Asian Zika Virus Isolate Significantly Changes the Transcriptional Profile and Alternative RNA Splicing Events in a Neuroblastoma Cell Line

 , and
, and 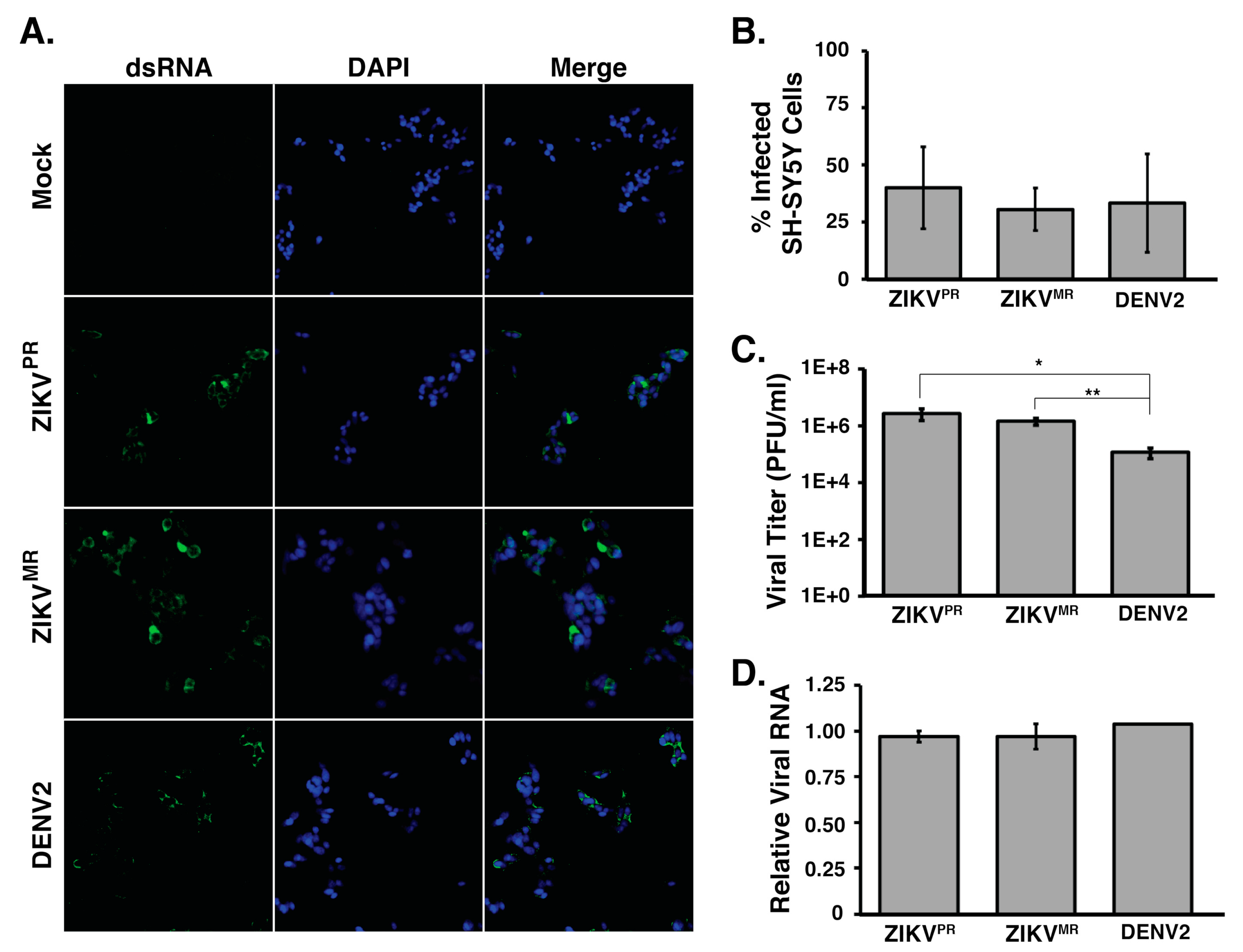{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Maintenance
2.2. Preparation of Virus Stocks
2.3. Viral Infections
2.4. Harvest of Virus-Infected Cells
2.5. Plaque Assays
2.6. Immunofluorescence Analysis
2.7. RT-qPCR
2.8. Preparation of Libraries for RNA-Seq
2.9. Bioinformatics Preprocessing
2.10. Differential Gene Expression (DGE) Analysis and Alternative Splicing
2.11. Alternative Splicing Analyses
2.12. Statistics
2.13. Data Access
3. Results
3.1. SH-SY5Y Cells are Permissive for Both Zika Virus Isolates and Dengue Virus
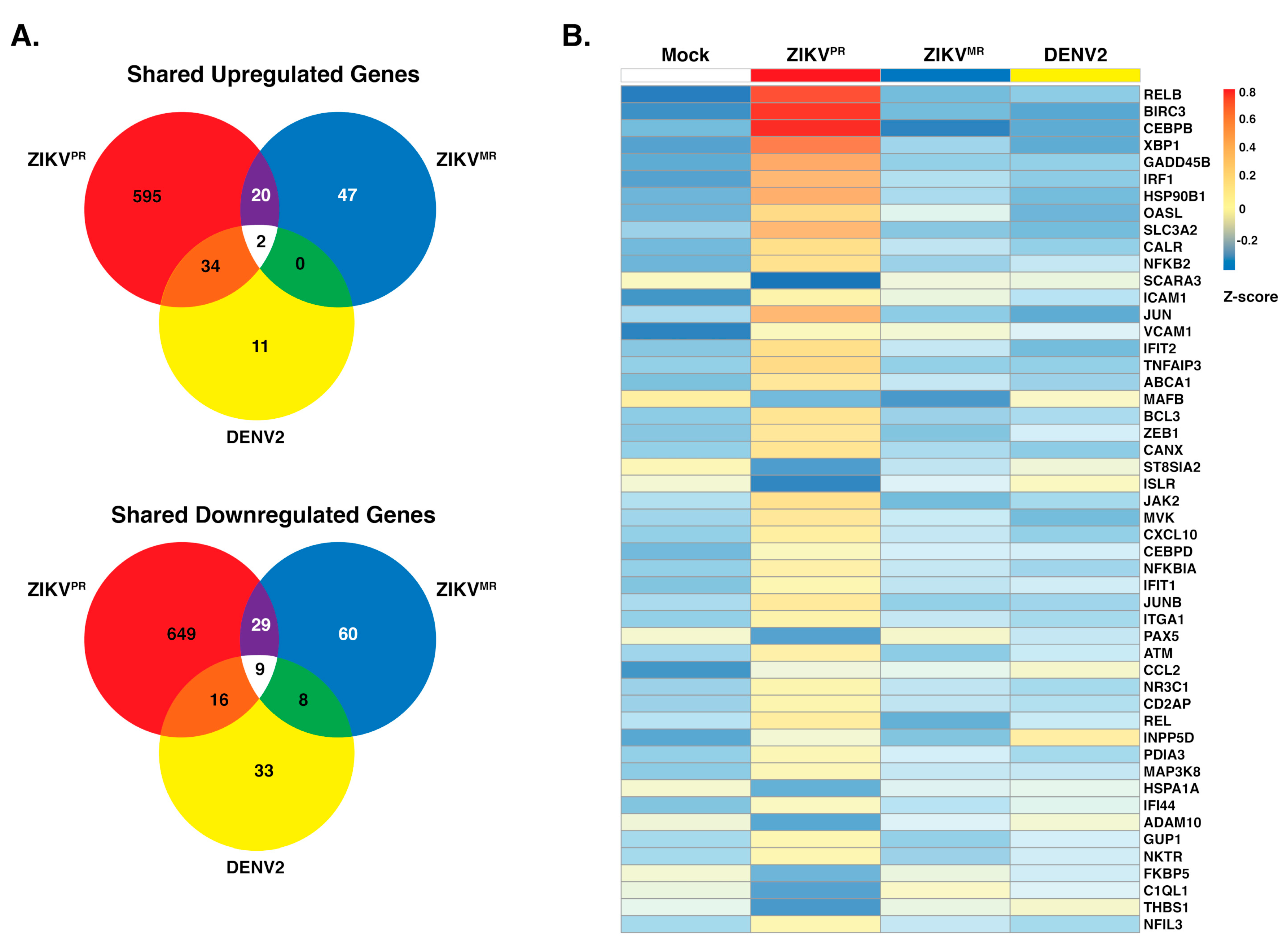
3.2. ZIKVPR Significantly Alters Differential Gene Expression Compared to ZIKVMR and DENV2
3.3. ZIKVPR Notably Upregulates Immune Response Genes Compared to ZIKVMR and DENV2
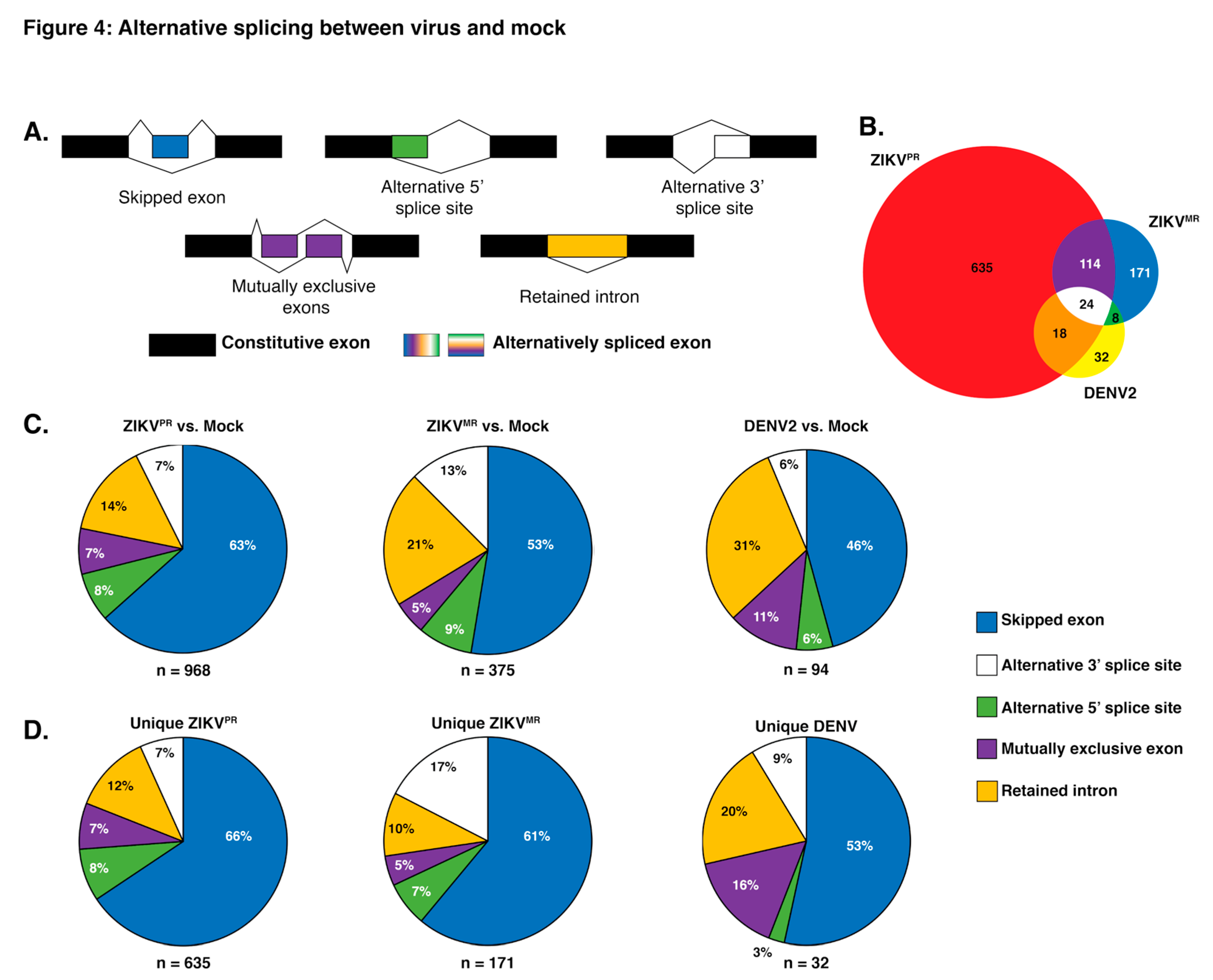
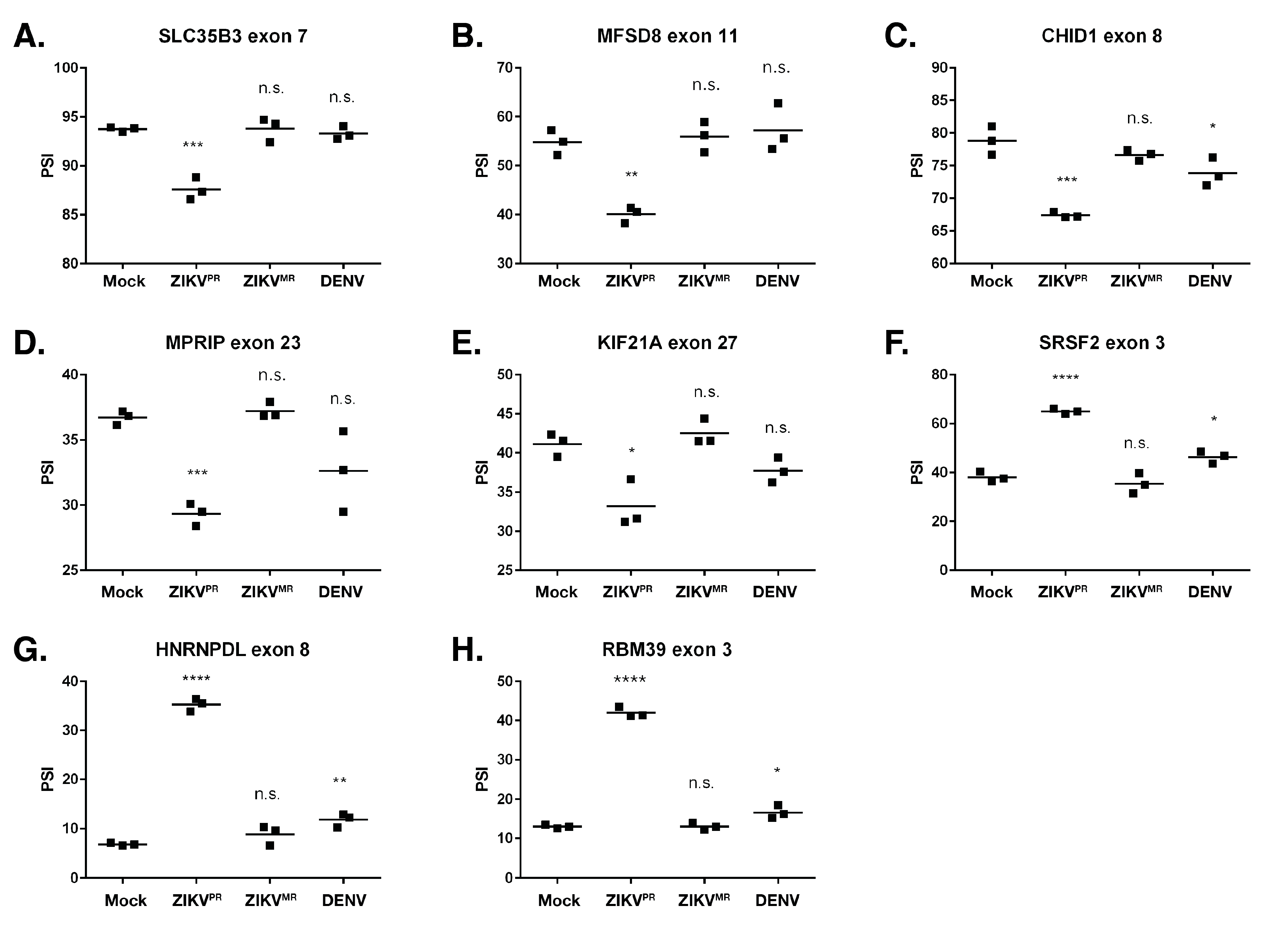
3.4. Exon Skipping is a Major Splicing Event during ZIKVPR, ZIKVMR, and DENV2 Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindenbach, B.D.; Murray, C.L.; Thiel, H.-J.; Rice, C.M. Flaviviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 712–746. ISBN 9781451105636. [Google Scholar]
- Gould, E.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef]
- Dick, G.W.A. Zika virus (II). Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 521–534. [Google Scholar] [CrossRef]
- Dick, G.W.A. Zika Virus (I). Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Fagbami, A.H. Zika virus infections in Nigeria: Virological and seroepidemiological investigations in Oyo State. J. Hyg. (Lond). 1979, 83, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, D.L.; Causey, O.R.; Carey, D.E.; Reddy, S.; Cooke, A.R.; Akinkugbe, F.M.; David-West, T.S.; Kemp, G.E. Arthropod-borne viral infections of man in Nigeria, 1964-1970. Ann. Trop. Med. Parasitol. 1975, 69, 49–64. [Google Scholar] [CrossRef]
- Olson, J.G.; Ksiazek, T.G.; Suhandiman, G.; Triwibowo, V. Zika virus, a cause of fever in Central Java, Indonesia. Trans. R. Soc. Trop. Med. Hyg. 1981, 75, 389–393. [Google Scholar] [CrossRef]
- Simpson, D.I.H. Zika virus infection in man. Trans. R. Soc. Trop. Med. Hyg. 1964, 58, 335–338. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.-H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef]
- Heang, V.; Yasuda, C.Y.; Sovann, L.; Haddow, A.D.; Travassos da Rosa, A.P.; Tesh, R.B.; Kasper, M.R. Zika Virus Infection, Cambodia, 2010. Emerg. Infect. Dis. 2012, 18, 349–351. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.M.; Roche, C.; Teissier, A.; Robin, E.; Berry, A.L.; Mallet, H.P.; Sall, A.A.; Musso, D. Zika virus, French Polynesia, South Pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1085–1086. [Google Scholar] [CrossRef]
- Alvarado, M.G.; Schwartz, D.A. Zika Virus Infection in Pregnancy, Microcephaly, and Maternal and Fetal Health: What We Think, What We Know, and What We Think We Know. Arch. Pathol. Lab. Med. 2017, 141, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martines, R.B.; Bhatnagar, J.; De Oliveira Ramos, A.M.; Davi, H.P.F.; Iglezias, S.D.A.; Kanamura, C.T.; Keating, M.K.; Hale, G.; Silva-Flannery, L.; Muehlenbachs, A.; et al. Pathology of congenital Zika syndrome in Brazil: A case series. Lancet 2016, 388, 898–904. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Xu, D.; Ye, Q.; Hong, S.; Jiang, Y.; Liu, X.; Zhang, N.; Shi, L.; Qin, C.-F.F.; Xu, Z.; et al. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell 2016, 19, 120–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miner, J.J.; Diamond, M.S. Understanding How Zika Virus Enters and Infects Neural Target Cells. Cell Stem Cell 2016, 18, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Da Silva, I.R.F.; Frontera, J.A.; De Filippis, A.M.B.; Nascimento, O.J.M.D.; RIO-GBS-ZIKV Research Group. Neurologic Complications Associated With the Zika Virus in Brazilian Adults. JAMA Neurol. 2017, 74, 1190–1198. [Google Scholar] [CrossRef]
- Nascimento, O.J.M.; Da Silva, I.R.F. Guillain–Barré syndrome and Zika virus outbreaks. Curr. Opin. Neurol. 2017, 30, 500–507. [Google Scholar] [CrossRef]
- Styczynski, A.R.; Malta, J.M.A.S.; Krow-Lucal, E.R.; Percio, J.; Nóbrega, M.E.; Vargas, A.; Lanzieri, T.M.; Leite, P.L.; Staples, J.E.; Fischer, M.X.; et al. Increased rates of Guillain-Barré syndrome associated with Zika virus outbreak in the Salvador metropolitan area, Brazil. PLoS Negl. Trop. Dis. 2017, 11, e0005869. [Google Scholar] [CrossRef]
- Salinas, J.L.; Walteros, D.M.; Styczynski, A.; Garzón, F.; Quijada, H.; Bravo, E.; Chaparro, P.; Madero, J.; Acosta-Reyes, J.; Ledermann, J.; et al. Zika virus disease-associated Guillain-Barré syndrome—Barranquilla, Colombia 2015–2016. J. Neurol. Sci. 2017, 381, 272–277. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Jacob, F.; Song, H.; Ming, G. Using brain organoids to understand Zika virus-induced microcephaly. Development 2017, 144, 952–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, G.l.; Tang, H.; Song, H. Advances in Zika Virus Research: Stem Cell Models, Challenges, and Opportunities. Cell Stem Cell 2016, 19, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Saucedo-Cuevas, L.; Regla-Nava, J.A.A.; Chai, G.; Sheets, N.; Tang, W.; Terskikh, A.V.V.; Shresta, S.; Gleeson, J.G.G. Zika Virus Infects Neural Progenitors in the Adult Mouse Brain and Alters Proliferation. Cell Stem Cell 2016, 19, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Hammack, C.; Ogden, S.C.; Cheng, Y.; Lee, E.M.; Wen, Z.; Qian, X.; Nguyen, H.N.; Li, Y.; Yao, B.; et al. Molecular signatures associated with ZIKV exposure in human cortical neural progenitors. Nucleic Acids Res. 2016, 44, 8610–8620. [Google Scholar] [CrossRef]
- Souza, B.S.F.; Sampaio, G.L.A.; Pereira, C.S.; Campos, G.S.; Sardi, S.I.; Freitas, L.A.R.; Figueira, C.P.; Paredes, B.D.; Nonaka, C.K.V.; Azevedo, C.M.; et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci. Rep. 2016, 6, 39775. [Google Scholar] [CrossRef]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Leon, W.R.M.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [Green Version]
- Simonin, Y.; Loustalot, F.; Desmetz, C.; Foulongne, V.; Constant, O.; Fournier-Wirth, C.; Leon, F.; Molès, J.-P.P.; Goubaud, A.; Lemaitre, J.-M.M.; et al. Zika Virus Strains Potentially Display Different Infectious Profiles in Human Neural Cells. EBioMedicine 2016, 12, 161–169. [Google Scholar] [CrossRef] [Green Version]
- McGrath, E.L.; Rossi, S.L.; Gao, J.; Widen, S.G.; Grant, A.C.; Dunn, T.J.; Azar, S.R.; Roundy, C.M.; Xiong, Y.; Prusak, D.J.; et al. Differential Responses of Human Fetal Brain Neural Stem Cells to Zika Virus Infection. Stem Cell Rep. 2017, 8, 715–727. [Google Scholar] [CrossRef]
- Goodfellow, F.T.; Willard, K.A.; Stice, S.L.; Brindley, M.A.; Wu, X.; Scoville, S. Strain-dependent consequences of zika virus infection and differential impact on neural development. Viruses 2018, 10, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brault, J.-B.B.; Khou, C.C.; Basset, J.; Coquand, L.; Fraisier, V.; Frenkiel, M.-P.P.; Goud, B.; Manuguerra, J.-C.C.; Pardigon, N.; Baffet, A.D. Comparative Analysis Between Flaviviruses Reveals Specific Neural Stem Cell Tropism for Zika Virus in the Mouse Developing Neocortex. EBioMedicine 2016, 10, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Bayless, N.L.; Greenberg, R.S.; Swigut, T.; Wysocka, J.; Blish, C.A. Zika Virus Infection Induces Cranial Neural Crest Cells to Produce Cytokines at Levels Detrimental for Neurogenesis. Cell Host Microbe 2016, 20, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonenfant, G.; Williams, N.; Netzband, R.; Schwarz, M.C.; Evans, M.J.; Pager, C.T. Zika Virus Subverts Stress Granules To Promote and Restrict Viral Gene Expression. J. Virol. 2019, 93, e00520-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo, J.M. Hu Antigen R (HuR) Functions as an Alternative Pre-mRNA Splicing Regulator of Fas Apoptosis-promoting Receptor on Exon Definition. J. Biol. Chem. 2008, 283, 19077–19084. [Google Scholar] [CrossRef] [Green Version]
- Barnhart, M.D.; Moon, S.L.; Emch, A.W.; Wilusz, C.J.; Wilusz, J. Changes in Cellular mRNA Stability, Splicing, and Polyadenylation through HuR Protein Sequestration by a Cytoplasmic RNA Virus. Cell Rep. 2013, 5, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Raj, B.; Blencowe, B.J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Mazin, P.; Xiong, J.; Liu, X.; Yan, Z.; Zhang, X.; Li, M.; He, L.; Somel, M.; Yuan, Y.; Phoebe Chen, Y.-P.; et al. Widespread splicing changes in human brain development and aging. Mol. Syst. Biol. 2014, 9, 633. [Google Scholar] [CrossRef]
- Dillman, A.A.; Hauser, D.N.; Gibbs, J.R.; Nalls, M.A.; McCoy, M.K.; Rudenko, I.N.; Galter, D.; Cookson, M.R. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat. Neurosci. 2013, 16, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Weyn-Vanhentenryck, S.M.; Wu, J.; Sloan, S.A.; Zhang, Y.; Chen, K.; Wu, J.Q.; Barres, B.A.; Zhang, C. Systematic discovery of regulated and conserved alternative exons in the mammalian brain reveals NMD modulating chromatin regulators. Proc. Natl. Acad. Sci. USA 2015, 112, 3445–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [Green Version]
- Blighe, K. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R package version 1.2.0. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 19 April 2020).
- Kolde, R. Pheatmap: Pretty Heatmaps; Software Version 1.0.8; University of Tartu: Tartu, Estonia, 2015; Available online: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed on 19 April 2020).
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Shen, S.; Park, J.W.; Lu, Z.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [Green Version]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Katz, Y.; Wang, E.T.; Silterra, J.; Schwartz, S.; Wong, B.; Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P.; Airoldi, E.M.; Burge, C.B. Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 2015, 31, 2400–2402. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Faye, O.; Freire, C.C.M.; Iamarino, A.; Faye, O.; De Oliveira, J.V.C.; Diallo, M.; Zanotto, P.M.A.; Sall, A.A. Molecular Evolution of Zika Virus during Its Emergence in the 20th Century. PLoS Negl. Trop. Dis. 2014, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.-I.; Song, B.-H.; Frank, J.C.; Julander, J.G.; Polejaeva, I.A.; Davies, C.J.; White, K.L.; Lee, Y.-M. Complete Genome Sequences of Three Historically Important, Spatiotemporally Distinct, and Genetically Divergent Strains of Zika Virus: MR-766, P6-740, and PRVABC-59: TABLE 1. Genome Announc. 2016, 4, e00800–e00816. [Google Scholar] [CrossRef] [Green Version]
- Lazear, H.M.; Diamond, M.S. Zika Virus: New Clinical Syndromes and Its Emergence in the Western Hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [Green Version]
- Lanciotti, R.S.; Lambert, A.J.; Holodniy, M.; Saavedra, S.; Del Carmen Castillo Signor, L. Phylogeny of zika virus in western Hemisphere, 2015. Emerg. Infect. Dis. 2016, 22, 933–935. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Shresta, S.; Gleeson, J.G. The Neurobiology of Zika Virus. Neuron 2016, 92, 949–958. [Google Scholar] [CrossRef]
- Yumoto, N.; Wakatsuki, S.; Kurisaki, T.; Hara, Y.; Osumi, N.; Frisén, J.; Sehara-Fujisawa, A. Meltrin β/ADAM19 Interacting with EphA4 in Developing Neural Cells Participates in Formation of the Neuromuscular Junction. PLoS ONE 2008, 3, e3322. [Google Scholar] [CrossRef] [Green Version]
- Park, J.G.; Tischfield, M.A.; Nugent, A.A.; Cheng, L.; Di Gioia, S.A.; Chan, W.-M.; Maconachie, G.; Bosley, T.M.; Summers, C.G.; Hunter, D.G.; et al. Loss of MAFB Function in Humans and Mice Causes Duane Syndrome, Aberrant Extraocular Muscle Innervation, and Inner-Ear Defects. Am. J. Hum. Genet. 2016, 98, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Huber, J.D.; Campos, C.R.; Mark, K.S.; Davis, T.P. Alterations in blood-brain barrier ICAM-1 expression and brain microglial activation after λ-carrageenan-induced inflammatory pain. Am. J. Physiol. Circ. Physiol. 2006, 290, H732–H740. [Google Scholar] [CrossRef] [Green Version]
- Haarmann, A.; Nowak, E.; Deiß, A.; Van der Pol, S.; Monoranu, C.-M.; Kooij, G.; Müller, N.; Van der Valk, P.; Stoll, G.; De Vries, H.E.; et al. Soluble VCAM-1 impairs human brain endothelial barrier integrity via integrin α-4-transduced outside-in signalling. Acta Neuropathol. 2015, 129, 639–652. [Google Scholar] [CrossRef] [Green Version]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef] [Green Version]
- Li, R.Z.; Hou, J.; Wei, Y.; Luo, X.; Ye, Y.; Zhang, Y. hnRNPDL extensively regulates transcription and alternative splicing. Gene 2019, 687, 125–134. [Google Scholar] [CrossRef]
- Jung, D.J.; Na, S.Y.; Na, D.S.; Lee, J.W. Molecular cloning and characterization of CAPER, a novel coactivator of activating protein-1 and estrogen receptors. J. Biol. Chem. 2002, 277, 1229–1234. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Cheng, Y.; Liu, Y.; Chen, L.; Liu, L.; Wei, N.; Xie, Z.; Wu, W.; Feng, Y. SRSF2 Regulates Alternative Splicing to Drive Hepatocellular Carcinoma Development. Cancer Res. 2017, 77, 1168–1178. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Zhao, Y.; Bai, X.; Liu, Y.; Green, T.J.; Luo, M.; Zheng, X. Structure of human stabilin-1 interacting chitinase-like protein (SI-CLP) reveals a saccharide-binding cleft with lower sugar-binding selectivity. J. Biol. Chem. 2010, 285, 39898–39904. [Google Scholar] [CrossRef] [Green Version]
- Van der Vaart, B.; Van Riel, W.E.; Doodhi, H.; Kevenaar, J.T.; Katrukha, E.A.; Gumy, L.; Bouchet, B.P.; Grigoriev, I.; Spangler, S.A.; Yu, K.L.; et al. CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev. Cell 2013, 27, 145–160. [Google Scholar] [CrossRef] [Green Version]
- Agol, V.I.; Drozdov, S.G.; Ivannikova, T.A.; Kolesnikova, M.S.; Korolev, M.B.; Tolskaya, E.A. Restricted growth of attenuated poliovirus strains in cultured cells of a human neuroblastoma. J. Virol. 1989, 63, 4034–4038. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.M.; Hixon, A.M.; Oldfield, L.M.; Zhang, Y.; Novotny, M.; Wang, W.; Das, S.R.; Shabman, R.S.; Tyler, K.L.; Scheuermann, R.H. Contemporary circulating enterovirus D68 strains have acquired the capacity for viral entry and replication in human neuronal cells. MBio 2018, 9, e01954-18. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.; Steain, M.; Slobedman, B.; Abendroth, A. Differentiated Neuroblastoma Cells Provide a Highly Efficient Model for Studies of Productive Varicella-Zoster Virus Infection of Neuronal Cells. J. Virol. 2011, 85, 8436–8442. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.H.; Fortunato, E.A. Long-Term Infection and Shedding of Human Cytomegalovirus in T98G Glioblastoma Cells. J. Virol. 2007, 81, 10424–10436. [Google Scholar] [CrossRef] [Green Version]
- Shipley, M.M.; Mangold, C.A.; Kuny, C.V.; Szpara, M.L. Differentiated Human SH-SY5Y Cells Provide a Reductionist Model of Herpes Simplex Virus 1 Neurotropism. J. Virol. 2017, 91, e00958-17. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.-I.; Choi, Y.-J.; Song, B.-H.; Lee, Y.-M. 3′ cis-Acting Elements That Contribute to the Competence and Efficiency of Japanese Encephalitis Virus Genome Replication: Functional Importance of Sequence Duplications, Deletions, and Substitutions. J. Virol. 2009, 83, 7909–7930. [Google Scholar] [CrossRef] [Green Version]
- Luplertlop, N.; Suwanmanee, S.; Muangkaew, W.; Ampawong, S.; Kitisin, T.; Poovorawan, Y. The impact of zika virus infection on human neuroblastoma (Sh-SY5Y) cell line. J. Vector Borne Dis. 2017, 54, 207–214. [Google Scholar] [CrossRef]
- Sánchez-San Martín, C.; Li, T.; Bouquet, J.; Streithorst, J.; Yu, G.; Paranjpe, A.; Chiu, C.Y. Differentiation enhances Zika virus infection of neuronal brain cells. Sci. Rep. 2018, 8, 14543. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Banerjee, S.; Wang, Y.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Silverman, R.H.; Weiss, S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.-J.; Yu, H.-P.; Chang, B.-L.; Tang, W.-C.; Liao, C.-L.; Lin, Y.-L. Distinct Antiviral Roles for Human 2′,5′-Oligoadenylate Synthetase Family Members against Dengue Virus Infection. J. Immunol. 2009, 183, 8035–8043. [Google Scholar] [CrossRef] [Green Version]
- Scherbik, S.V.; Paranjape, J.M.; Stockman, B.M.; Silverman, R.H.; Brinton, M.A. RNase L Plays a Role in the Antiviral Response to West Nile Virus. J. Virol. 2006, 80, 2987–2999. [Google Scholar] [CrossRef] [Green Version]
- Rolfe, A.J.; Bosco, D.B.; Wang, J.; Nowakowski, R.S.; Fan, J.; Ren, Y. Bioinformatic analysis reveals the expression of unique transcriptomic signatures in Zika virus infected human neural stem cells. Cell Biosci. 2016, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Caplen, F.V.; Krug, R.M. Regulation of the extent of splicing of influenza virus NS1 mRNA: Role of the rates of splicing and of the nucleocytoplasmic transport of NS1 mRNA. Mol. Cell. Biol. 1991, 11, 1092–1098. [Google Scholar] [CrossRef] [Green Version]
- Kreivi, J.P.; Zerivitz, K.; Akusjärvi, G. Sequences involved in the control of adenovirus L1 alternative RNA splicing. Nucleic Acids Res. 1991, 19, 2379–2386. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.K.; Flanagan, W.M.; Devi-Rao, G.; Zhang, Y.F.; Hill, J.M.; Anderson, K.P.; Stevens, J.G. The herpes simplex virus latency-associated transcript is spliced during the latent phase of infection. J. Virol. 1988, 62, 4577–4585. [Google Scholar] [CrossRef] [Green Version]
- De Maio, F.A.; Risso, G.; Iglesias, N.G.; Shah, P.; Pozzi, B.; Gebhard, L.G.; Mammi, P.; Mancini, E.; Yanovsky, M.J.; Andino, R.; et al. The Dengue Virus NS5 Protein Intrudes in the Cellular Spliceosome and Modulates Splicing. PLoS Pathog. 2016, 12, e1005841. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Boudreault, S.; Martenon-Brodeur, C.; Caron, M.; Garant, J.M.; Tremblay, M.P.; Armero, V.E.S.; Durand, M.; Lapointe, E.; Thibault, P.; Tremblay-Létourneau, M.; et al. Global profiling of the cellular alternative RNA splicing landscape during virus-host interactions. PLoS ONE 2016, 11, e0161914. [Google Scholar] [CrossRef] [Green Version]
- Braunschweig, U.; Barbosa-Morais, N.L.; Pan, Q.; Nachman, E.N.; Alipanahi, B.; Gonatopoulos-Pournatzis, T.; Frey, B.; Irimia, M.; Blencowe, B.J. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014, 24, 1774–1786. [Google Scholar] [CrossRef]
- Kamiyama, S.; Sasaki, N.; Goda, E.; Ui-Tei, K.; Saigo, K.; Narimatsu, H.; Jigami, Y.; Kannagi, R.; Irimura, T.; Nishihara, S. Molecular cloning and characterization of a novel 3′-phosphoadenosine 5′-phosphosulfate transporter, PAPST2. J. Biol. Chem. 2006, 281, 10945–10953. [Google Scholar] [CrossRef] [Green Version]
- Siintola, E.; Topcu, M.; Aula, N.; Lohi, H.; Minassian, B.A.; Paterson, A.D.; Liu, X.-Q.; Wilson, C.; Lahtinen, U.; Anttonen, A.-K.; et al. The Novel Neuronal Ceroid Lipofuscinosis Gene MFSD8 Encodes a Putative Lysosomal Transporter. Am. J. Hum. Genet. 2007, 81, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Santavuori, P. Neuronal ceroid-lipofuscinoses in childhood. Brain Dev. 1988, 10, 80–83. [Google Scholar] [CrossRef]
- Koga, Y.; Ikebe, M. p116Rip decreases myosin II phosphorylation by activating myosin light chain phosphatase and by inactivating RhoA. J. Biol. Chem. 2005, 280, 4983–4991. [Google Scholar] [CrossRef] [Green Version]
- Surks, H.K.; Riddick, N.; Ohtani, K. M-RIP Targets Myosin Phosphatase to Stress Fibers to Regulate Myosin Light Chain Phosphorylation in Vascular Smooth Muscle Cells. J. Biol. Chem. 2005, 280, 42543–42551. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.; Kumar, A.; Xu, Z.; Airo, A.M.; Stryapunina, I.; Wong, C.P.; Branton, W.; Tchesnokov, E.; Götte, M.; Power, C.; et al. Zika Virus Hijacks Stress Granule Proteins and Modulates the Host Stress Response. J. Virol. 2017, 91, e00474-17. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.-Y.; Ding, J.-H.; Adams, J.A.; Ghosh, G.; Fu, X.-D. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009, 23, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Huo, Y.; Yang, L.; Chen, G.; Luo, M.; Yang, J.; Zhou, J. ZIKV infection effects changes in gene splicing, isoform composition and lncRNA expression in human neural progenitor cells. Virol. J. 2017, 14, 217. [Google Scholar] [CrossRef] [Green Version]
- Kemmerer, K.; Fischer, S.; Weigand, J.E. Auto- and cross-regulation of the hnRNPs D and DL. RNA 2018, 24, 324–331. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonenfant, G.; Meng, R.; Shotwell, C.; Badu, P.; Payne, A.F.; Ciota, A.T.; Sammons, M.A.; Berglund, J.A.; Pager, C.T. Asian Zika Virus Isolate Significantly Changes the Transcriptional Profile and Alternative RNA Splicing Events in a Neuroblastoma Cell Line. Viruses 2020, 12, 510. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050510
Bonenfant G, Meng R, Shotwell C, Badu P, Payne AF, Ciota AT, Sammons MA, Berglund JA, Pager CT. Asian Zika Virus Isolate Significantly Changes the Transcriptional Profile and Alternative RNA Splicing Events in a Neuroblastoma Cell Line. Viruses. 2020; 12(5):510. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050510
Chicago/Turabian StyleBonenfant, Gaston, Ryan Meng, Carl Shotwell, Pheonah Badu, Anne F. Payne, Alexander T. Ciota, Morgan A. Sammons, J. Andrew Berglund, and Cara T. Pager. 2020. "Asian Zika Virus Isolate Significantly Changes the Transcriptional Profile and Alternative RNA Splicing Events in a Neuroblastoma Cell Line" Viruses 12, no. 5: 510. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050510