Phylogenetic, Evolutionary and Structural Analysis of Canine Parvovirus (CPV-2) Antigenic Variants Circulating in Colombia



Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Selection and Sampling
2.2. DNA Extraction and Quantification
2.3. VP2 Amplification Using PCR
2.4. Sequencing and Sequence Analysis
2.5. Phylogenetic Analysis
2.6. Evolutionary Analysis
2.7. Structural Analysis
2.8. Analysis of Selection Pressure
2.9. Statistical Analyses
3. Results
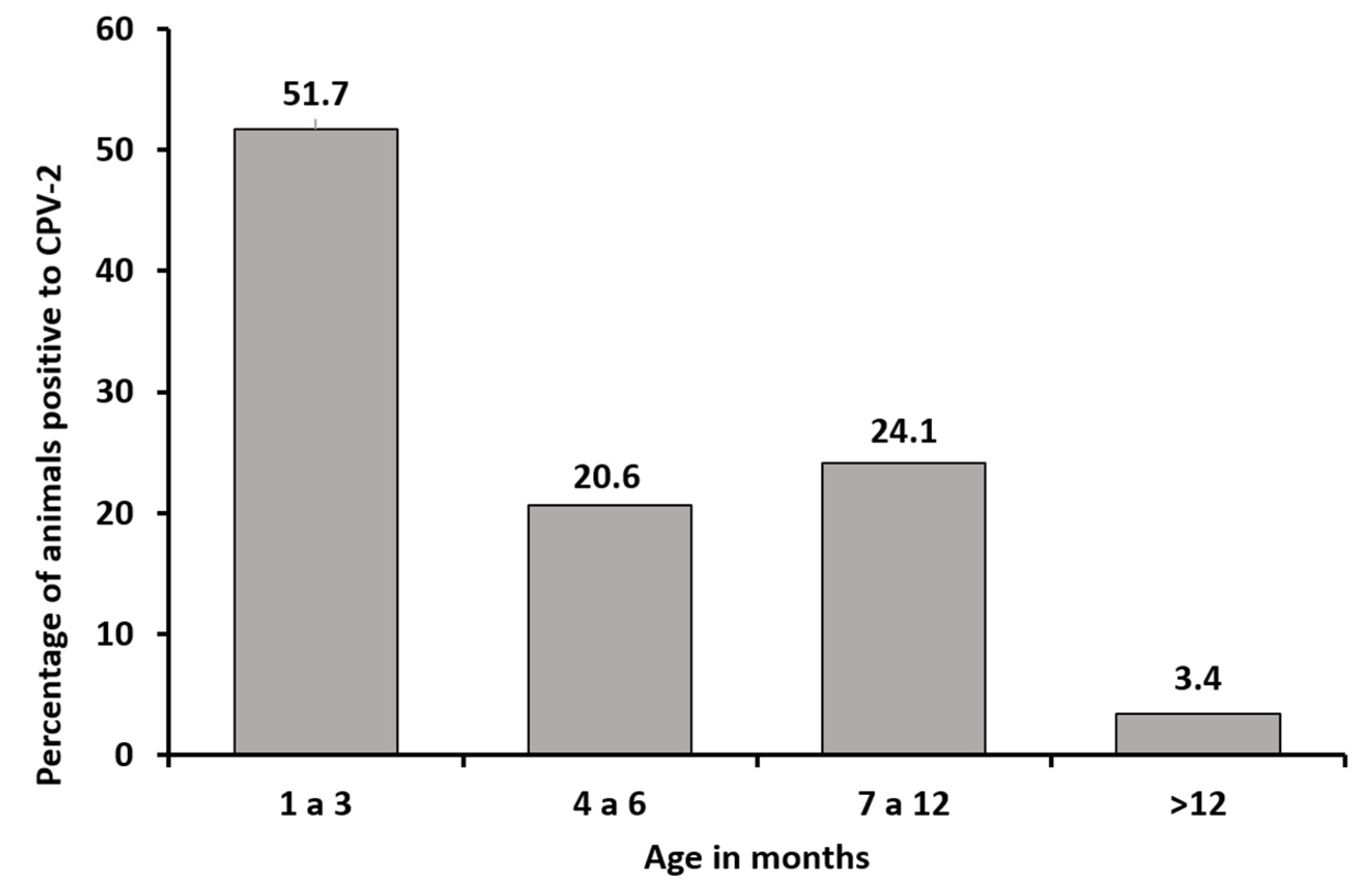
3.1. Sequence Analysis
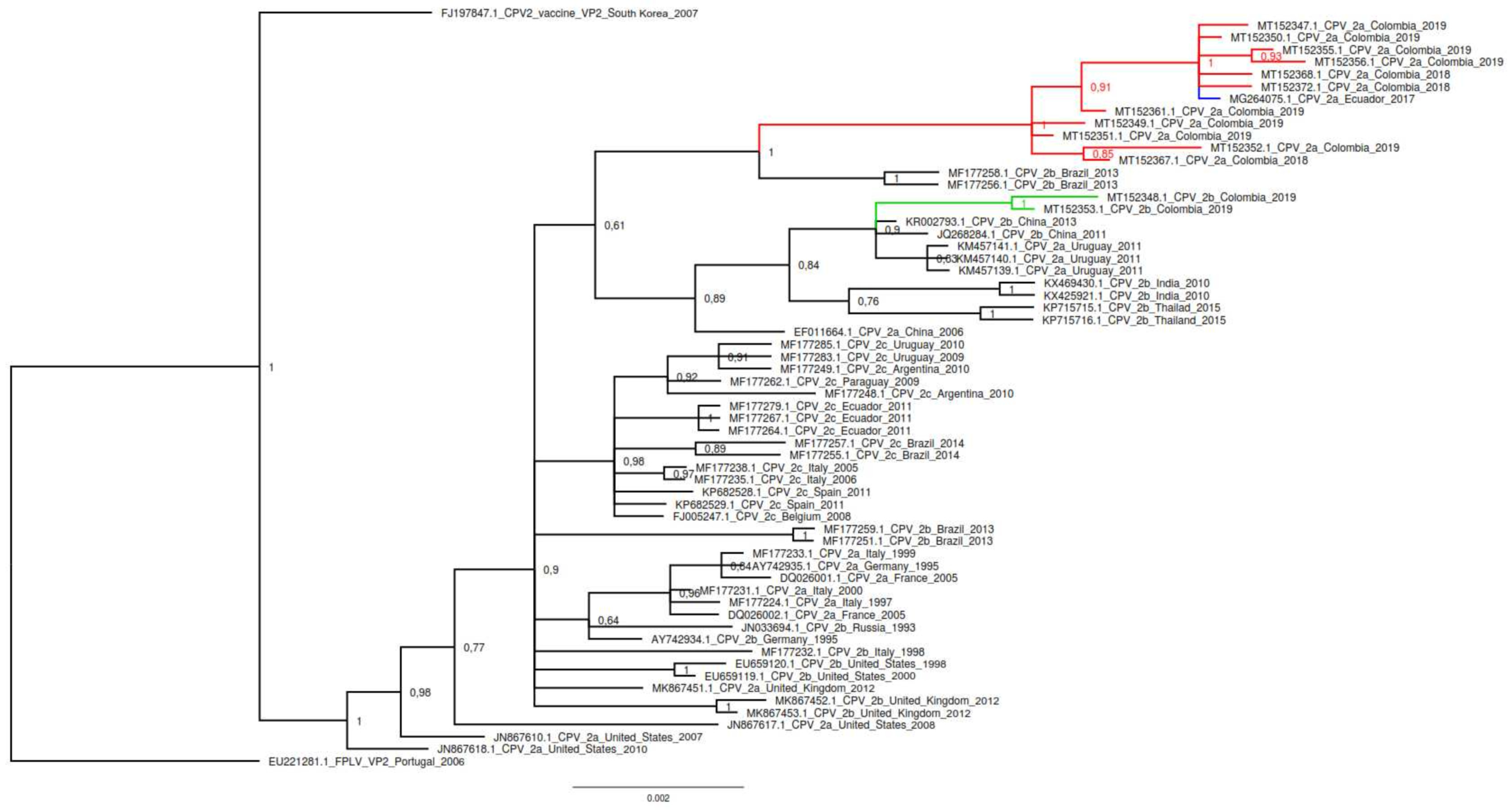
3.2. Phylogenetic Analysis
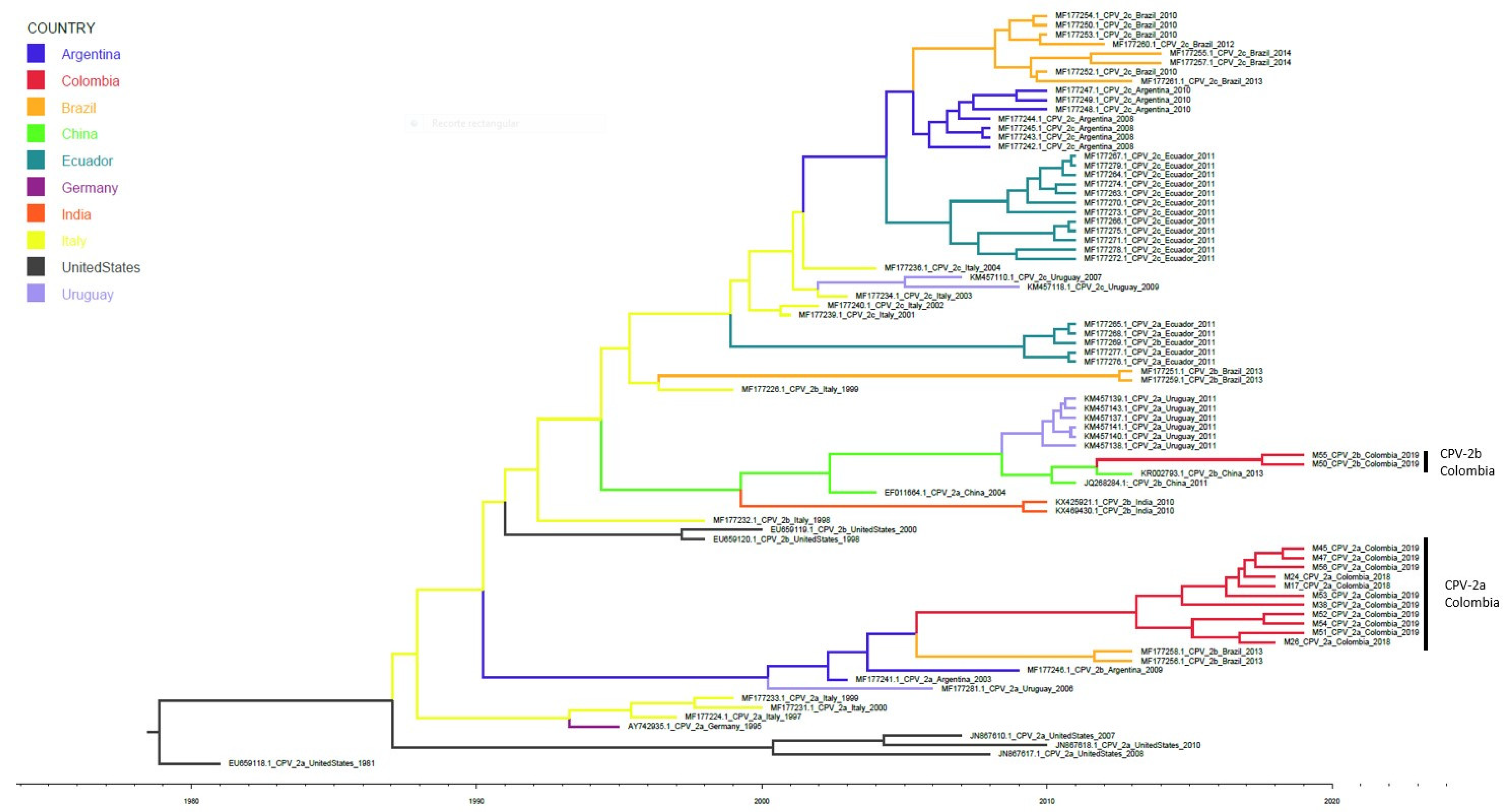
3.3. Evolutionary Analysis
3.4. Structural Analysis
3.5. Sites under Positive Selection
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Goddard, A.; Leisewitz, A.L. Canine parvovirus. Vet. Clin. N. Am. Small Anim. Pract. 2010, 40, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.P.; Jones, E.V.; Miller, T.J. Nucleotide sequence and genome organization of canine parvovirus. J. Virol. 1988, 62, 266–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hueffer, K.; Parker, J.S.; Weichert, W.S.; Geisel, R.E.; Sgro, J.Y.; Parrish, C.R. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J. Virol. 2003, 77, 1718–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.S.; Parrish, C.R. Cellular uptake and infection by canine parvovirus involves rapid dynamin-regulated clathrin-mediated endocytosis, followed by slower intracellular trafficking. J. Virol. 2000, 74, 1919–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, C.; Thompson, G. Canine parvovirus: The worldwide occurrence of antigenic variants. J. Gen. Virol. 2016, 97, 2043–2057. [Google Scholar] [CrossRef]
- Chang, S.F.; Sgro, J.Y.; Parrish, C.R. Multiple amino acids in the capsid structure of canine parvovirus coordinately determine the canine host range and specific antigenic and hemagglutination properties. J. Virol. 1992, 66, 6858–6867. [Google Scholar] [CrossRef] [Green Version]
- Truyen, U.; Evermann, J.F.; Vieler, E.; Parrish, C.R. Evolution of canine parvovirus involved loss and gain of feline host range. Virology 1996, 215, 186–189. [Google Scholar] [CrossRef] [Green Version]
- Parrish, C.R. Host range relationships and the evolution of canine parvovirus. Vet. Microbiol. 1999, 69, 29–40. [Google Scholar] [CrossRef]
- Parrish, C.R.; Aquadro, C.F.; Strassheim, M.L.; Evermann, J.F.; Sgro, J.Y.; Mohammed, H.O. Rapid antigenic-type replacement and DNA sequence evolution of canine parvovirus. J. Virol. 1991, 65, 6544–6552. [Google Scholar] [CrossRef] [Green Version]
- Tsao, J.; Chapman, M.S.; Agbandje, M.; Keller, W.; Smith, K.; Wu, H.; Luo, M.; Smith, T.J.; Rossmann, M.G.; Compans, R.W.; et al. The three-dimensional structure of canine parvovirus and its functional implications. Science 1991, 251, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonavoglia, C.; Martella, V.; Pratelli, A.; Tempesta, M.; Cavalli, A.; Buonavoglia, D.; Bozzo, G.; Elia, G.; Decaro, N.; Carmichael, L. Evidence for evolution of canine parvovirus type 2 in Italy. J. Gen. Virol. 2001, 82 Pt 12, 3021–3025. [Google Scholar] [CrossRef]
- Decaro, N.; Buonavoglia, C. Canine parvovirus—A review of epidemiological and diagnostic aspects, with emphasis on type 2c. Vet. Microbiol. 2012, 155, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Duque-Garcia, Y.; Echeverri-Zuluaga, M.; Trejos-Suarez, J.; Ruiz-Saenz, J. Prevalence and molecular epidemiology of Canine parvovirus 2 in diarrheic dogs in Colombia, South America: A possible new CPV-2a is emerging? Vet. Microbiol. 2017, 201, 56–61. [Google Scholar] [CrossRef]
- Zhou, P.; Zeng, W.; Zhang, X.; Li, S. The genetic evolution of canine parvovirus—A new perspective. PLoS ONE 2017, 12, e0175035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grecco, S.; Iraola, G.; Decaro, N.; Alfieri, A.; Alfieri, A.; Gallo Calderon, M.; da Silva, A.P.; Name, D.; Aldaz, J.; Calleros, L.; et al. Inter- and intracontinental migrations and local differentiation have shaped the contemporary epidemiological landscape of canine parvovirus in South America. Virus Evol. 2018, 4, vey011. [Google Scholar] [CrossRef] [Green Version]
- Faz, M.; Martinez, J.S.; Gomez, L.B.; Quijano-Hernandez, I.; Fajardo, R.; Del Angel-Caraza, J. Origin and genetic diversity of canine parvovirus 2c circulating in Mexico. Arch. Virol. 2019, 164, 371–379. [Google Scholar] [CrossRef]
- Ikeda, Y.; Mochizuki, M.; Naito, R.; Nakamura, K.; Miyazawa, T.; Mikami, T.; Takahashi, E. Predominance of canine parvovirus (CPV) in unvaccinated cat populations and emergence of new antigenic types of CPVs in cats. Virology 2000, 278, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Rossmann, M.G. Interpretation of electron density with stereographic roadmap projections. J. Struct. Biol. 2007, 158, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Mylonakis, M.E.; Kalli, I.; Rallis, T.S. Canine parvoviral enteritis: An update on the clinical diagnosis, treatment, and prevention. Vet. Med. (Auckland) 2016, 7, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Desario, C.; Decaro, N.; Campolo, M.; Cavalli, A.; Cirone, F.; Elia, G.; Martella, V.; Lorusso, E.; Camero, M.; Buonavoglia, C. Canine parvovirus infection: Which diagnostic test for virus? J. Virol. Methods 2005, 126, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Desario, C.; Elia, G.; Martella, V.; Mari, V.; Lavazza, A.; Nardi, M.; Buonavoglia, C. Evidence for immunisation failure in vaccinated adult dogs infected with canine parvovirus type 2c. New Microbiol. 2008, 31, 125–130. [Google Scholar] [PubMed]
- Nakamura, M.; Tohya, Y.; Miyazawa, T.; Mochizuki, M.; Phung, H.T.; Nguyen, N.H.; Huynh, L.M.; Nguyen, L.T.; Nguyen, P.N.; Nguyen, P.V.; et al. A novel antigenic variant of Canine parvovirus from a Vietnamese dog. Arch. Virol. 2004, 149, 2261–2269. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Bianchi, P.; Calleros, L.; Francia, L.; Hernandez, M.; Maya, L.; Panzera, Y.; Sosa, K.; Zoller, S. Recent spreading of a divergent canine parvovirus type 2a (CPV-2a) strain in a CPV-2c homogenous population. Vet. Microbiol. 2012, 155, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Palermo, L.M.; Hafenstein, S.L.; Parrish, C.R. Purified feline and canine transferrin receptors reveal complex interactions with the capsids of canine and feline parvoviruses that correspond to their host ranges. J. Virol. 2006, 80, 8482–8492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, L.M.; Hueffer, K.; Parrish, C.R. Residues in the apical domain of the feline and canine transferrin receptors control host-specific binding and cell infection of canine and feline parvoviruses. J. Virol. 2003, 77, 8915–8923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeoung, S.Y.; Ahn, S.J.; Kim, D. Genetic analysis of VP2 gene of canine parvovirus isolates in Korea. J. Vet. Med. Sci. 2008, 70, 719–722. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Yang, S.; Zhang, W.; Zhang, T.; Xie, Z.; Feng, H.; Wang, S.; Xia, X. Phylogenetic analysis of the VP2 gene of canine parvoviruses circulating in China. Virus Genes 2010, 40, 397–402. [Google Scholar] [CrossRef]
- Hong, C.; Decaro, N.; Desario, C.; Tanner, P.; Pardo, M.C.; Sanchez, S.; Buonavoglia, C.; Saliki, J.T. Occurrence of canine parvovirus type 2c in the United States. J. Vet. Diagn. Investig. 2007, 19, 535–539. [Google Scholar] [CrossRef] [Green Version]
- Decaro, N.; Desario, C.; Addie, D.D.; Martella, V.; Vieira, M.J.; Elia, G.; Zicola, A.; Davis, C.; Thompson, G.; Thiry, E.; et al. The study molecular epidemiology of canine parvovirus, Europe. Emerg. Infect. Dis. 2007, 13, 1222–1224. [Google Scholar] [CrossRef]
- Wang, H.; Jin, H.; Li, Q.; Zhao, G.; Cheng, N.; Feng, N.; Zheng, X.; Wang, J.; Zhao, Y.; Li, L.; et al. Isolation and sequence analysis of the complete NS1 and VP2 genes of canine parvovirus from domestic dogs in 2013 and 2014 in China. Arch. Virol. 2016, 161, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Mira, F.; Purpari, G.; Lorusso, E.; Di Bella, S.; Gucciardi, F.; Desario, C.; Macaluso, G.; Decaro, N.; Guercio, A. Introduction of Asian canine parvovirus in Europe through dog importation. Transbound. Emerg. Dis. 2018, 65, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Mira, F.; Purpari, G.; Di Bella, S.; Colaianni, M.L.; Schiro, G.; Chiaramonte, G.; Gucciardi, F.; Pisano, P.; Lastra, A.; Decaro, N.; et al. Spreading of canine parvovirus type 2c mutants of Asian origin in southern Italy. Transbound. Emerg. Dis. 2019, 66, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, I.E.H.; Lee, H.; Allison, A.B.; Lopez-Astacio, R.; Goodman, L.B.; Oyesola, O.O.; Omobowale, O.; Fagbohun, O.; Dubovi, E.J.; Hafenstein, S.L.; et al. Limited intra-host diversity and background evolution accompany 40 years of canine parvovirus host adaptation and spread. J. Virol. 2019, 94. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Callaway, H.M.; Cifuente, J.O.; Bator, C.M.; Parrish, C.R.; Hafenstein, S.L. Transferrin receptor binds virus capsid with dynamic motion. Proc. Natl. Acad. Sci. USA 2019, 116, 20462–20471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Torre, D.; Mafla, E.; Puga, B.; Erazo, L.; Astolfi-Ferreira, C.; Ferreira, A.P. Molecular characterization of canine parvovirus variants (CPV-2a, CPV-2b, and CPV-2c) based on the VP2 gene in affected domestic dogs in Ecuador. Vet. World 2018, 11, 480–487. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, H.K.; Matta, S.L.; Amsaveni, S.; Antony, P.X.; Thanislass, J.; Pillai, R.M. Phylogenetic analysis of canine parvovirus partial VP2 gene in India. Virus Genes 2014, 48, 89–95. [Google Scholar] [CrossRef]
- Mukhopadhyay, H.K.; Nookala, M.; Thangamani, N.R.; Sivaprakasam, A.; Antony, P.X.; Thanislass, J.; Srinivas, M.V.; Pillai, R.M. Molecular characterisation of parvoviruses from domestic cats reveals emergence of newer variants in India. J. Feline Med. Surg. 2017, 19, 846–852. [Google Scholar] [CrossRef]
- Magee, D.; Taylor, J.E.; Scotch, M. The Effects of Sampling Location and Predictor Point Estimate Certainty on Posterior Support in Bayesian Phylogeographic Generalized Linear Models. Sci. Rep. 2018, 8, 5905. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Sample | Variant | Age | Sex | Race | Vaccination |
|---|---|---|---|---|---|
| 1 | CPV-2a | 4 months | Male | Mixed-breed | Without vaccination |
| 5 | CPV-2a | 3 months | Female | French Bull Dog | Incomplete |
| 7 | CPV-2a | 1 month | Female | Mixed-breed | Without vaccination |
| 17 | CPV-2a | 3 months | Female | Golden Retriever | Incomplete |
| 19 | CPV-2a | 2 months | Female | French Bulldog | Incomplete |
| 20 | CPV-2a | 2 months | Female | French Bulldog | Incomplete |
| 21 | CPV-2a | 2 months | Male | French Bulldog | Incomplete |
| 24 | CPV-2a | 2 months | Female | Beagle | Without vaccination |
| 26 | CPV-2a | 3 months | Female | Mixed-breed | Without vaccination |
| 29 | CPV-2a | 2 months | Male | Mixed-breed | Without vaccination |
| 32 | CPV-2a | 9 months | Female | Pinscher | Without vaccination |
| 33 | CPV-2a | 2 months | Male | Cocker Spaniel | Incomplete |
| 36 | CPV-2a | 2 months | Male | Siberian Husky | Without vaccination |
| 37 | CPV-2a | 7 months | Male | Mixed-breed | Incomplete |
| 38 | CPV-2a | 6 months | Male | Cocker Spaniel | Complete |
| 40 | CPV-2a | 4 months | Female | Mixed-breed | Incomplete |
| 41 | CPV-2a | 4 months | Female | Mixed-breed | Incomplete |
| 43 | CPV-2a | 9 months | Male | Mixed-breed | Incomplete |
| 44 | CPV-2a | 3 months | Male | French Bulldog | Incomplete |
| 45 | CPV-2a | 12 months | Female | Mixed-breed | Without vaccination |
| 47 | CPV-2a | 7 months | Male | Mixed-breed | Without vaccination |
| 48 | CPV-2a | 6 months | Male | Mixed-breed | Complete |
| 50 | CPV-2b | 9 months | Male | Mixed-breed | Incomplete |
| 51 | CPV-2a | 2 months | Male | Mixed-breed | Without vaccination |
| 52 | CPV-2a | 2 months | Female | Mixed-breed | Incomplete |
| 53 | CPV-2a | 4 months | Male | Mixed-breed | Without vaccination |
| 54 | CPV-2a | 24 months | Male | Mixed-breed | Without vaccination |
| 55 | CPV-2b | 3 months | Male | German Shepherd | Without vaccination |
| 56 | CPV-2a | 7 months | Male | Shih Tzu | Incomplete |
| Sample | Variant | Amino Acid Position | |||||
|---|---|---|---|---|---|---|---|
| 267 | 297 | 324 | 426 | 440 | 514 | ||
| MF177231 | CPV-2a | F | A | Y | N | T | A |
| EU659119 | CPV-2b | F | A | Y | D | T | A |
| MF177238 | CPV-2c | F | A | Y | E | T | A |
| 1 | CPV-2a | F | N | I | N | T | S |
| 5 | CPV-2a | F | N | I | N | T | S |
| 7 | CPV-2a | F | N | I | N | T | S |
| 17 | CPV-2a | F | N | I | N | T | S |
| 19 | CPV-2a | F | N | I | N | T | S |
| 20 | CPV-2a | F | N | I | N | T | S |
| 21 | CPV-2a | F | N | I | N | T | S |
| 24 | CPV-2a | F | N | I | N | T | S |
| 26 | CPV-2a | F | N | I | N | T | S |
| 29 | CPV-2a | F | N | I | N | T | S |
| 32 | CPV-2a | F | N | I | N | T | S |
| 33 | CPV-2a | F | N | I | N | T | S |
| 36 | CPV-2a | F | N | I | N | T | S |
| 37 | CPV-2a | F | N | I | N | T | S |
| 38 | CPV-2a | F | N | I | N | T | S |
| 40 | CPV-2a | F | N | I | N | T | S |
| 41 | CPV-2a | F | N | I | N | T | S |
| 43 | CPV-2a | F | N | I | N | T | S |
| 44 | CPV-2a | F | N | I | N | T | S |
| 45 | CPV-2a | F | N | I | N | T | S |
| 47 | CPV-2a | F | N | I | N | T | S |
| 48 | CPV-2a | F | N | I | N | T | S |
| 50 | CPV-2b | Y | A | I | D | A | A |
| 51 | CPV-2a | F | N | I | N | T | S |
| 52 | CPV-2a | F | N | I | N | T | S |
| 53 | CPV-2a | F | N | I | N | T | S |
| 54 | CPV-2a | F | N | I | N | T | S |
| 55 | CPV-2b | Y | A | I | D | A | A |
| 56 | CPV-2a | F | N | I | N | T | S |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giraldo-Ramirez, S.; Rendon-Marin, S.; Ruiz-Saenz, J. Phylogenetic, Evolutionary and Structural Analysis of Canine Parvovirus (CPV-2) Antigenic Variants Circulating in Colombia. Viruses 2020, 12, 500. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050500
Giraldo-Ramirez S, Rendon-Marin S, Ruiz-Saenz J. Phylogenetic, Evolutionary and Structural Analysis of Canine Parvovirus (CPV-2) Antigenic Variants Circulating in Colombia. Viruses. 2020; 12(5):500. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050500
Chicago/Turabian StyleGiraldo-Ramirez, Sebastián, Santiago Rendon-Marin, and Julián Ruiz-Saenz. 2020. "Phylogenetic, Evolutionary and Structural Analysis of Canine Parvovirus (CPV-2) Antigenic Variants Circulating in Colombia" Viruses 12, no. 5: 500. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050500