In Silico Analysis of Genetic Diversity of Human Hepatitis B Virus in Southeast Asia, Australia and New Zealand

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
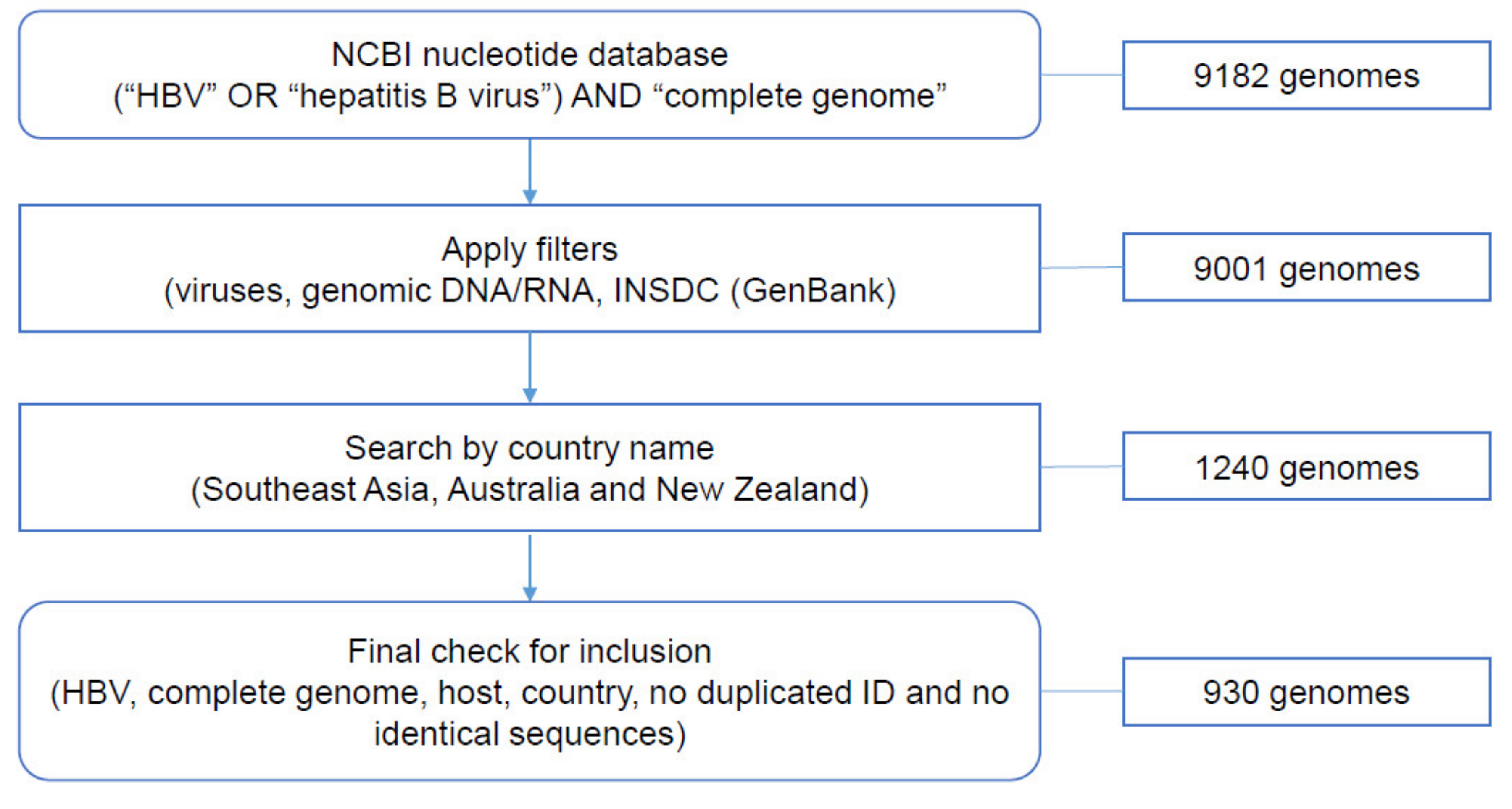
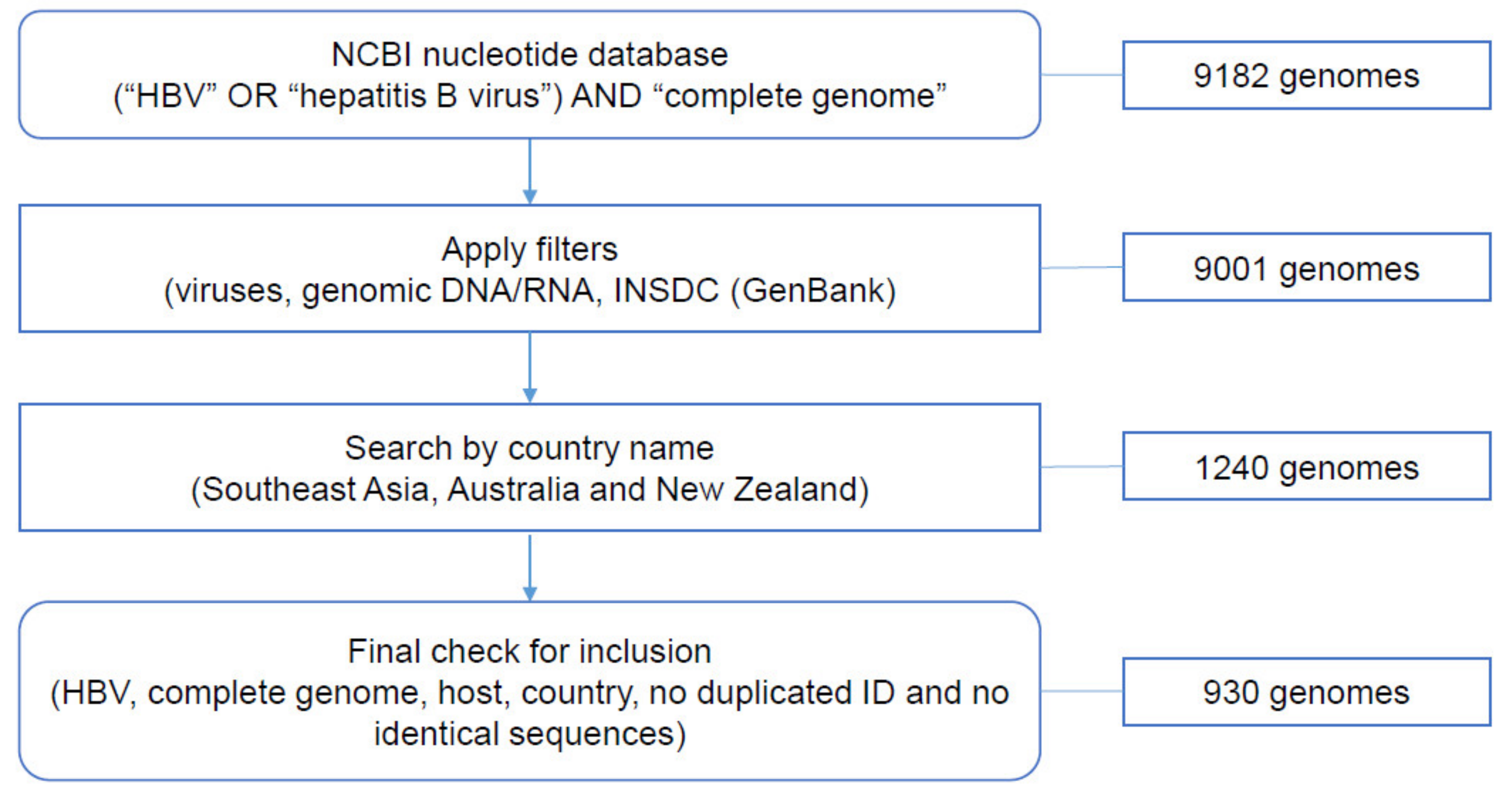
2.1. Data Collection from NCBI Nucleotide Database
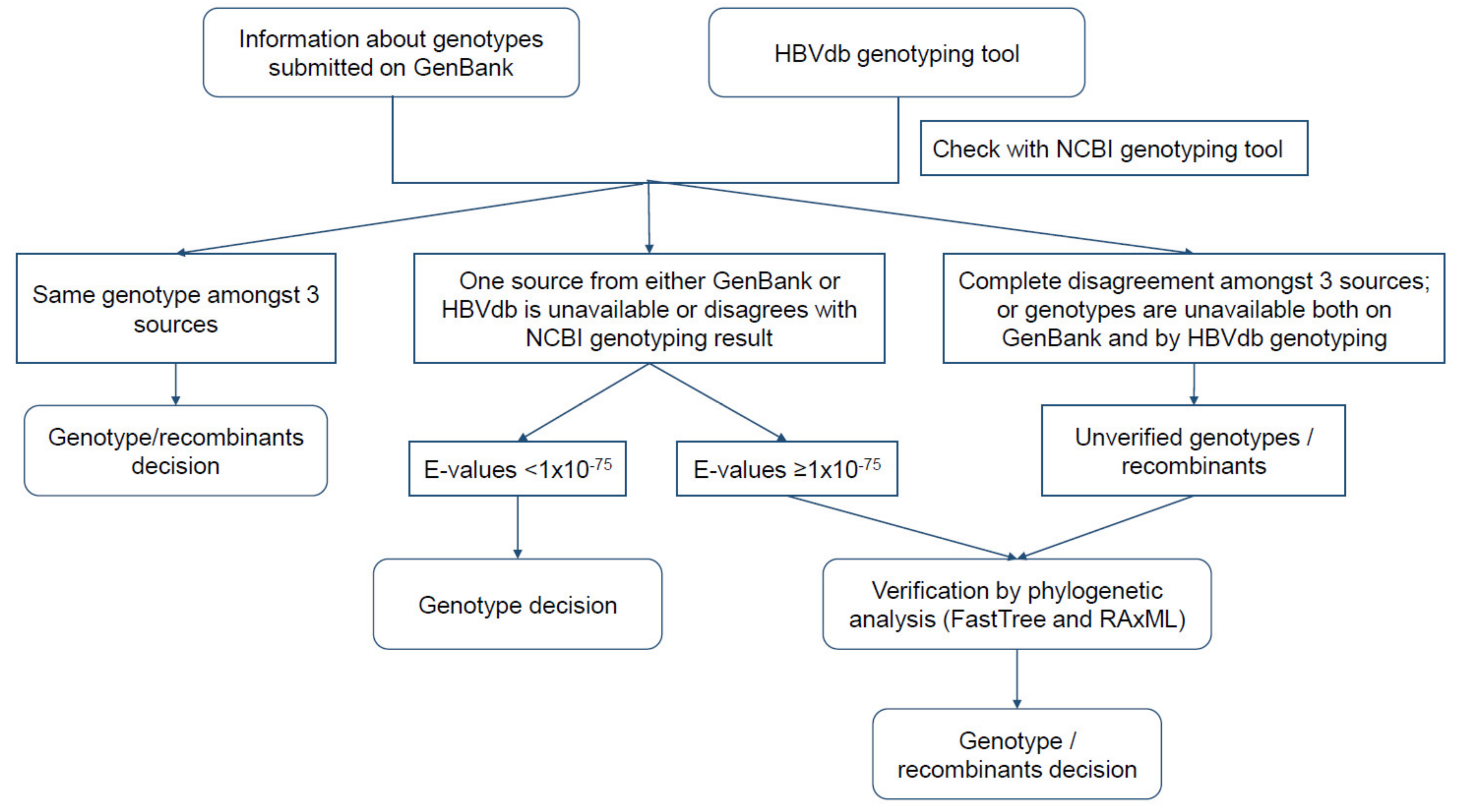
2.2. Analysis of HBV Genotypes
2.3. HBV Recombination Analysis
2.4. Representative Phylogenetic Analysis
2.5. HBV Serotype Prediction
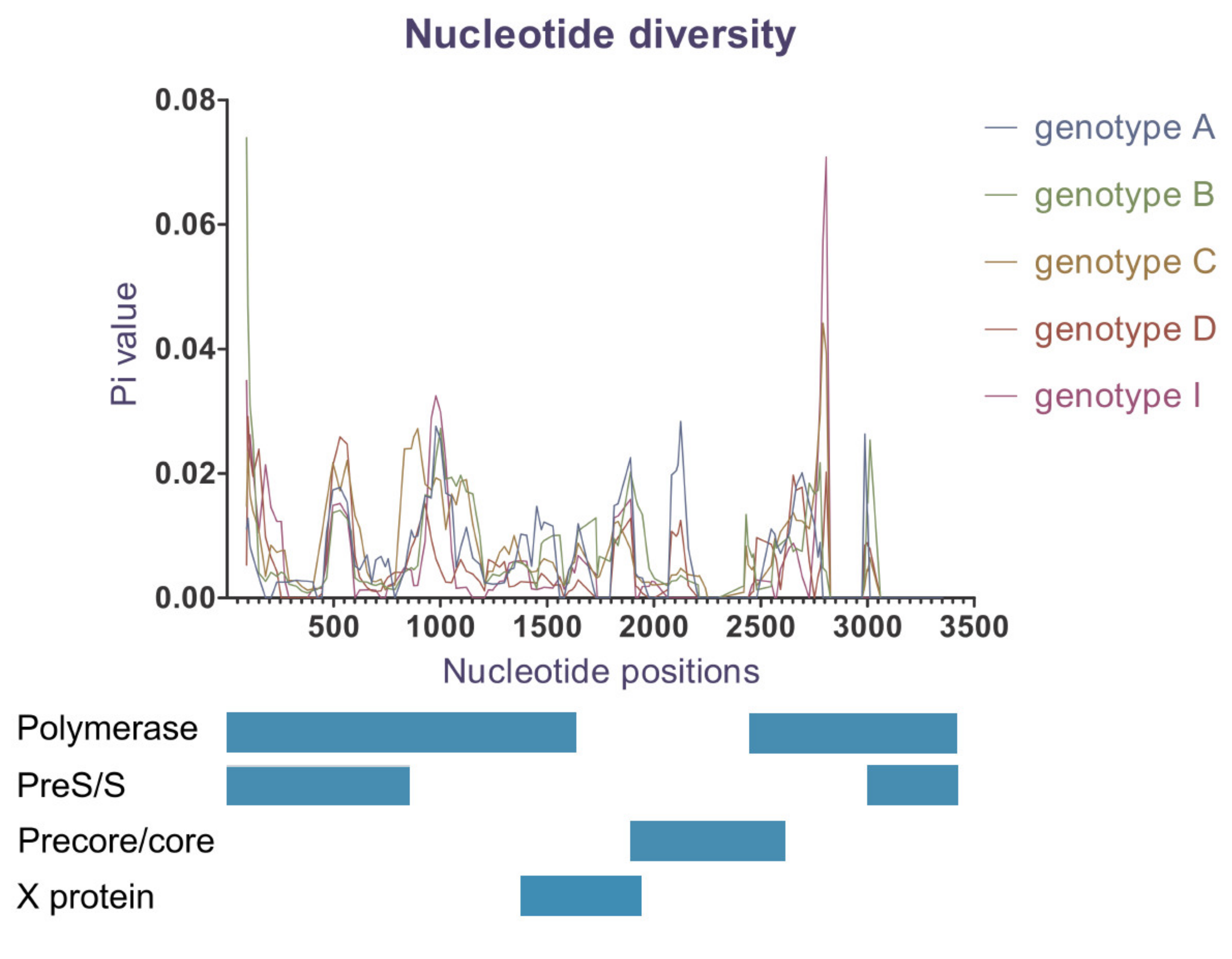
2.6. Nucleotide Diversity
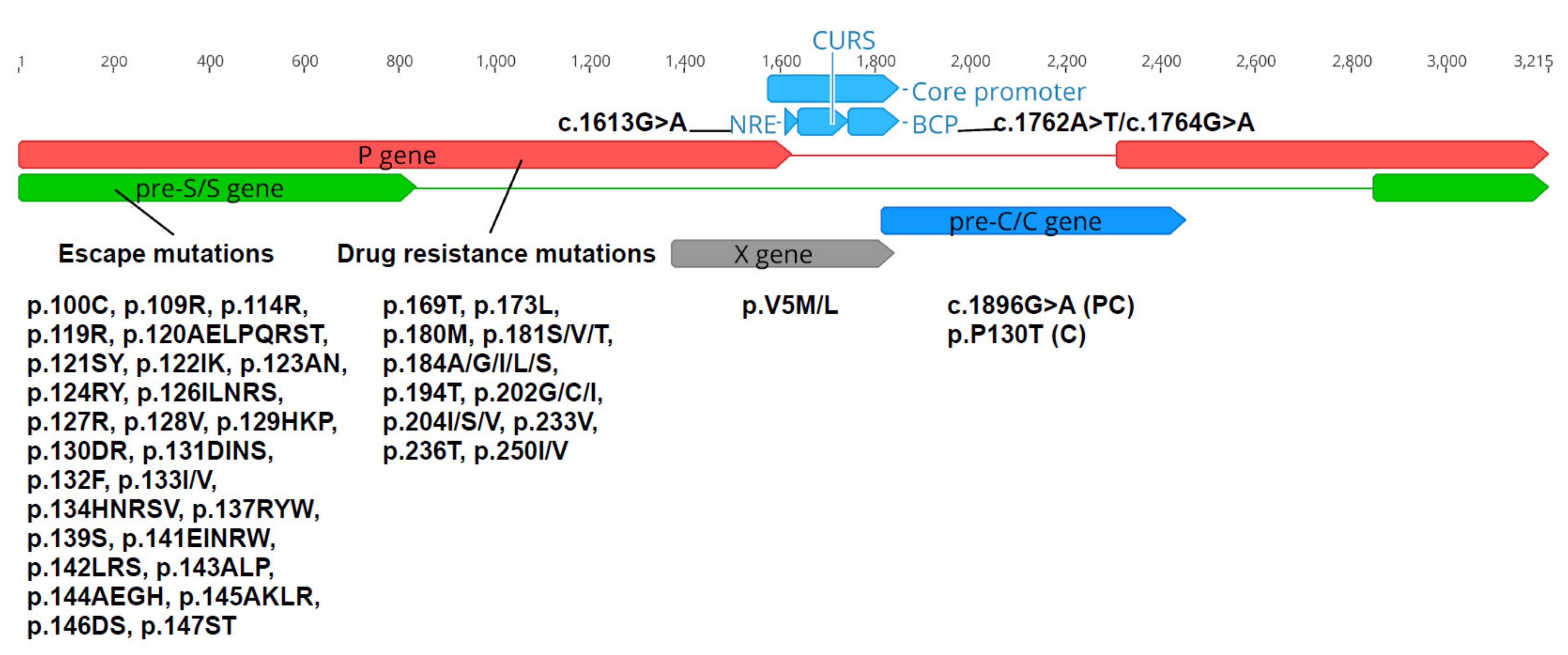
2.7. Analysis of Nucleotide and Amino Acid Variations
3. Results
3.1. Data Collection from NCBI Nucleotide Database
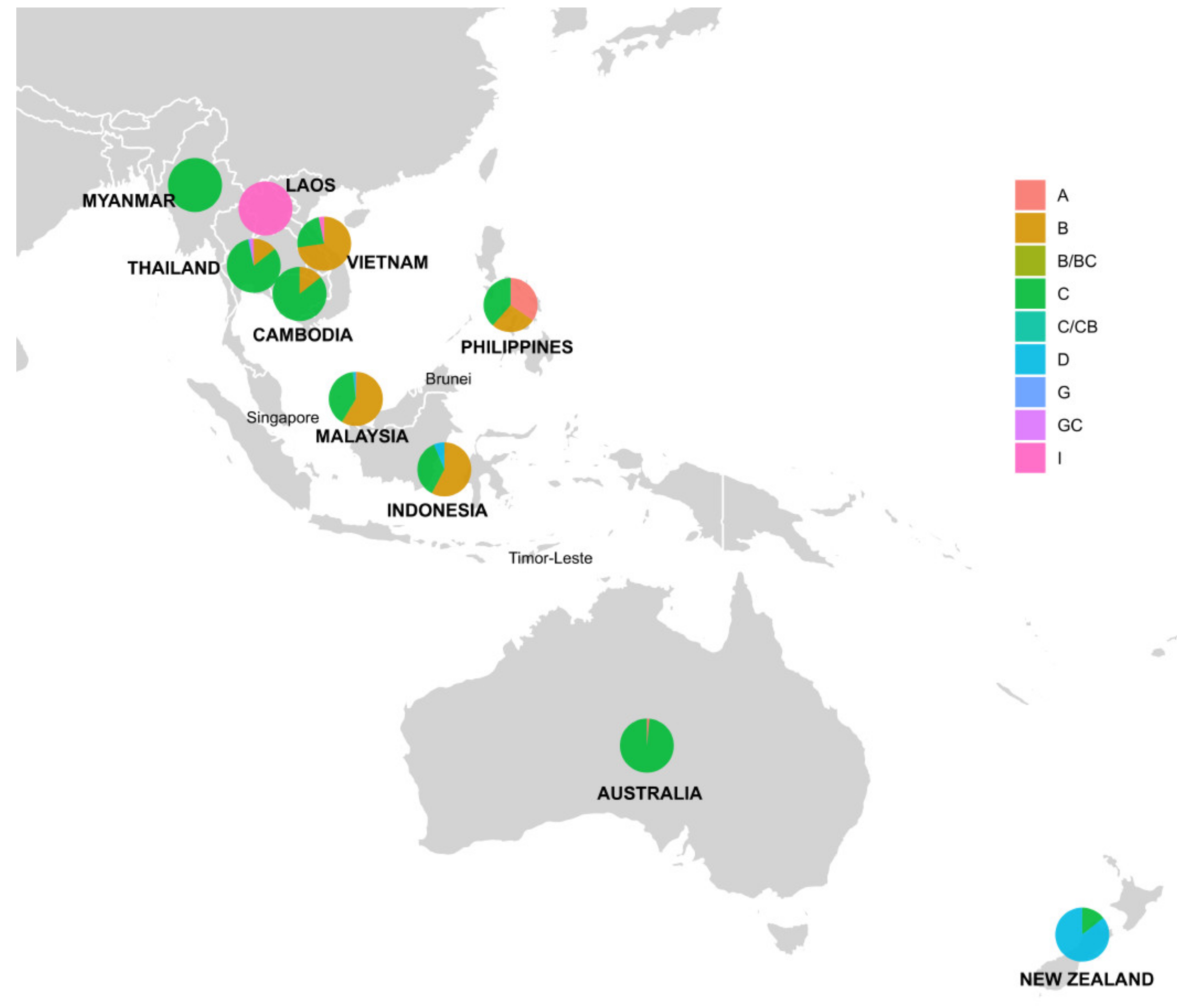
3.2. Analysis of HBV Genotypes
3.3. HBV Recombination Analysis
3.4. Representative Phylogenetic Analysis
3.5. HBV Serotype Prediction
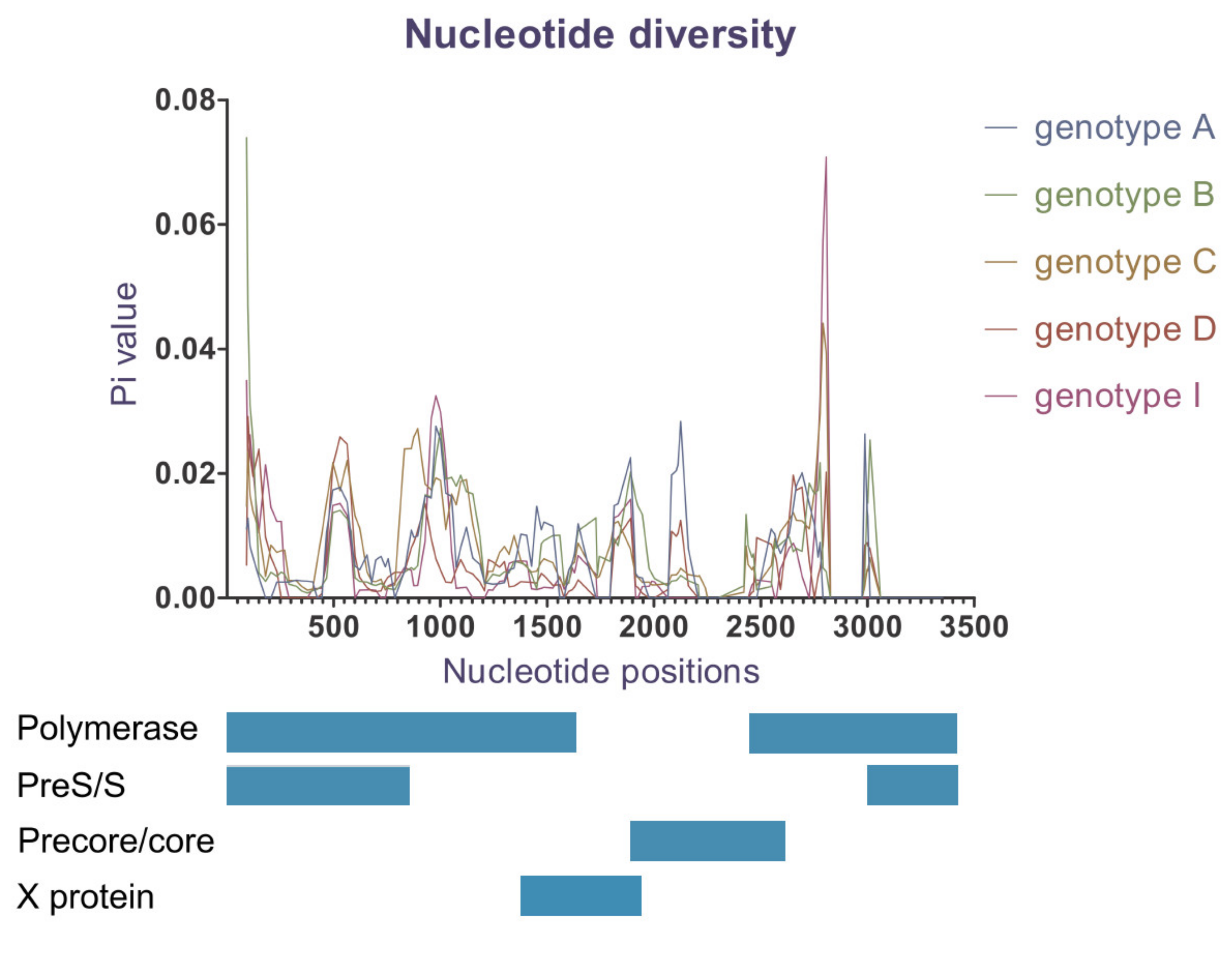
3.6. Nucleotide Diversity
3.7. Analysis of Single Nucleotide and Amino Acid Variations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Hepatitis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- McNaughton, A.L.; D’arienzo, V.; Ansari, M.A.; Lumley, S.F.; Littlejohn, M.; Revill, P.; McKeating, J.A.; Matthews, P.C. Insights from deep sequencing of the hbv genome—Unique, tiny, and misunderstood. Gastroenterology 2019, 156, 384–399. [Google Scholar] [CrossRef] [Green Version]
- Cento, V.; Mirabelli, C.; Dimonte, S.; Salpini, R.; Han, Y.; Trimoulet, P.; Bertoli, A.; Micheli, V.; Gubertini, G.; Cappiello, G.; et al. Overlapping structure of hepatitis b virus (hbv) genome and immune selection pressure are critical forces modulating hbv evolution. J. Gen. Virol. 2013, 94, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schödel, F.; Peterson, D.; Zheng, J.; Jones, J.E.; Hughes, J.L.; Milich, D.R. Structure of hepatitis b virus core and e-antigen. A single precore amino acid prevents nucleocapsid assembly. J. Biol. Chem. 1993, 268, 1332. [Google Scholar] [PubMed]
- Kim, H.; Lee, S.A.; Do, S.Y.; Kim, B.J. Precore/core region mutations of hepatitis b virus related to clinical severity. World J. Gastroenterol. 2016, 22, 4287–4296. [Google Scholar] [CrossRef] [PubMed]
- Li, M.S.; LauTerrence, C.K.; ChanSophie, K.P.; Wong, C.H.; NgPatrick, K.S.; SungJoseph, J.Y.; ChanHenry, L.Y.; TsuiStephen, K.W. The g1613a mutation in the hbv genome affects hbeag expression and viral replication through altered core promoter activity. PLoS ONE 2011, 6, e21856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, C.; Chen, X.; Li, X.; Li, J.; Lu, M. Genetic variation of hepatitis b virus and its significance for pathogenesis. World J. Gastroenterol. 2016, 22, 126–144. [Google Scholar] [CrossRef]
- Wang, W.; Shi, Y.E.; Bai, G.; Tang, Y.; Yuan, Y.; Zhang, T.; Li, C. Hbxag suppresses apoptosis of human placental trophoblastic cell lines via activation of the pi3k/akt pathway. Cell Biol. Int. 2016, 40, 708. [Google Scholar] [CrossRef]
- Cha, M.-Y.; Kim, C.-M.; Park, Y.-M.; Ryu, W.-S. Hepatitis b virus x protein is essential for the activation of wnt/beta-catenin signaling in hepatoma cells. Hepatol. (Baltim. Md.) 2004, 39, 1683. [Google Scholar] [CrossRef]
- Osiowy, C.; Giles, E.; Tanaka, Y.; Mizokami, M.; Minuk, G.Y. Molecular evolution of hepatitis b virus over 25 years. J. Virol. 2006, 80, 10307. [Google Scholar] [CrossRef] [Green Version]
- Park, S.G.; Kim, Y.; Park, E.; Ryu, H.M.; Jung, G. Fidelity of hepatitis b virus polymerase. Eur. J. Biochem. 2003, 270, 2929–2936. [Google Scholar] [CrossRef] [Green Version]
- Nowak, M.A.; Bonhoeffer, S.; Hill, A.M.; Boehme, R.; Thomas, H.C.; Mcdade, H. Viral dynamics in hepatitis b virus infection. Proc. Natl. Acad. Sci. USA 1996, 93, 4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norder, H.; Couroucé, A.-M.; Coursaget, P.; Echevarria, J.M.; Lee, S.-D.; Mushahwar, I.K.; Robertson, B.H.; Locarnini, S.; Magnius, L.O. Genetic diversity of hepatitis b virus strains derived worldwide: Genotypes, subgenotypes, and hbsag subtypes. Intervirology 2004, 47, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Tsuda, F.; Sakugawa, H.; Sastrosoewignjo, R.I.; Imai, M.; Miyakawa, Y.; Mayumi, M. Typing hepatitis b virus by homology in nucleotide sequence: Comparison of surface antigen subtypes. J. Gen. Virol. 1988, 69, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Velkov, S.; Ott, J.; Protzer, U.; Michler, T. The global hepatitis b virus genotype distribution approximated from available genotyping data. Genes 2018, 9, 495. [Google Scholar] [CrossRef] [Green Version]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis b virus divergent from known human and ape genotypes isolated from a japanese patient and provisionally assigned to new genotype j. J. Virol. 2009, 83, 10538. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Carr, M.J.; Dunford, L.; Zhu, C.; Hall, W.W.; Higgins, D.G. Identification of novel inter-genotypic recombinants of human hepatitis b viruses by large-scale phylogenetic analysis. Virology 2012, 427, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.; Li, X.; Liu, Y.; Xu, Z.; Huang, P.; Nian, X.; Liu, X.; Xu, D. Intergenotype recombinant analysis of full-length hepatitis b virus genomes from 516 chinese patients with different illness categories. J. Med. Virol. 2017, 89, 139–145. [Google Scholar] [CrossRef]
- Boyce, C.; Ganova-Raeva, L.; Archampong, T.; Lartey, M.; Sagoe, K.; Obo-Akwa, A.; Kenu, E.; Kwara, A.; Blackard, J. Identification and comparative analysis of hepatitis b virus genotype d/e recombinants in africa. Virus Genes 2017, 53, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Norder, H.; Courouce, A.M.; Magnius, L.O. Molecular basis of hepatitis b virus serotype variations within the four major subtypes. J. Gen. Virol. 1992, 73, 3141–3145. [Google Scholar] [CrossRef]
- Purdy, M.A.; Talekar, G.; Swenson, P.; Araujo, A.; Fields, H. A new algorithm for deduction of hepatitis b surface antigen subtype determinants from the amino acid sequence. Intervirology 2007, 50, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Norder, H.; Hammas, B.; Lofdahl, S.; Courouce, A.M.; Magnius, L.O. Comparison of the amino acid sequences of nine different serotypes of hepatitis b surface antigen and genomic classification of the corresponding hepatitis b virus strains. J. Gen. Virol. 1992, 73, 1201–1208. [Google Scholar] [CrossRef]
- Lin, C.-L.; Kao, J.-H. Hepatitis b virus genotypes and variants. Cold Spring Harb. Perspect. Med. 2015, 5, a021436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Yotsuyanagi, H.; Yatsuhashi, H.; Karino, Y.; Takikawa, Y.; Saito, T.; Arase, Y.; Imazeki, F.; Kurosaki, M.; Umemura, T.; et al. Risk factors for long-term persistence of serum hepatitis b surface antigen following acute hepatitis b virus infection in japanese adults. Hepatology 2014, 59, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Mayerat, C.; Mantegani, A.; Frei, P.C. Does hepatitis b virus (hbv) genotype influence the clinical outcome of hbv infection? J. Viral Hepat. 1999, 6, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.-W.; Yeh, S.-H.; Chen, P.-J.; Liaw, Y.-F.; Lin, C.-L.; Liu, C.-J.; Shih, W.-L.; Kao, J.-H.; Chen, D.-S.; Chen, C.-J. Hepatitis b virus genotype and DNA level and hepatocellular carcinoma: A prospective study in men. J. Natl. Cancer Inst. 2005, 97, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wu, Y.; Ye, S.; Wang, T.; Zhao, R.; Chen, F.; Abe, K.; Jin, X. The response to interferon is influenced by hepatitis b virus genotype in vitro and in vivo. Virus Res. 2013, 171, 65–70. [Google Scholar] [CrossRef]
- Cassidy, A.; Mossman, S.; Olivieri, A.; Ridder, M.D.; Leroux-Roels, G. Hepatitis b vaccine effectiveness in the face of global hbv genotype diversity. Expert Rev. Vaccines 2011, 10, 1709–1715. [Google Scholar] [CrossRef] [Green Version]
- Hamada-Tsutsumi, S.; Iio, E.; Watanabe, T.; Murakami, S.; Isogawa, M.; Iijima, S.; Inoue, T.; Matsunami, K.; Tajiri, K.; Ozawa, T.; et al. Validation of cross-genotype neutralization by hepatitis b virus-specific monoclonal antibodies by in vitro and in vivo infection. PLoS ONE 2015, 10, e0118062. [Google Scholar] [CrossRef] [Green Version]
- Shokrgozar, M.A.; Shokri, F. Subtype specificity of anti-hbs antibodies produced by human b-cell lines isolated from normal individuals vaccinated with recombinant hepatitis b vaccine. Vaccine 2002, 20, 2215–2220. [Google Scholar] [CrossRef]
- Stramer, S.L.; Wend, U.; Candotti, D.; Foster, G.A.; Hollinger, F.B.; Dodd, R.Y.; Allain, J.-P.; Gerlich, W. Nucleic acid testing to detect hbv infection in blood donors. N. Engl. J. Med. 2011, 364, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, E.; Reeve, C.; Marley, J.V. Hepatitis b notifications in a vaccinated cohort of aboriginal people in the kimberley region. Med. J. Aust. 2014, 201, 343–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seed, C.R.; Jones, N.T.; Pickworth, A.M.; Graham, W.R. Two cases of asymptomatic hbv “vaccine breakthrough” infection detected in blood donors screened for hbv DNA. Med. J. Aust. 2012, 196, 651–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purdy, M. Hepatitis b virus s gene escape mutants. Asian J. Transfus. Sci. 2007, 1, 62–70. [Google Scholar] [CrossRef]
- Xiao, Y.; Sun, K.; Duan, Z.; Liu, Z.; Li, Y.; Yan, L.; Song, Y.; Zou, H.; Zhuang, H.; Wang, J.; et al. Quasispecies characteristic in “a” determinant region is a potential predictor for the risk of immunoprophylaxis failure of mother-to-child-transmission of sub-genotype c2 hepatitis b virus: A prospective nested case–control study. Gut 2020, 69, 933–941. [Google Scholar] [CrossRef]
- Huang, X.; Lu, D.; Ji, G.; Sun, Y.; Ma, L.; Chen, Z.; Zhang, L.; Huang, J.; Yu, L. Hepatitis b virus (hbv) vaccine-induced escape mutants of hbv s gene among children from qidong area, china. Virus Res. 2004, 99, 63–68. [Google Scholar] [CrossRef]
- Oon, C.J.; Chen, W. Current aspects of hepatitis b surface antigen mutants in singapore. J. Viral Hepat. 1998, 5, 17–23. [Google Scholar] [CrossRef]
- Kato, H.; Sugiyama, M.; Mizokami, M. Hepatitis b virus genotypes. In Hepatitis b Virus in Human Diseases, 1st ed.; Liaw, Y.-F., Zoulim, F., Eds.; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Ngui, S.L.; O’Connell, S.; Eglin, R.P.; Heptonstall, J.; Teo, C.G. Low detection rate and maternal provenance of hepatitis b virus s gene mutants in cases of failed postnatal immunoprophylaxis in england and wales. J. Infect. Dis. 1997, 176, 1360–1365. [Google Scholar] [CrossRef] [Green Version]
- Haruki, K.; Ayano, I.; Shuichiro, U.; Tomoyuki, T.; Tsuyoshi, S.; Yasuhiro, K.; Tomoo, F. Evaluation of the g145r mutant of the hepatitis b virus as a minor strain in mother-to-child transmission. PLoS ONE 2016, 11, e0165674. [Google Scholar]
- Mokaya, J.; McNaughton, A.L.; Hadley, M.; Beloukas, A.; Geretti, A.; Goedhals, D.; Matthews, P.C. A systematic review of hepatitis b virus (hbv) drug and vaccine escape mutations in africa: A call for urgent action. PLoS Negl. Trop. Dis. 2018, 12, e0006629. [Google Scholar] [CrossRef] [Green Version]
- Shaw, T.; Bartholomeusz, A.; Locarnini, S. Hbv drug resistance: Mechanisms, detection and interpretation. J. Hepatol. 2006, 44, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, P.; Cerruti, R.; Icardi, G.; Bruzzone, B. Overview of hepatitis b virus mutations and their implications in the management of infection. World J. Gastroenterol. 2016, 22, 145. [Google Scholar] [CrossRef] [PubMed]
- Colombatto, P.; Barbera, C.; Bortolotti, F.; Maina, A.M.; Moriconi, F.; Cavallone, D.; Calvo, P.; Oliveri, F.; Bonino, F.; Brunetto, M.R. Hbv pre-core mutant in genotype-d infected children is selected during hbeag/anti-hbe seroconversion and leads to hbeag negative chronic hepatitis b in adulthood. J. Med. Virol. 2018, 90, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Funk, M.L.; Rosenberg, D.M.; Lok, A.S.F. World-wide epidemiology of hbeag-negative chronic hepatitis b and associated precore and core promoter variants. J. Viral Hepat. 2002, 9, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Liu, S.; Zhao, Y.; Zhang, L.; Liu, B.; Guo, Z. Precore/core region mutations in hepatitis b virus DNA predict postoperative survival in hepatocellular carcinoma. PLoS ONE 2015, 10, e0133393. [Google Scholar] [CrossRef]
- Wu, S.; Chang, Y.; Chu, T.; Shih, C. Persistence of hepatitis b virus DNA and the tempos between virion secretion and genome maturation in a mouse model. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Nie, H.; Evans, A.A.; London, W.T.; Block, T.M.; Ren, X.D. Quantitative dynamics of hepatitis b basal core promoter and precore mutants before and after hbeag seroconversion. J. Hepatol. 2012, 56, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhuang, L.; Lu, Y.; Xu, Q.N.; Tang, B.; Chen, X. Naturally occurring basal core promoter a1762t/g1764a dual mutations increase the risk of hbv-related hepatocellular carcinoma: A meta-analysis. Oncotarget 2016, 7, 12525–12536. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Park, J.H.; Jee, Y.; Lee, S.A.; Kim, H.; Song, B.C.; Yang, S.; Lee, M.; Yoon, J.H.; Kim, Y.J.; et al. Hepatitis b virus x mutations occurring naturally associated with clinical severity of liver disease among korean patients with chronic genotype c infection. J. Med. Virol. 2008, 80, 1337–1343. [Google Scholar] [CrossRef]
- Rozanov, M.; Plikat, U.; Chappey, C.; Kochergin, A.; Tatusova, T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004, 32, W654–W659. [Google Scholar] [CrossRef] [Green Version]
- Hayer, J.; Jadeau, F.; Deléage, G.; Kay, A.; Zoulim, F.; Combet, C. Hbvdb: A knowledge database for hepatitis b virus. Nucleic Acids Res. 2013, 41, D566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree 2—Approximately maximum-likelihood trees for large alignments (fasttree 2). PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Stamatakis, A. Raxml-vi-hpc: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.-I.; Miyata, T. Mafft: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Rybicki, E. Rdp: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Smith, J. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- David, P.; Keith, A.C. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757. [Google Scholar]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved algorithmic complexity for the 3seq recombination detection algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, T.G.; Kramvis, A. Mutation reporter tool: An online tool to interrogate loci of interest, with its utility demonstrated using hepatitis b virus. Virol. J. 2013, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, T.G.; Kramvis, A. Bioinformatics tools for small genomes, such as hepatitis b virus. Viruses 2015, 7, 781–797. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. Dnasp 6: DNA sequence ppolymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Bayliss, J.; Yuen, L.; Rosenberg, G.; Wong, D.; Littlejohn, M.; Jackson, K.; Gaggar, A.; Kitrinos, K.M.; Subramanian, G.M.; Marcellin, P.; et al. Deep sequencing shows that hbv basal core promoter and precore variants reduce the likelihood of hbsag loss following tenofovir disoproxil fumarate therapy in hbeag-positive chronic hepatitis b. Gut 2017, 66, 2013. [Google Scholar] [CrossRef]
- Neumann-Fraune, M.; Beggel, B.; Kaiser, R.; Obermeier, M. Hepatitis B virus drug resistance tools: One sequence, two predictions. Intervirology 2014, 57, 232–236. [Google Scholar] [CrossRef]
- Chen, X.; Gao, J.; Ji, Z.; Zhang, W.; Zhang, L.; Xu, R.; Zhang, J.; Li, F.; Li, S.; Hu, S.; et al. A description of the hepatitis b virus genomic background in a high-prevalence area in China. Virol. J. 2014, 11, 101. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | A | B | C | D | G | I | B/BC or C/CB | G/GC | Total | |
|---|---|---|---|---|---|---|---|---|---|---|
| Country | ||||||||||
| Australia | 1 | 66 | 67 (7.2%) | |||||||
| Cambodia | 4 | 24 | 28 (3%) | |||||||
| Indonesia | 82 | 51 | 9 | 142 (15.3%) | ||||||
| Laos | 14 | 14 (1.5%) | ||||||||
| Malaysia | 2 | 170 | 116 | 4 | 4 | 296 (31.8%) | ||||
| Myanmar | 18 | 18 (1.9%) | ||||||||
| New Zealand | 6 | 36 | 42 (4.5%) | |||||||
| Philippines | 9 | 7 | 10 | 26 (2.8%) | ||||||
| Thailand | 16 | 96 | 1 | 1 | 1 | 2 | 117 (12.6%) | |||
| Vientam | 131 | 43 | 6 | 180 (19.4%) | ||||||
| Total | 12 (1.3%) | 410 (44.1%) | 430 (46.2%) | 49 (5.3%) | 1 (0.1%) | 21 (2.3%) | 5 (0.5%) | 2 (0.2%) | 930 (100%) | |
| Detection Method | RDP | 3SEQ | Chimaera | Geneconv | Maxchi | |
|---|---|---|---|---|---|---|
| Events/Recombinants | ||||||
| Recombination events | 83 | 80 | 49 | 87 | 41 | |
| Potential recombinants (possibly present in different events) | 467 | 613 | 744 | 1199 | 1355 | |
| Potential recombinants (after removing replicate recombinants) | 380 | 566 | 510 | 857 | 548 | |
| Recombinants identified by all methods: 83 (65 sequences from our data and 18 reference sequences) | ||||||
| + with overlapping breakpoint positions: 35 (31 sequences from our data and 4 reference sequences) | ||||||
| + with at least one same major parent: 10 (8 sequences from our data and 2 reference sequences) | ||||||
| Serotype | adr (n = 335, 36%) | adw2 (n = 270, 29%) | adw3 (n = 7, 0.8%) | ayr (n = 6, 0.7%) | ayw1 (n = 185, 19.9%) | ayw2 (n = 45, 4.8%) | ayw3 (n = 70, 7.5%) | Unassigned (n = 12, 1.3%) | |
|---|---|---|---|---|---|---|---|---|---|
| Genotype/ Country | |||||||||
| A | 9 | 2 | 1 | ||||||
| Australia | 1 | ||||||||
| Malaysia | 1 | 1 | |||||||
| Philippines | 7 | 2 | |||||||
| B | 218 | 5 | 172 | 1 | 6 | 8 | |||
| Cambodia | 2 | 2 | |||||||
| Indonesia | 46 | 1 | 34 | 1 | |||||
| Malaysia | 141 | 4 | 20 | 1 | 4 | ||||
| Philippines | 1 | 6 | |||||||
| Thailand | 12 | 4 | |||||||
| Vietnam | 16 | 106 | 6 | 3 | |||||
| C | 331 | 29 | 1 | 6 | 60 | 3 | |||
| Australia | 6 | 60 | |||||||
| Cambodia | 23 | 1 | |||||||
| Indonesia | 45 | 3 | 3 | ||||||
| Malaysia | 107 | 5 | 1 | 3 | |||||
| Myanmar | 17 | 1 | |||||||
| New Zealand | 6 | ||||||||
| Philippines | 1 | 9 | |||||||
| Thailand | 86 | 7 | 3 | ||||||
| Vietnam | 40 | 3 | |||||||
| D | 1 | 44 | 4 | ||||||
| Indonesia | 1 | 8 | |||||||
| Malaysia | 4 | ||||||||
| New Zealand | 36 | ||||||||
| G | 1 | ||||||||
| Thailand | 1 | ||||||||
| I | 11 | 10 | |||||||
| Laos | 4 | 10 | |||||||
| Thailand | 1 | ||||||||
| Vietnam | 6 | ||||||||
| B/BC | 1 | 1 | 1 | ||||||
| Thailand | 1 | ||||||||
| Malaysia | 1 | 1 | |||||||
| C/CB | 1 | 1 | |||||||
| Malaysia | 1 | 1 | |||||||
| G/GC | 2 | ||||||||
| Thailand | 2 | ||||||||
| Genotype | Number of Polymorphic (Segregating) Sites | Average Number of Nucleotide Differences (k) | Nucleotide Diversity (Pi) |
|---|---|---|---|
| Genotype A (n = 12) | 44 | 10.788 | 0.00775 |
| Genotype B (n = 410) | 454 | 11.515 | 0.00827 |
| Genotype C (n = 430) | 446 | 12.548 | 0.00901 |
| Genotype D (n = 49) | 68 | 7.770 | 0.00558 |
| Genotype I (n = 21) | 37 | 8.148 | 0.00585 |
| Total N = 930 (including genotype G and recombinants) | 676 | 20.944 | 0.01505 |
| Mutation Type (Region) | A (n = 12) | B (n = 410) | C (n = 430) | D (n = 49) | G (n = 1) | I (n = 21) | B/BC, C/CB, G/GC (n = 7) | Total (n = 930) |
|---|---|---|---|---|---|---|---|---|
| Drug resistance mutations (polymerase) | 1 (8%) | 15 (3.7%) | 22 (5%) | 2 (4%) | 0 | 0 | 0 | 40 (4.3%) |
| Escape mutations (pre-S/S) | 2 (16%) | 41 (10%) | 79 (18%) | 8 (16%) | 0 | 2 (10%) | 2 (27%) | 134 (14.4%) |
| c.1613G>A (NRE) | 2 (16%) | 44 (11%) | 67 (15%) | 0 | 1 (100%) | 4 (19%) | 1 (14%) | 119 (12.8%) |
| Only c.1762A>T (BCP) | 0 | 7 (2%) | 3 (0.7%) | 0 | 0 | 0 | 0 | 10 (1.1%) |
| Only c.1764G>A (BCP) | 1 (8%) | 3 (0.7%) | 20 (5%) | 1 (2%) | 0 | 0 | 0 | 25 (2.7%) |
| Double c.1762A>T/ c.1764G>A (BCP) | 5 (42%) | 60 (15%) | 159 (37%) | 7 (14%) | 1 (100%) | 5 (24%) | 2 (27%) | 239 (25.7%) |
| c.1896G>A (PC) | 0 | 193 (47%) | 77 (18%) | 13 (27%) | 0 | 4 (19%) | 2 (27%) | 289 (31.1%) |
| p.P130T (C) | 0 | 28 (6.8%) | 60 (14%) | 0 | 1 (100%) | 0 | 0 | 89 (9.6%) |
| p.V5M (X) | 0 | 8 (2%) | 11 (2.6%) | 0 | 0 | 0 | 1 (14%) | 20 (2.2%) |
| p.V5L (X) | 10 (83%) | 401 (97%) | 395 (92%) | 48 (97%) | 1 (100%) | 21 (100%) | 6 (86%) | 882 (95%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, N.M.H.; Faddy, H.; Flower, R.; Spann, K.; Roulis, E. In Silico Analysis of Genetic Diversity of Human Hepatitis B Virus in Southeast Asia, Australia and New Zealand. Viruses 2020, 12, 427. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040427
Phan NMH, Faddy H, Flower R, Spann K, Roulis E. In Silico Analysis of Genetic Diversity of Human Hepatitis B Virus in Southeast Asia, Australia and New Zealand. Viruses. 2020; 12(4):427. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040427
Chicago/Turabian StylePhan, Ngoc Minh Hien, Helen Faddy, Robert Flower, Kirsten Spann, and Eileen Roulis. 2020. "In Silico Analysis of Genetic Diversity of Human Hepatitis B Virus in Southeast Asia, Australia and New Zealand" Viruses 12, no. 4: 427. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040427