Glucose-Regulated Protein 78 Interacts with Zika Virus Envelope Protein and Contributes to a Productive Infection

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Strains
2.2. Cells
2.3. Virus Growth and Titration
2.4. Small Molecule Inhibitor Assays
2.5. Transfection of Nucleic Acids and Silencing of Gene Expression
2.6. Freeze/Thaw Assay
2.7. Dual Luciferase Assay
2.8. Protein Immunoprecipitation (IP)
2.9. Peptide Analysis by LC-MS/MS
2.10. Data Analysis
2.11. Immunoblot Analysis
2.12. Immunofluorescence Microscopy
2.13. cDNA Synthesis and RT-qPCR
2.14. Statistical Analysis
2.15. Data Availability
3. Results
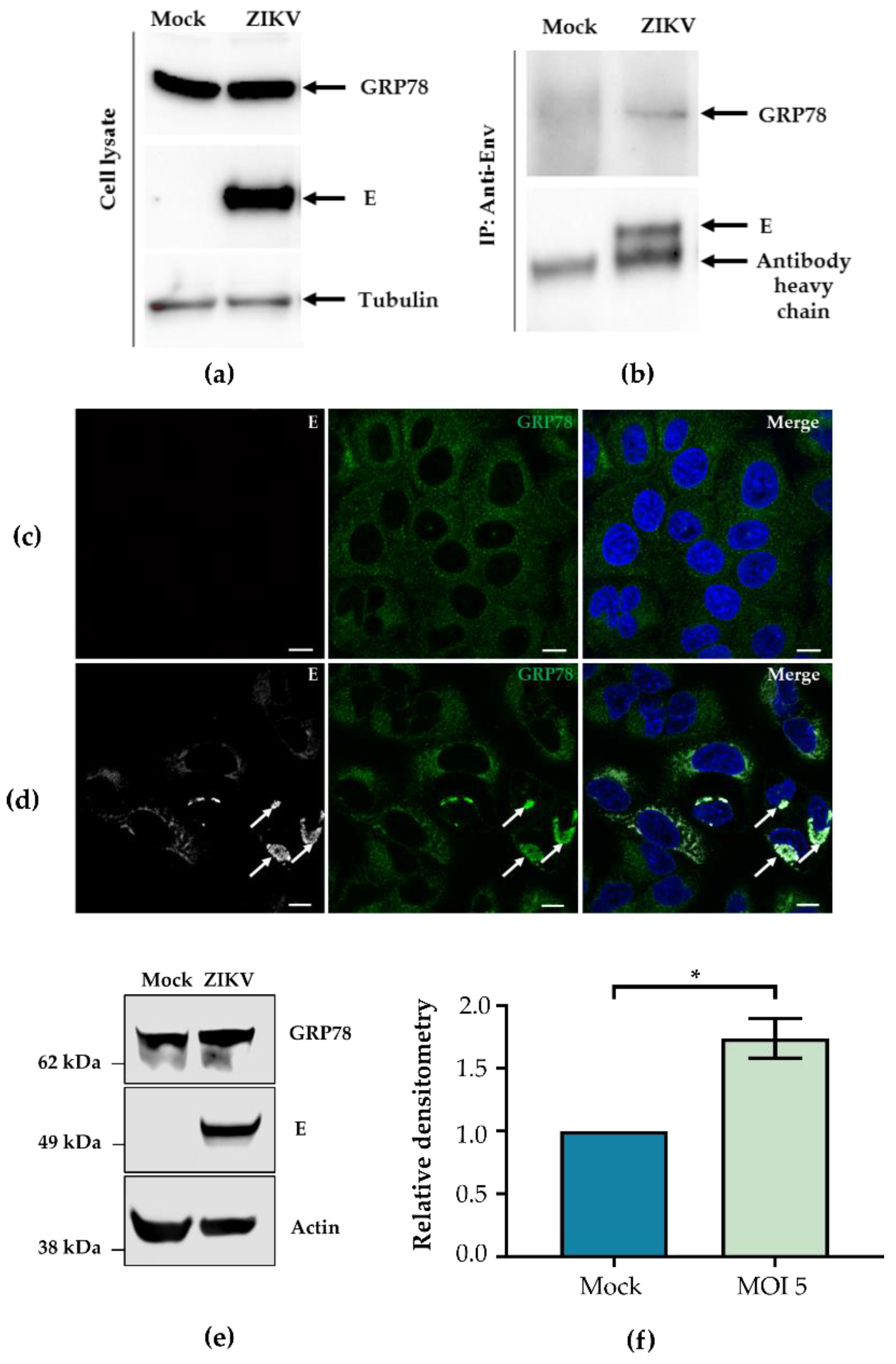
3.1. ZIKV E Interacts with GRP78
3.2. Small Molecule Inhibitors of GRP78-Mediated UPR do not Affect Viral Replication
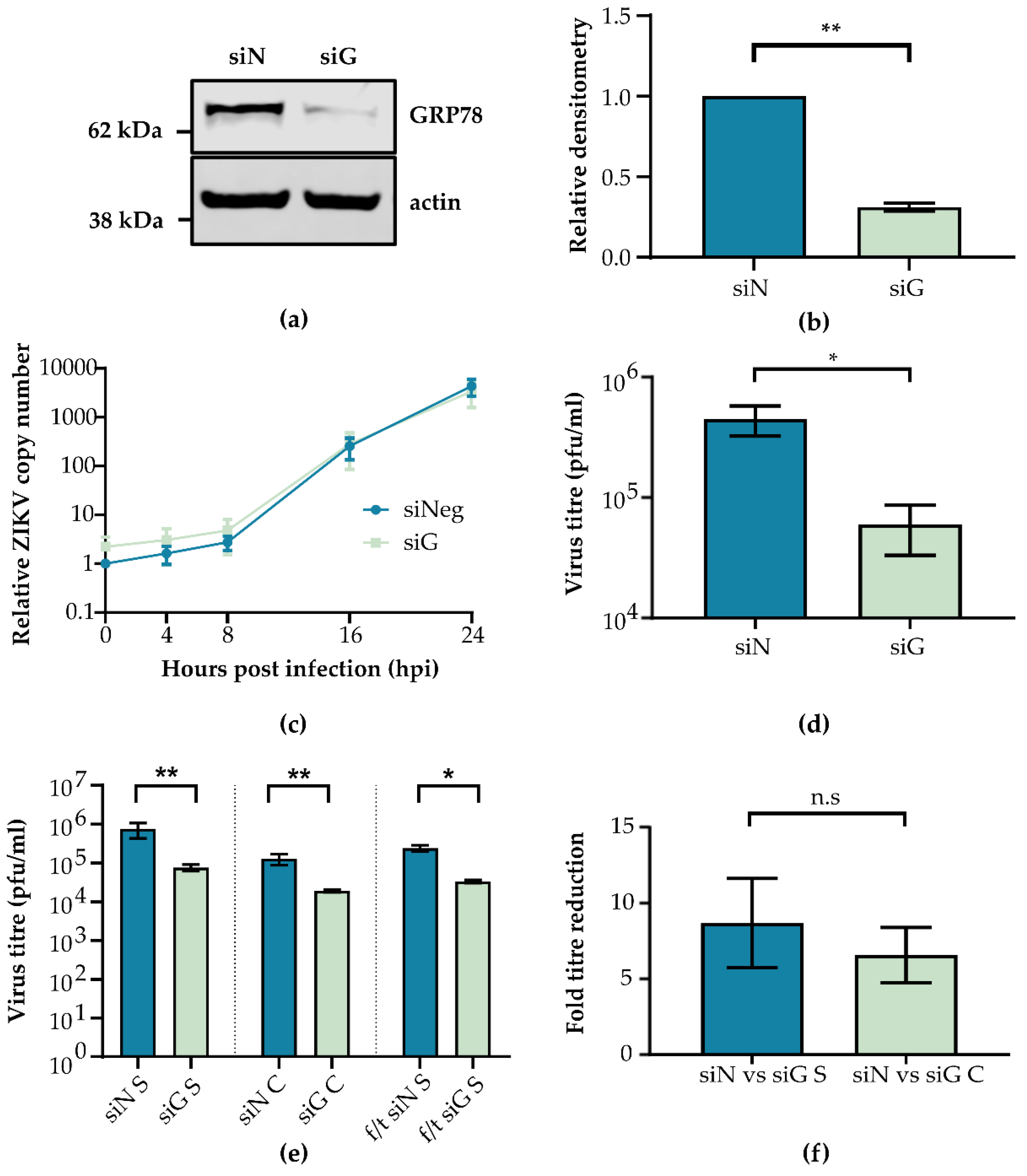
3.3. GRP78 Depletion Reduces ZIKV Infectious Titre
3.4. GRP78 Knockdown Specifically Impacts Viral Translation
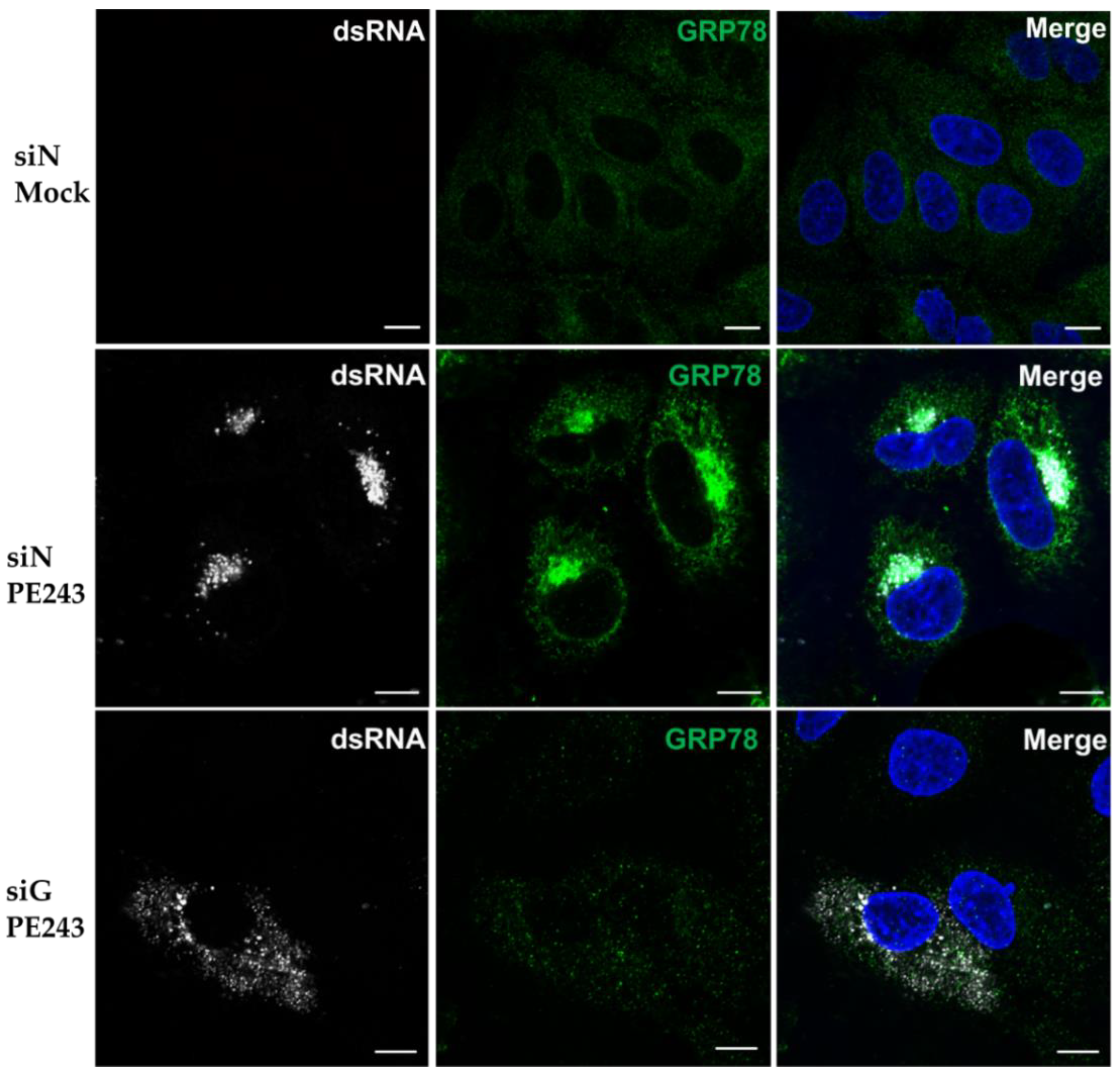
3.5. Viral dsRNA Localisation is Disrupted Following GRP78 Depletion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Simpson, D.I. Zika Virus Infection in Man. Trans. R. Soc. Trop. Med. Hyg. 1964, 58, 335–338. [Google Scholar] [CrossRef]
- Haddow, A.D.; Schuh, A.J.; Yasuda, C.Y.; Kasper, M.R.; Heang, V.; Huy, R.; Guzman, H.; Tesh, R.B.; Weaver, S.C. Genetic Characterization of Zika Virus Strains: Geographic Expansion of the Asian Lineage. PLoS Negl. Trop. Dis. 2012, 6, e1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, R.; Nguyen, D.; Debs, L.H.; Patel, A.P.; Liu, G.; Jhaveri, V.M.; S Kay, S.-I.; Mittal, J.; Bandstra, E.S.; Younis, R.T.; et al. Zika Virus: An Emerging Global Health Threat. Front. Cell. Infect. Microbiol. 2017, 7, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud, D.; Gubler, D.J.; Schaub, B.; Lanteri, M.C.; Musso, D. An update on Zika virus infection. Lancet 2017, 390, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Russo, F.B.; Jungmann, P.; Beltrão-Braga, P.C.B. Zika infection and the development of neurological defects. Cell. Microbiol. 2017, 19, e12744. [Google Scholar] [CrossRef] [Green Version]
- Barbeito-Andrés, J.; Schuler-Faccini, L.; Garcez, P.P. Why is congenital Zika syndrome asymmetrically distributed among human populations? PLoS Biol. 2018, 16, e2006592. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, G.F. Structural Biology of the Zika Virus. Trends Biochem. Sci. 2017, 42, 443–456. [Google Scholar] [CrossRef]
- Long, F.; Doyle, M.; Fernandez, E.; Miller, A.S.; Klose, T.; Sevvana, M.; Bryan, A.; Davidson, E.; Doranz, B.J.; Kuhn, R.J.; et al. Structural basis of a potent human monoclonal antibody against Zika virus targeting a quaternary epitope. Proc. Natl. Acad. Sci. USA 2019, 116, 1591–1596. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar]
- Sirohi, D.; Kuhn, R.J. Zika Virus Structure, Maturation, and Receptors. J. Infect. Dis. 2017, 216, S935–S944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrelli, A.; De Moura, R.R.; Crovella, S.; Brandão, L.A.C. ZIKA virus entry mechanisms in human cells. Infect. Genet. Evol. 2019, 69, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Laureti, M.; Narayanan, D.; Rodriguez-Andres, J.; Fazakerley, J.K.; Kedzierski, L. Flavivirus Receptors: Diversity, Identity, and Cell Entry. Front. Immunol. 2018, 9, 2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [Green Version]
- Rey, F.A.; Stiasny, K.; Heinz, F.X. Flavivirus structural heterogeneity: Implications for cell entry. Curr. Opin. Virol. 2017, 24, 132–139. [Google Scholar] [CrossRef]
- Ma, J.; Ketkar, H.; Geng, T.; Lo, E.; Wang, L.; Xi, J.; Sun, Q.; Zhu, Z.; Cui, Y.; Yang, L.; et al. Zika Virus Non-structural Protein 4A Blocks the RLR-MAVS Signaling. Front. Microbiol. 2018, 9, 1350. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef]
- Shah, P.S.; Link, N.; Jang, G.M.; Sharp, P.P.; Zhu, T.; Swaney, D.L.; Johnson, J.R.; Von Dollen, J.; Ramage, H.R.; Satkamp, L.; et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175, 1931–1945. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, K.A.; Leon, K.E.; Khalid, M.M.; Tomar, S.; Jimenez-Morales, D.; Dunlap, M.; Kaye, J.A.; Shah, P.S.; Finkbeiner, S.; Krogan, N.J.; et al. The cellular NMD pathway restricts zika virus infection and is targeted by the viral capsid protein. MBio 2018, 9, e02126-18. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Bos, S.; Li, G.; Wang, S.; Gadea, G.; Desprès, P.; Zhao, R.Y. Probing Molecular Insights into Zika Virus–Host Interactions. Viruses 2018, 10, 233. [Google Scholar] [CrossRef] [Green Version]
- Srisutthisamphan, K.; Jirakanwisal, K.; Ramphan, S.; Tongluan, N.; Kuadkitkan, A.; Smith, D.R. Hsp90 interacts with multiple dengue virus 2 proteins. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, S.S.; Sevvana, M.; Kuhn, R.J.; Rossmann, M.G. Structural biology of Zika virus and other flaviviruses. Nat. Struct. Mol. Biol. 2018, 25, 13–20. [Google Scholar] [CrossRef]
- Carbaugh, D.L.; Baric, R.S.; Lazear, H.M. Envelope Protein Glycosylation Mediates Zika Virus Pathogenesis. J. Virol. 2019, 93, e00113-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontes-Garfias, C.R.; Shan, C.; Luo, H.; Muruato, A.E.; Medeiros, D.B.A.; Mays, E.; Xie, X.; Zou, J.; Roundy, C.M.; Wakamiya, M.; et al. Functional Analysis of Glycosylation of Zika Virus Envelope Protein. Cell Rep. 2017, 21, 1180–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ni, M.; Lee, B.; Barron, E.; Hinton, D.R.; Lee, A.S. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008, 15, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Quinones, Q.J.; De Ridder, G.G.; Pizzo, S. V GRP78: A chaperone with diverse roles beyond the endoplasmic reticulum. Histol. Histopathol. 2008, 23, 1409–1416. [Google Scholar]
- Alfano, C.; Gladwyn-Ng, I.; Couderc, T.; Lecuit, M.; Nguyen, L. The unfolded protein response: A key player in zika virus-associated congenital microcephaly. Front. Cell. Neurosci. 2019, 13, 94. [Google Scholar] [CrossRef]
- Tan, Z.; Zhang, W.; Sun, J.; Fu, Z.; Ke, X.; Zheng, C.; Zhang, Y.; Li, P.; Liu, Y.; Hu, Q.; et al. ZIKV infection activates the IRE1-XBP1 and ATF6 pathways of unfolded protein response in neural cells. J. Neuroinflammation 2018, 15, 275. [Google Scholar] [CrossRef] [Green Version]
- Donald, C.L.; Brennan, B.; Cumberworth, S.L.; Rezelj, V.V.; Clark, J.J.; Cordeiro, M.T.; Freitas de Oliveira França, R.; Pena, L.J.; Wilkie, G.S.; Da Silva Filipe, A.; et al. Full Genome Sequence and sfRNA Interferon Antagonist Activity of Zika Virus from Recife, Brazil. PLoS Negl. Trop. Dis. 2016, 10, e0005048. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Magri, A.; Hill, M.; Lai, A.G.; Kumar, A.; Rambhatla, S.B.; Donald, C.L.; Lopez-Clavijo, A.F.; Rudge, S.; Pinnick, K.; et al. The circadian clock components BMAL1 and REV-ERBα regulate flavivirus replication. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutso, M.; Saul, S.; Rausalu, K.; Susova, O.; Žusinaite, E.; Mahalingam, S.; Merits, A. Reverse genetic system, genetically stable reporter viruses and packaged subgenomic replicon based on a Brazilian Zika virus isolate. J. Gen. Virol. 2017, 98, 2712–2724. [Google Scholar] [CrossRef] [PubMed]
- Hilton, L.; Moganeradj, K.; Zhang, G.; Chen, Y.-H.; Randall, R.E.; McCauley, J.W.; Goodbourn, S. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J. Virol. 2006, 80, 11723–11732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Nain, M.; Mukherjee, S.; Karmakar, S.P.; Paton, A.W.; Paton, J.C.; Abdin, M.Z.; Basu, A.; Kalia, M.; Vrati, S. GRP78 Is an Important Host Factor for Japanese Encephalitis Virus Entry and Replication in Mammalian Cells. J. Virol. 2017, 91, e02274-16. [Google Scholar] [CrossRef] [Green Version]
- Wati, S.; Soo, M.-L.; Zilm, P.; Li, P.; Paton, A.W.; Burrell, C.J.; Beard, M.; Carr, J.M. Dengue virus infection induces upregulation of GRP78, which acts to chaperone viral antigen production. J. Virol. 2009, 83, 12871–12880. [Google Scholar] [CrossRef] [Green Version]
- Buchkovich, N.J.; Maguire, T.G.; Yu, Y.; Paton, A.W.; Paton, J.C.; Alwine, J.C. Human Cytomegalovirus Specifically Controls the Levels of the Endoplasmic Reticulum Chaperone BiP/GRP78, Which Is Required for Virion Assembly. J. Virol. 2008, 82, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Gurusinghe, K.R.D.S.N.S.; Mishra, A.; Mishra, S. Glucose-regulated protein 78 substrate-binding domain alters its conformation upon EGCG inhibitor binding to nucleotide-binding domain: Molecular dynamics studies. Sci. Rep. 2018, 8, 5487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Tam, A.B.; Roberts, L.S.; Chandra, V.; Rivera, G.; Nomura, D.K.; Forbes, D.J. The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev. Cell 2018, 46, 327–343. [Google Scholar] [CrossRef] [Green Version]
- C Bell, M.; E Meier, S.; L Ingram, A.; F Abisambra, J. PERK-opathies: An Endoplasmic Reticulum Stress Mechanism Underlying Neurodegeneration. Curr. Alzheimer Res. 2015, 13, 150–163. [Google Scholar] [CrossRef]
- Martin, S.; Lamb, H.K.; Brady, C.; Lefkove, B.; Bonner, M.Y.; Thompson, P.; Lovat, P.E.; Arbiser, J.L.; Hawkins, A.R.; Redfern, C.P.F. Inducing apoptosis of cancer cells using small-molecule plant compounds that bind to GRP78. Br. J. Cancer 2013, 109, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, B.M.; Batista, M.N.; Braga, A.C.S.; Nogueira, M.L.; Rahal, P. The green tea molecule EGCG inhibits Zika virus entry. Virology 2016, 496, 215–218. [Google Scholar] [CrossRef]
- Sharma, N.; Murali, A.; Singh, S.K.; Giri, R. Epigallocatechin gallate, an active green tea compound inhibits the Zika virus entry into host cells via binding the envelope protein. Int. J. Biol. Macromol. 2017, 104, 1046–1054. [Google Scholar] [CrossRef]
- Hierholzer, J.C.; Killington, R.A.; Stokes, A. Preparation of antigens. In Virology Methods Manual; Elsevier: Amsterdam, The Netherlands, 1996; pp. 47–70. [Google Scholar]
- Harak, C.; Lohmann, V. Ultrastructure of the replication sites of positive-strand RNA viruses. Virology 2015, 479, 418–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Bartenschlager, R. Membranous replication factories induced by plus-strand RNA viruses. Viruses 2014, 6, 2826–2857. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Kumar, M. Role of Endoplasmic Reticulum-Associated Proteins in Flavivirus Replication and Assembly Complexes. Pathogens 2019, 8, 148. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, M.; Wakita, T.; Esumi, M. 2′,5′-Oligoadenylate synthetase-like gene highly induced by hepatitis C virus infection in human liver is inhibitory to viral replication in vitro. Biochem. Biophys. Res. Commun. 2010, 392, 397–402. [Google Scholar] [CrossRef]
- Choi, U.Y.; Kang, J.S.; Hwang, Y.S.; Kim, Y.J. Oligoadenylate synthase-like (OASL) proteins: Dual functions and associations with diseases. Exp. Mol. Med. 2015, 47, e144. [Google Scholar] [CrossRef] [Green Version]
- Corry, J.; Arora, N.; Good, C.A.; Sadovsky, Y.; Coyne, C.B. Organotypic models of type III interferon-mediated protection from Zika virus infections at the maternal–fetal interface. Proc. Natl. Acad. Sci. USA 2017, 114, 9433–9438. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Liu, Q.; Han, K.; Wang, H.; Yang, J.; Bi, K.; Liu, Y.; Liu, N.; Tian, Y.; Li, Y. Identification of Glucose-Regulated Protein 78 (GRP78) as a Receptor in BHK-21 Cells for Duck Tembusu Virus Infection. Front. Microbiol. 2018, 9, 694. [Google Scholar] [CrossRef]
- Jindadamrongwech, S.; Thepparit, C.; Smith, D.R. Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch. Virol. 2004, 149, 915–927. [Google Scholar] [CrossRef]
- Limjindaporn, T.; Wongwiwat, W.; Noisakran, S.; Srisawat, C.; Netsawang, J.; Puttikhunt, C.; Kasinrerk, W.; Avirutnan, P.; Thiemmeca, S.; Sriburi, R.; et al. Interaction of dengue virus envelope protein with endoplasmic reticulum-resident chaperones facilitates dengue virus production. Biochem. Biophys. Res. Commun. 2009, 379, 196–200. [Google Scholar] [CrossRef]
- Kumar, A.; Buhler, S.; Selisko, B.; Davidson, A.; Mulder, K.; Canard, B.; Miller, S.; Bartenschlager, R. Nuclear Localization of Dengue Virus Nonstructural Protein 5 Does Not Strictly Correlate with Efficient Viral RNA Replication and Inhibition of Type I Interferon Signaling. J. Virol. 2013, 87, 4545–4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.; Luo, G. Zika virus NS5 nuclear accumulation is protective of protein degradation and is required for viral RNA replication. Virology 2020, 541, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Roth, H.; Magg, V.; Uch, F.; Mutz, P.; Klein, P.; Haneke, K.; Lohmann, V.; Bartenschlager, R.; Fackler, O.T.; Locker, N.; et al. Flavivirus infection uncouples translation suppression from cellular stress responses. MBio 2017, 8, e02150-16. [Google Scholar] [CrossRef] [Green Version]
- Bushell, M.; Sarnow, P. Hijacking the translation apparatus by RNA viruses. J. Cell Biol. 2002, 158, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [Green Version]
- Bílý, T.; Palus, M.; Eyer, L.; Elsterová, J.; Vancová, M.; Růžek, D. Electron Tomography Analysis of Tick-Borne Encephalitis Virus Infection in Human Neurons. Sci. Rep. 2015, 5, 10745. [Google Scholar] [CrossRef] [Green Version]
- Junjhon, J.; Pennington, J.G.; Edwards, T.J.; Perera, R.; Lanman, J.; Kuhn, R.J. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J. Virol. 2014, 88, 4687–4697. [Google Scholar] [CrossRef] [Green Version]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef] [Green Version]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.E.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and Three-Dimensional Architecture of the Dengue Virus Replication and Assembly Sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Scaturro, P.; Kastner, A.L.; Pichlmair, A. Chasing intracellular Zika virus using proteomics. Viruses 2019, 11, 878. [Google Scholar] [CrossRef] [Green Version]
- Coyaud, E.; Ranadheera, C.; Cheng, D.; Gonçalves, J.; Dyakov, B.J.A.; Laurent, E.M.N.; St-Germain, J.; Pelletier, L.; Gingras, A.C.; Brumell, J.H.; et al. Global interactomics uncovers extensive organellar targeting by Zika Virus. Mol. Cell. Proteom. 2018, 17, 2242–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaturro, P.; Stukalov, A.; Haas, D.A.; Cortese, M.; Draganova, K.; Płaszczyca, A.; Bartenschlager, R.; Götz, M.; Pichlmair, A. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature 2018, 561, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Cash, D.R.; Tavallai, S.; Jean, S.; Fidanza, A.; Cruz-Luna, T.; Siembiba, P.; Cycon, K.A.; Cornelissen, C.N.; et al. GRP78/BiP/HSPA5/Dna K is a universal therapeutic target for human disease. J. Cell. Physiol. 2015, 230, 1661–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia-Rong, J.; Shin-Chyang, W.; Chao-Rih, J.; Jim-Tong, H. Enterovirus 71 induces dsRNA/PKR-dependent cytoplasmic redistribution of GRP78/BiP to promote viral replication. Emerg. Microbes Infect. 2016, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Roux, L. Selective and transient association of sendai virus HN glycoprotein with BiP. Virology 1990, 175, 161–166. [Google Scholar] [CrossRef]
- Anderson, K.; Stott, E.J.; Wertz, G.W. Intracellular processing of the human respiratory syncytial virus fusion glycoprotein: Amino acid substitutions affecting folding, transport and cleavage. J. Gen. Virol. 1992, 73, 1177–1188. [Google Scholar] [CrossRef]
- Hogue, B.G.; Nayak, D.P. Synthesis and processing of the influenza virus neuraminidase, a type II transmembrane glycoprotein. Virology 1992, 188, 510–517. [Google Scholar] [CrossRef]
- Xu, Z.; Jensen, G.; Yen, T.S. Activation of hepatitis B virus S promoter by the viral large surface protein via induction of stress in the endoplasmic reticulum. J. Virol. 1997, 71, 7387–7392. [Google Scholar] [CrossRef] [Green Version]
- Bolt, G. The measles virus (MV) glycoproteins interact with cellular chaperones in the endoplasmic reticulum and MV infection upregulates chaperone expression. Arch. Virol. 2001, 146, 2055–2068. [Google Scholar] [CrossRef]
- Dimcheff, D.E.; Faasse, M.A.; McAtee, F.J.; Portis, J.L. Endoplasmic reticulum (ER) stress induced by a neurovirulent mouse retrovirus is associated with prolonged BiP binding and retention of a viral protein in the ER. J. Biol. Chem. 2004, 279, 33782–33790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.Y.L.; Wu, Y.J.; Chen, H.S.; Chen, C.J. A KDEL retrieval system for ER-golgi transport of Japanese encephalitis viral particles. Viruses 2016, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, C.-M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.-H.; Sze, K.-H.; Yang, D.; Shuai, H.; et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | UniProt Accession | Infection, Sample 1 | Infection, Sample 2 | Infection, Sample 3 | Control, Sample 4 | Control, Sample 5 | Control, Sample 6 |
|---|---|---|---|---|---|---|---|
| LMNA | P02545 | Yes | Yes | Yes | No | Yes | No |
| PGAM5 | Q96HS1 | Yes | Yes | No | Yes | No | No |
| GRP78 | P11021 | Yes | Yes | Yes | No | No | No |
| OASL | Q15646 | Yes | Yes | No | No | No | No |
| TAO1 | Q7L7X3 | Yes | Yes | Yes | No | No | No |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Royle, J.; Ramírez-Santana, C.; Akpunarlieva, S.; Donald, C.L.; Gestuveo, R.J.; Anaya, J.-M.; Merits, A.; Burchmore, R.; Kohl, A.; Varjak, M. Glucose-Regulated Protein 78 Interacts with Zika Virus Envelope Protein and Contributes to a Productive Infection. Viruses 2020, 12, 524. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050524
Royle J, Ramírez-Santana C, Akpunarlieva S, Donald CL, Gestuveo RJ, Anaya J-M, Merits A, Burchmore R, Kohl A, Varjak M. Glucose-Regulated Protein 78 Interacts with Zika Virus Envelope Protein and Contributes to a Productive Infection. Viruses. 2020; 12(5):524. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050524
Chicago/Turabian StyleRoyle, Jamie, Carolina Ramírez-Santana, Snezhana Akpunarlieva, Claire L. Donald, Rommel J. Gestuveo, Juan-Manuel Anaya, Andres Merits, Richard Burchmore, Alain Kohl, and Margus Varjak. 2020. "Glucose-Regulated Protein 78 Interacts with Zika Virus Envelope Protein and Contributes to a Productive Infection" Viruses 12, no. 5: 524. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050524