin A are potent inhibitors of chikungunya virus replication through their direct effect on the viral non-structural protein 1. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef] [Green Version]
- Levitt, N.H.; Ramsburg, H.H.; Hasty, S.E.; Repik, P.M.; Cole, F.E.; Lupton, H.W. Development of an attenuated strain of chikungunya virus for use in vaccine production. Vaccine 1986, 4, 157–162. [Google Scholar] [CrossRef]
- Gorchakov, R.; Wang, E.; Leal, G.; Forrester, N.L.; Plante, K.; Rossi, S.L.; Borland, E.M. Attenuation of Chikungunya virus vaccine strain 181/clone 25 is determined by two amino acid substitutions in the E2 envelope glycoprotein. J. Virol. 2012, 86, 6084–6096. [Google Scholar] [CrossRef] [Green Version]
- Gardner, C.L.; Hritz, J.; Sun, C.; VanLandingham, D.L.; Song, T.Y.; Ghedin, E.; Higgs, S.; Klimstra, W.B.; Ryman, K.D. Deliberate Attenuation of Chikungunya Virus by Adaptation to Heparan Sulfate-Dependent Infectivity: A Model for Rational Arboviral Vaccine Design. PLoS Negl. Trop. Dis. 2014, 8, e2719. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Xiang, Y.; Akahata, W.; Holdaway, H.; Pal, P.; Zhang, X.; Diamond, M.S.; Nabel, G.J.; Rossmann, M.G. Structural analyses at pseudo atomic resolution of Chikungunya virus and antibodies show mechanisms of neutralization. ELife 2013, 2, e00435. [Google Scholar] [CrossRef]
- Weber, C.; Berberich, E.; Von Rhein, C.; Henß, L.; Hildt, E.; Schnierle, B.S. Identification of Functional Determinants in the Chikungunya Virus E2 Protein. PLoS Negl. Trop. Dis. 2017, 11, e0005318. [Google Scholar] [CrossRef]
- World Health Organization. Control and Surveillance of Human African Trypanosomiasis; World Health Organization Technical Report Series; 2013; Volume 984, pp. 1–237. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24552089 (accessed on 22 January 2020).
- Van Genderen, P.J.J. Side Effects of Drugs, Annual 28; Elsevier B.V.: Amsterdam, The Netherlands, 2005; pp. 346–355. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation in | At P5 (150 μM Suramin) | At P7 (300 μM Suramin) | ||
|---|---|---|---|---|
| nt Substitution | AA Substitution | nt Substitution | AA Substitution | |
| nsP1 | G588A | R171Q | G588A A979G G980A | R171Q T301K G302R |
| nsP3 | U5645C | opal524R | ΔU5645-A5650 | Δopal524L525 |
| E2 | A8554G | N5R | A8554G C8595A | N5R H18Q |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albulescu, I.C.; White-Scholten, L.; Tas, A.; Hoornweg, T.E.; Ferla, S.; Kovacikova, K.; Smit, J.M.; Brancale, A.; Snijder, E.J.; van Hemert, M.J. Suramin Inhibits Chikungunya Virus Replication by Interacting with Virions and Blocking the Early Steps of Infection. Viruses 2020, 12, 314. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030314
Albulescu IC, White-Scholten L, Tas A, Hoornweg TE, Ferla S, Kovacikova K, Smit JM, Brancale A, Snijder EJ, van Hemert MJ. Suramin Inhibits Chikungunya Virus Replication by Interacting with Virions and Blocking the Early Steps of Infection. Viruses. 2020; 12(3):314. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030314
Chicago/Turabian StyleAlbulescu, Irina C., Leonie White-Scholten, Ali Tas, Tabitha E. Hoornweg, Salvatore Ferla, Kristina Kovacikova, Jolanda M. Smit, Andrea Brancale, Eric J. Snijder, and Martijn J. van Hemert. 2020. "Suramin Inhibits Chikungunya Virus Replication by Interacting with Virions and Blocking the Early Steps of Infection" Viruses 12, no. 3: 314. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030314