Genetic Adaptations, Biases, and Evolutionary Analysis of Canine Distemper Virus Asia-4 Lineage in a Fatal Outbreak of Wild-Caught Civets in Thailand

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Postmortem Examination
2.3. General Virologic Testing
2.4. Full-Length Genomic Characterization of the Civet CDV
2.5. Genome, Phylogeny, and Recombination Analyses of the Full-Length Civet CDV Genome
2.6. Evolutionary Analysis of the CDV H Gene
2.7. Codon Usage Analysis of the Asian CDV H Gene
2.7.1. Sequence Information and Recombination Sequence Removal
2.7.2. Nucleotide and Codon Usage Composition of CDVs
2.7.3. Relative Synonymous Codon Usage (RSCU) of Asian CDVs and between CDV-Susceptible Host Species
2.8. Detection of CDV in Civet Tissues by IHC Analysis
2.9. Ethics Statement
2.10. Data Availability Statement
3. Results
3.1. Animal Habitats and Potential History
3.2. Pathological and General Virologic Findings
3.3. CDV Localization in Tissues
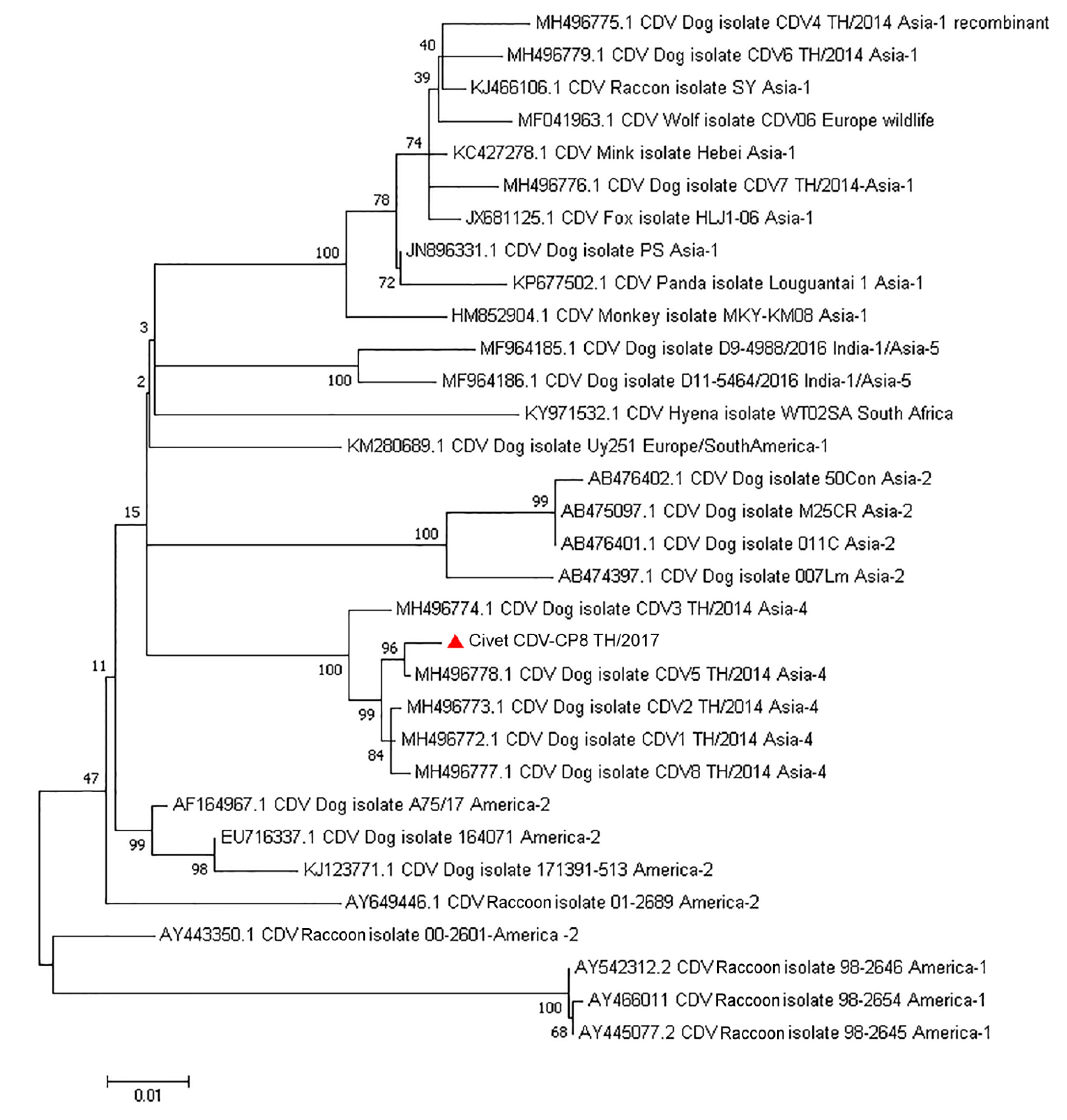
3.4. Genetic Characterization and Phylogenetic, Recombination, and Evolutionary Analyses of the Complete Genome of Civet CDV
3.5. Frequent Nucleotide Composition and the RSCU of the Asian CDV H Gene
3.6. Relative Synonymous Codon Usage between Asian CDV and Selective Hosts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Uhl, E.W.; Kelderhouse, C.; Buikstra, J.; Blick, J.P.; Bolon, B.; Hogan, R.J. New world origin of canine distemper: Interdisciplinary insights. Int. J. Paleopathol. 2019, 24, 266–278. [Google Scholar] [CrossRef]
- Duque-Valencia, J.; Sarute, N.; Olarte-Castillo, X.A.; Ruiz-Saenz, J. Evolution and Interspecies Transmission of Canine Distemper Virus-An Outlook of the Diverse Evolutionary Landscapes of a Multi-Host Virus. Viruses 2019, 11, 582. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Shrimpton, S.B.; Russell, S.E. Nucleotide sequence of the entire protein coding region of canine distemper virus polymerase-associated (P) protein mRNA. Virus Res. 1985, 3, 367–372. [Google Scholar] [CrossRef]
- Bellini, W.J.; Englund, G.; Richardson, C.D.; Rozenblatt, S.; Lazzarini, R.A. Matrix genes of measles virus and canine distemper virus: Cloning, nucleotide sequences, and deduced amino acid sequences. J. Virol. 1986, 58, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, M.S.; Husar, W.; Cook, S.D.; Dowling, P.C.; Udem, S.A. Canine distemper terminal and intergenic non-protein coding nucleotide sequences: Completion of the entire CDV genome sequence. Virology 1993, 193, 66–72. [Google Scholar] [CrossRef]
- Loots, A.K.; Mitchell, E.; Dalton, D.L.; Kotze, A.; Venter, E.H. Advances in canine distemper virus pathogenesis research: A wildlife perspective. J. Gen. Virol. 2017, 98, 311–321. [Google Scholar] [CrossRef]
- Beineke, A.; Baumgärtner, W.; Wohlsein, P. Cross-species transmission of canine distemper virus-an update. One Health (Amsterdam, Netherlands) 2015, 1, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Munson, L.; Terio, K.A.; Kock, R.; Mlengeya, T.; Roelke, M.E.; Dubovi, E.; Summers, B.; Sinclair, A.R.E.; Packer, C. Climate extremes promote fatal co-infections during canine distemper epidemics in African lions. PLoS ONE 2008, 3, e2545. [Google Scholar] [CrossRef]
- Sarkar, J.; Balamurugan, V.; Sen, A.; Saravanan, P.; Sahay, B.; Rajak, K.K.; Rasool, T.J.; Bhanuprakash, V.; Singh, R.K. Sequence analysis of morbillivirus CD150 receptor-Signaling Lymphocyte Activation Molecule (SLAM) of different animal species. Virus Genes 2009, 39, 335–341. [Google Scholar] [CrossRef]
- Tatsuo, H.; Yanagi, Y. The morbillivirus receptor SLAM (CD150). Microbiol. Immunol. 2002, 46, 135–142. [Google Scholar] [CrossRef]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.-T.; Sisson, G.; Tsao, M.-S.; Richardson, C.D. Tumor Cell Marker PVRL4 (Nectin 4) Is an Epithelial Cell Receptor for Measles Virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef]
- Pratakpiriya, W.; Seki, F.; Otsuki, N.; Sakai, K.; Fukuhara, H.; Katamoto, H.; Hirai, T.; Maenaka, K.; Techangamsuwan, S.; Lan, N.T.; et al. Nectin4 is an epithelial cell receptor for canine distemper virus and involved in neurovirulence. J. Virol. 2012, 86, 10207–10210. [Google Scholar] [CrossRef] [Green Version]
- Pratakpiriya, W.; Ping Teh, A.P.; Radtanakatikanon, A.; Pirarat, N.; Thi Lan, N.; Takeda, M.; Techangamsuwan, S.; Yamaguchi, R. Expression of canine distemper virus receptor nectin-4 in the central nervous system of dogs. Sci. Rep. 2017, 7, 349. [Google Scholar] [CrossRef]
- Bringolf, F.; Herren, M.; Wyss, M.; Vidondo, B.; Langedijk, J.P.; Zurbriggen, A.; Plattet, P. Dimerization Efficiency of Canine Distemper Virus Matrix Protein Regulates Membrane-Budding Activity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Russell, C.J.; Jardetzky, T.S.; Lamb, R.A. Membrane fusion machines of paramyxoviruses: Capture of intermediates of fusion. EMBO J. 2001, 20, 4024–4034. [Google Scholar] [CrossRef] [Green Version]
- Winters, K.A.; Mathes, L.E.; Krakowka, S.; Olsen, R.G. Immunoglobulin class response to canine distemper virus in gnotobiotic dogs. Vet. Immunol. Immunopathol. 1983, 5, 209–215. [Google Scholar] [CrossRef]
- Krakowka, S.; Cockerell, G.; Koestner, A. Effects of canine distemper virus infection on lymphoid function in vitro and in vivo. Infect. Immun. 1975, 11, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Leisewitz, A.L.; Carter, A.; van Vuuren, M.; van Blerk, L. Canine distemper infections, with special reference to South Africa, with a review of the literature. J. S. Afr. Vet. Assoc. 2001, 72, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Pomeroy, L.W.; Bjornstad, O.N.; Holmes, E.C. The evolutionary and epidemiological dynamics of the paramyxoviridae. J. Mol. Evol. 2008, 66, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Panzera, Y.; Sarute, N.; Iraola, G.; Hernandez, M.; Perez, R. Molecular phylogeography of canine distemper virus: Geographic origin and global spreading. Mol. Phylogenetics Evo.l 2015, 92, 147–154. [Google Scholar] [CrossRef]
- Martinez-Gutierrez, M.; Ruiz-Saenz, J. Diversity of susceptible hosts in canine distemper virus infection: A systematic review and data synthesis. BMC Vet. Res. 2016, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Ke, G.-M.; Ho, C.-H.; Chiang, M.-J.; Sanno-Duanda, B.; Chung, C.-S.; Lin, M.-Y.; Shi, Y.-Y.; Yang, M.-H.; Tyan, Y.-C.; Liao, P.-C.; et al. Phylodynamic analysis of the canine distemper virus hemagglutinin gene. BMC Vet. Res. 2015, 11, 164. [Google Scholar] [CrossRef] [Green Version]
- Piewbang, C.; Radtanakatikanon, A.; Puenpa, J.; Poovorawan, Y.; Techangamsuwan, S. Genetic and evolutionary analysis of a new Asia-4 lineage and naturally recombinant canine distemper virus strains from Thailand. Sci. Rep. 2019, 9, 3198. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, M.; Rajak, K.K.; Chakravarti, S.; Yadav, A.K.; Kumar, A.; Gupta, V.; Chander, V.; Mathesh, K.; Chandramohan, S.; Sharma, A.K.; et al. Phylogenetic analysis of haemagglutinin gene deciphering a new genetically distinct lineage of canine distemper virus circulating among domestic dogs in India. Transbound Emerg. Dis. 2019, 66, 1252–1267. [Google Scholar] [CrossRef]
- Jo, W.K.; Peters, M.; Kydyrmanov, A.; van de Bildt, M.W.G.; Kuiken, T.; Osterhaus, A.; Ludlow, M. The Canine Morbillivirus Strain Associated with An Epizootic in Caspian Seals Provides New Insights into the Evolutionary History of this Virus. Viruses 2019, 11, 894. [Google Scholar] [CrossRef] [Green Version]
- Techangamsuwan, S.; Banlunara, W.; Radtanakatikanon, A.; Sommanustweechai, A.; Siriaroonrat, B.; Lombardini, E.D.; Rungsipipat, A. Pathologic and Molecular Virologic Characterization of a Canine Distemper Outbreak in Farmed Civets. Vet. Pathol. 2015, 52, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Nakano, H.; Kameo, Y.; Sato, H.; Mochizuki, M.; Yokoyama, M.; Uni, S.; Shibasaki, T.; Maeda, K. Detection of Antibody to Canine Distemper Virus in Wild Raccoons (Procyon lotor) in Japan. J. Vet. Med. Sci. 2009, 71, 1661–1663. [Google Scholar] [CrossRef] [Green Version]
- Bush, M.; Montali, R.J.; Brownstein, D.; James, A.E., Jr.; Appel, M.J. Vaccine-induced canine distemper in a lesser panda. J. Am. Vet. Med. Assoc. 1976, 169, 959–960. [Google Scholar]
- Di Francesco, C.E.; Gentile, L.; Di Pirro, V.; Ladiana, L.; Tagliabue, S.; Marsilio, F. Serologic evidence for selected infectious diseases in Marsican brown bears (Ursus arctos marsicanus) in Italy (2004-09). J. Wildl. Dis. 2015, 51, 209–213. [Google Scholar] [CrossRef]
- Perpiñán, D.; Ramis, A.; Tomás, A.; Carpintero, E.; Bargalló, F. Outbreak of canine distemper in domestic ferrets (Mustela putorius furo). Vet. Rec. 2008, 163, 246–250. [Google Scholar] [CrossRef]
- Terio, K.A.; Craft, M.E. Canine Distemper Virus (CDV) in Another Big Cat: Should CDV Be Renamed Carnivore Distemper Virus? mBio 2013, 4, e00702–e00713. [Google Scholar] [CrossRef] [Green Version]
- Berentsen, A.R.; Dunbar, M.R.; Becker, M.S.; M’Soka, J.; Droge, E.; Sakuya, N.M.; Matandiko, W.; McRobb, R.; Hanlon, C.A. Rabies, Canine Distemper, and Canine Parvovirus Exposure in Large Carnivore Communities from Two Zambian Ecosystems. Vector Borne Zoonotic Dis. 2013, 13, 643–649. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Ochikubo, F.; Matsubara, Y.; Tsuruoka, H.; Ishii, M.; Shirota, K.; Nomura, Y.; Sugiyama, M.; Yamanouchi, K. Natural infection with canine distemper virus in a Japanese monkey (Macaca fuscata). Vet. Microbiol. 1989, 20, 193–205. [Google Scholar] [CrossRef]
- Origgi, F.C.; Sattler, U.; Pilo, P.; Waldvogel, A.S. Fatal Combined Infection With Canine Distemper Virus and Orthopoxvirus in a Group of Asian Marmots (Marmota caudata). Vet. Pathol. 2013, 50, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Lunardi, M.; Darold, G.M.; Amude, A.M.; Headley, S.A.; Sonne, L.; Yamauchi, K.C.I.; Boabaid, F.M.; Alfieri, A.F.; Alfieri, A.A. Canine distemper virus active infection in order Pilosa, family Myrmecophagidae, species Tamandua tetradactyla. Vet. Microbiol. 2018, 220, 7–11. [Google Scholar] [CrossRef]
- Rudd, P.A.; Cattaneo, R.; von Messling, V. Canine distemper virus uses both the anterograde and the hematogenous pathway for neuroinvasion. J. Virol. 2006, 80, 9361–9370. [Google Scholar] [CrossRef] [Green Version]
- VanDevanter, D.R.; Warrener, P.; Bennett, L.; Schultz, E.R.; Coulter, S.; Garber, R.L.; Rose, T.M. Detection and analysis of diverse herpesviral species by consensus primer PCR. J. Clin. Microbiol. 1996, 34, 1666–1671. [Google Scholar] [CrossRef] [Green Version]
- Van Boheemen, S.; Bestebroer, T.M.; Verhagen, J.H.; Osterhaus, A.D.M.E.; Pas, S.D.; Herfst, S.; Fouchier, R.A.M. A family-wide RT-PCR assay for detection of paramyxoviruses and application to a large-scale surveillance study. PLoS ONE 2012, 7, e34961. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.; Chern, S.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, M.; Horiuchi, M.; Hiragi, H.; San Gabriel, M.C.; Yasuda, N.; Uno, T. Isolation of canine parvovirus from a cat manifesting clinical signs of feline panleukopenia. J. Clin. Microbiol. 1996, 34, 2101–2105. [Google Scholar] [CrossRef] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. New Engl J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Piewbang, C.; Techangamsuwan, S. Phylogenetic evidence of a novel lineage of canine pneumovirus and a naturally recombinant strain isolated from dogs with respiratory illness in Thailand. BMC Vet. Res. 2019, 15, 300. [Google Scholar] [CrossRef]
- Piewbang, C.; Rungsipipat, A.; Poovorawan, Y.; Techangamsuwan, S. Development and application of multiplex PCR assays for detection of virus-induced respiratory disease complex in dogs. J. Vet. Med. Sci. 2017, 78, 1847–1854. [Google Scholar] [CrossRef] [Green Version]
- Piewbang, C.; Jo, W.K.; Puff, C.; van der Vries, E.; Kesdangsakonwut, S.; Rungsipipat, A.; Kruppa, J.; Jung, K.; Baumgärtner, W.; Techangamsuwan, S.; et al. Novel canine circovirus strains from Thailand: Evidence for genetic recombination. Sci. Rep. 2018, 8, 7524. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhang, W.; Wang, R.; Xing, G.; Wang, S.; Ji, X.; Wang, N.; Su, S.; Zhou, J. Genetic Analysis and Evolutionary Changes of the Torque teno sus Virus. Int. J. Mol. Sci. 2019, 20, 2881. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Wang, R.; Zhang, C.; Wang, S.; He, W.; Zhang, J.; Liu, J.; Cai, Y.; Zhou, J.; Su, S. Genetic and evolutionary analysis of emerging H3N2 canine influenza virus. Emerg. Microbes Infect. 2018, 7, 73. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Gan, H.; Liang, X. Analysis of Synonymous Codon Usage Bias in Potato Virus M and Its Adaption to Hosts. Viruses 2019, 11, 752. [Google Scholar] [CrossRef] [Green Version]
- López-Peña, M.; Vázquez, S.; Alemañ, N.; López-Beceiro, A.; Muñoz, F.; Pereira, J.L.; Nieto, J.M. Canine Distemper in a Genet (Gennetta gennetta), Associated with Endogenous Lipid Pneumonia. J. Comp. Pathol. 2001, 124, 207–211. [Google Scholar] [CrossRef]
- Takayama, I.; Kubo, M.; Takenaka, A.; Fujita, K.; Sugiyama, T.; Arai, T.; Yoneda, M.; Sato, H.; Yanai, T.; Kai, C. Pathological and phylogenetic features of prevalent canine distemper viruses in wild masked palm civets in Japan. Comp. Immunol. Microbiol. Infect. Dis. 2009, 32, 539–549. [Google Scholar] [CrossRef]
- Radtanakatikanon, A.; Keawcharoen, J.; Charoenvisal, N.T.; Poovorawan, Y.; Prompetchara, E.; Yamaguchi, R.; Techangamsuwan, S. Genotypic lineages and restriction fragment length polymorphism of canine distemper virus isolates in Thailand. Vet. Microbiol. 2013, 166, 76–83. [Google Scholar] [CrossRef]
- Bi, Z.; Wang, Y.; Wang, X.; Xia, X. Phylogenetic analysis of canine distemper virus in domestic dogs in Nanjing, China. Arch. Virol. 2015, 160, 523–527. [Google Scholar] [CrossRef]
- Gilbert, M. Understanding and managing canine distemper virus as a disease threat to Amur tigers. Ph.D. Thesis, University of Glasgow, Glasgow, UK, 2016. [Google Scholar]
- Nikolin, V.M.; Olarte-Castillo, X.A.; Osterrieder, N.; Hofer, H.; Dubovi, E.; Mazzoni, C.J.; Brunner, E.; Goller, K.V.; Fyumagwa, R.D.; Moehlman, P.D.; et al. Canine distemper virus in the Serengeti ecosystem: Molecular adaptation to different carnivore species. Mol. Ecol. 2017, 26, 2111–2130. [Google Scholar] [CrossRef]
- Kapil, S.; Yeary, T.J. Canine distemper spillover in domestic dogs from urban wildlife. Vet. Clin. North. Am. Small Anim. Pract. 2011, 41, 1069–1086. [Google Scholar] [CrossRef]
- Goñi, N.; Iriarte, A.; Comas, V.; Soñora, M.; Moreno, P.; Moratorio, G.; Musto, H.; Cristina, J. Pandemic influenza A virus codon usage revisited: Biases, adaptation and implications for vaccine strain development. Virol. J. 2012, 9, 263. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S.; Acevedo, A.; Cooper, S.B.; Andino, R. Codon usage determines the mutational robustness, evolutionary capacity, and virulence of an RNA virus. Cell Host Microbe 2012, 12, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, N.; Freire, C.C.d.M.; Zanotto, P.M.d.A. Does adaptation to vertebrate codon usage relate to flavivirus emergence potential? PLoS ONE 2018, 13, e0191652. [Google Scholar] [CrossRef] [Green Version]
- Freire, C.C.d.M.; Palmisano, G.; Braconi, C.T.; Cugola, F.R.; Russo, F.B.; Beltrão-Braga, P.C.; Iamarino, A.; Lima Neto, D.F.d.; Sall, A.A.; Rosa-Fernandes, L.; et al. NS1 codon usage adaptation to humans in pandemic Zika virus. Memorias do Instituto Oswaldo Cruz 2018, 113, e170385. [Google Scholar] [CrossRef]
- Behura, S.K.; Severson, D.W. Codon usage bias: Causative factors, quantification methods and genome-wide patterns: With emphasis on insect genomes. Biol. Rev. 2013, 88, 49–61. [Google Scholar] [CrossRef]
- Tatsuo, H.; Ono, N.; Yanagi, Y. Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J. Virol. 2001, 75, 5842–5850. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Codon | CDV Strain | Potential Host | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Asia-1 | Asia-2 | Asia-3 | Asia-4 | Asia-5 | C. familiaris | P. hermaphroditus | ||
| Phe (F) | UUU | 0.98 | 1.04 | 0.98 | 0.92 | 0.85 | 0.98 | 1.09 | 1.06 |
| UUC | 1.02 | 0.96 | 1.02 | 1.08 | 1.15 | 1.02 | 0.91 | 0.94 | |
| Leu (L) | UUA | 0.98 | 0.98 | 1.2 | 1.22 | 0.9 | 0.94 | 1.32 | 1.78 |
| UUG | 1.69 | 1.69 | 1.37 | 1.13 | 1.87 | 1.64 | 0.51 | 0.4 | |
| CUU | 0.87 | 0.81 | 0.88 | 0.94 | 0.89 | 0.89 | 1.22 | 1.04 | |
| CUC | 0.65 | 0.69 | 0.67 | 0.56 | 0.47 | 0.66 | 1.01 | 0.74 | |
| CUA | 0.91 | 0.97 | 0.79 | 0.84 | 1.02 | 0.94 | 1.42 | 1.66 | |
| CUG | 0.91 | 0.86 | 1.09 | 1.31 | 0.85 | 0.94 | 0.51 | 0.37 | |
| Ile (I) | AUU | 1.03 | 1.01 | 1.06 | 1.08 | 1.12 | 0.98 | 1.05 | 1.17 |
| AUC | 0.88 | 0.8 | 0.94 | 0.96 | 0.86 | 0.91 | 0.94 | 0.65 | |
| AUA | 1.09 | 1.19 | 0.99 | 0.96 | 1.02 | 1.11 | 1.01 | 1.19 | |
| Val (V) | GUU | 0.71 | 0.64 | 0.44 | 0.27 | 0.65 | 0.63 | 1.12 | 1.17 |
| GUC | 1.02 | 0.99 | 1.05 | 0.98 | 1.1 | 1.08 | 0.57 | 0.62 | |
| GUA | 0.98 | 0.99 | 1.1 | 1.24 | 1.03 | 1.03 | 1.67 | 1.81 | |
| GUG | 1.29 | 1.37 | 1.41 | 1.51 | 1.23 | 1.26 | 0.64 | 0.4 | |
| Ser (S) | UCU | 0.83 | 0.93 | 0.75 | 0.58 | 0.72 | 0.94 | 1.35 | 1.61 |
| UCC | 1.36 | 1.47 | 1.49 | 1.58 | 1.4 | 1.18 | 1.04 | 1.1 | |
| UCA | 2.02 | 2.03 | 1.85 | 1.85 | 2.02 | 2 | 1.27 | 1.37 | |
| UCG | 0.37 | 0.27 | 0.6 | 0.58 | 0.42 | 0.35 | 0.39 | 0.28 | |
| AGU | 0.7 | 0.49 | 0.81 | 0.81 | 0.74 | 0.88 | 0.91 | 0.72 | |
| AGC | 0.72 | 0.81 | 0.51 | 0.58 | 0.7 | 0.65 | 1.05 | 0.92 | |
| Pro (P) | CCU | 1.39 | 1.59 | 1.21 | 1.44 | 1.31 | 1.13 | 1.41 | 1.3 |
| CCC | 0.81 | 0.77 | 0.85 | 0.85 | 0.87 | 0.81 | 1.24 | 1.05 | |
| CCA | 1.23 | 0.92 | 1.24 | 1.05 | 1.33 | 1.63 | 0.92 | 1.28 | |
| CCG | 0.58 | 0.71 | 0.7 | 0.66 | 0.48 | 0.44 | 0.43 | 0.37 | |
| Thr (T) | ACU | 1.06 | 1.18 | 0.89 | 0.86 | 1.12 | 0.9 | 1.35 | 1.18 |
| ACC | 0.84 | 0.79 | 0.99 | 0.97 | 0.73 | 0.9 | 1.06 | 0.91 | |
| ACA | 1.74 | 1.82 | 1.92 | 1.84 | 1.63 | 1.7 | 1.16 | 1.54 | |
| ACG | 0.36 | 0.21 | 0.2 | 0.32 | 0.51 | 0.5 | 0.43 | 0.38 | |
| Ala (A) | GCU | 1.22 | 1.14 | 1.17 | 1.28 | 1.51 | 1.25 | 1.07 | 1.16 |
| GCC | 0.83 | 0.99 | 0.6 | 0.64 | 0.67 | 0.86 | 1.27 | 1.36 | |
| GCA | 1.68 | 1.68 | 1.92 | 1.6 | 1.65 | 1.49 | 1.18 | 1.32 | |
| GCG | 0.27 | 0.19 | 0.3 | 0.48 | 0.17 | 0.39 | 0.48 | 0.16 | |
| Tyr (Y) | UAU | 1.4 | 1.49 | 1.18 | 1.16 | 1.48 | 1.53 | 1.15 | 1.17 |
| UAC | 0.6 | 0.51 | 0.82 | 0.84 | 0.52 | 0.47 | 0.85 | 0.83 | |
| His (H) * | CAU | 1.02 | 1 | 1.22 | 0.94 | 1.05 | 0.96 | 1.2 | 1.03 |
| CAC | 0.98 | 1 | 0.78 | 1.06 | 0.95 | 1.04 | 0.8 | 0.97 | |
| Gln (Q) | CAA | 1.53 | 1.51 | 1.43 | 0.9 | 1.54 | 1.47 | 1.25 | 1.52 |
| CAG | 0.47 | 0.49 | 0.57 | 1.1 | 0.46 | 0.53 | 0.75 | 0.48 | |
| Asn (N) | AAU | 1.14 | 1.01 | 1.35 | 1.43 | 1.1 | 1.14 | 1.18 | 1.28 |
| AAC | 0.86 | 0.99 | 0.65 | 0.57 | 0.9 | 0.86 | 0.82 | 0.72 | |
| Lys (K) | AAA | 1.28 | 1.28 | 1.31 | 0.84 | 1.32 | 1.07 | 1.37 | 1.42 |
| AAG | 0.72 | 0.72 | 0.69 | 1.16 | 0.68 | 0.93 | 0.63 | 0.58 | |
| Asp (D) | GAU | 1.22 | 1.19 | 1.24 | 1.18 | 1.15 | 1.24 | 1.13 | 1.15 |
| GAC | 0.78 | 0.81 | 0.76 | 0.82 | 0.85 | 0.76 | 0.87 | 0.85 | |
| Glu (E) | GAA | 0.75 | 0.68 | 0.85 | 1.11 | 0.79 | 0.87 | 1.17 | 1.35 |
| GAG | 1.25 | 1.32 | 1.15 | 0.89 | 1.21 | 1.13 | 0.83 | 0.65 | |
| Cys (C) | UGU | 1.45 | 1.5 | 1.46 | 1.43 | 1.5 | 1.13 | 0.89 | 0.92 |
| UGC | 0.55 | 0.5 | 0.54 | 0.57 | 0.5 | 0.87 | 1.11 | 1.08 | |
| Arg (R) | CGU | 0.58 | 0.57 | 0.57 | 0.46 | 0.6 | 0.59 | 1.17 | 0.78 |
| CGC | 0.22 | 0.2 | 0.33 | 0.46 | 0.2 | 0.3 | 0.92 | 0.69 | |
| CGA | 0.56 | 0.64 | 0.33 | 1.08 | 0.6 | 0.59 | 0.71 | 1.18 | |
| CGG | 1.02 | 1.14 | 0.94 | 0.77 | 1 | 1.08 | 0.48 | 0.46 | |
| AGA | 2.69 | 2.77 | 2.79 | 2 | 2.56 | 2.75 | 1.29 | 1.47 | |
| AGG | 0.92 | 0.69 | 1.04 | 1.23 | 1.04 | 0.69 | 1.42 | 1.41 | |
| Gly (G) | GGU | 0.94 | 0.93 | 0.92 | 1.1 | 0.89 | 0.94 | 1.02 | 1.07 |
| GGC | 0.42 | 0.29 | 0.55 | 0.28 | 0.45 | 0.41 | 1.05 | 0.73 | |
| GGA | 1.71 | 1.7 | 1.87 | 1.79 | 1.77 | 1.88 | 1.27 | 1.68 | |
| GGG | 0.92 | 1.07 | 0.66 | 0.83 | 0.89 | 0.76 | 0.66 | 0.52 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piewbang, C.; Chansaenroj, J.; Kongmakee, P.; Banlunara, W.; Poovorawan, Y.; Techangamsuwan, S. Genetic Adaptations, Biases, and Evolutionary Analysis of Canine Distemper Virus Asia-4 Lineage in a Fatal Outbreak of Wild-Caught Civets in Thailand. Viruses 2020, 12, 361. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040361
Piewbang C, Chansaenroj J, Kongmakee P, Banlunara W, Poovorawan Y, Techangamsuwan S. Genetic Adaptations, Biases, and Evolutionary Analysis of Canine Distemper Virus Asia-4 Lineage in a Fatal Outbreak of Wild-Caught Civets in Thailand. Viruses. 2020; 12(4):361. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040361
Chicago/Turabian StylePiewbang, Chutchai, Jira Chansaenroj, Piyaporn Kongmakee, Wijit Banlunara, Yong Poovorawan, and Somporn Techangamsuwan. 2020. "Genetic Adaptations, Biases, and Evolutionary Analysis of Canine Distemper Virus Asia-4 Lineage in a Fatal Outbreak of Wild-Caught Civets in Thailand" Viruses 12, no. 4: 361. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040361