Development, Characterization, and Application of Two Reporter-Expressing Recombinant Zika Viruses

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. DNA Cloning
2.3. In Vitro Transcription
2.4. RNA Transfection
2.5. Infectious Center Assay
2.6. Immunoblot Analysis
2.7. Fluorescence Microscopy
2.8. Luciferase Assay
2.9. Antiviral Testing
3. Results
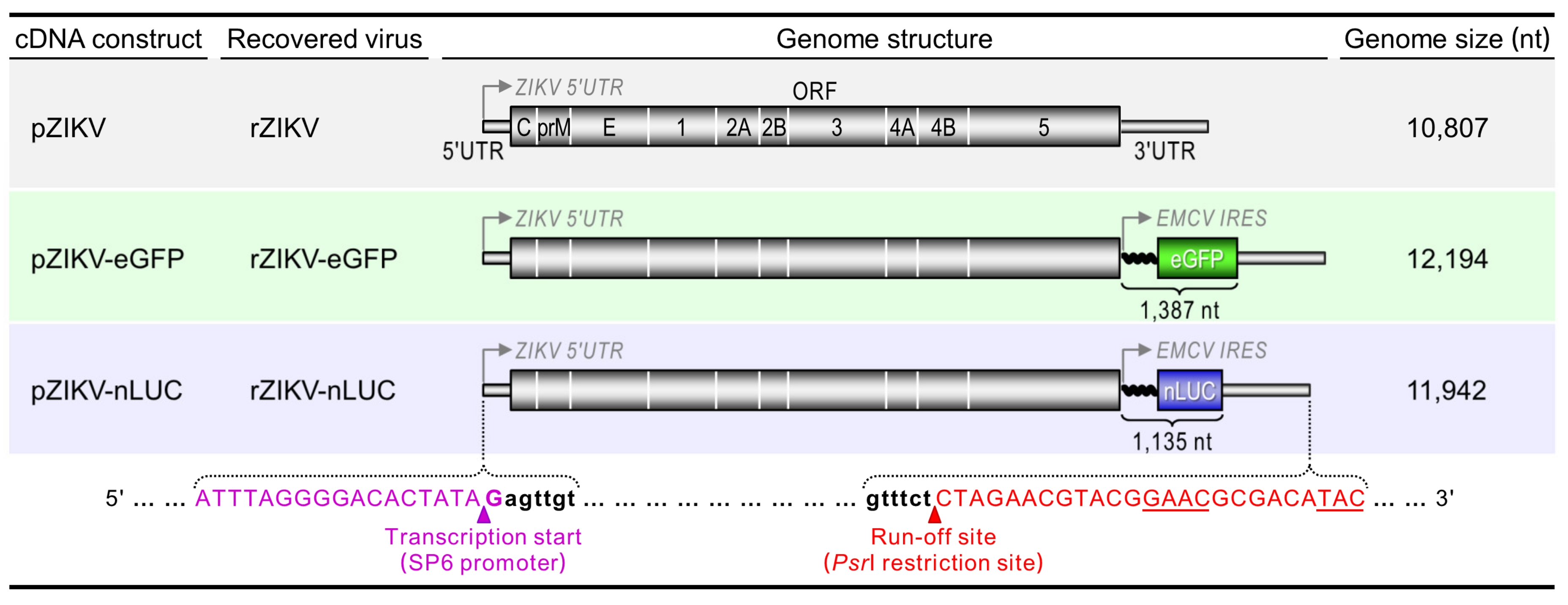
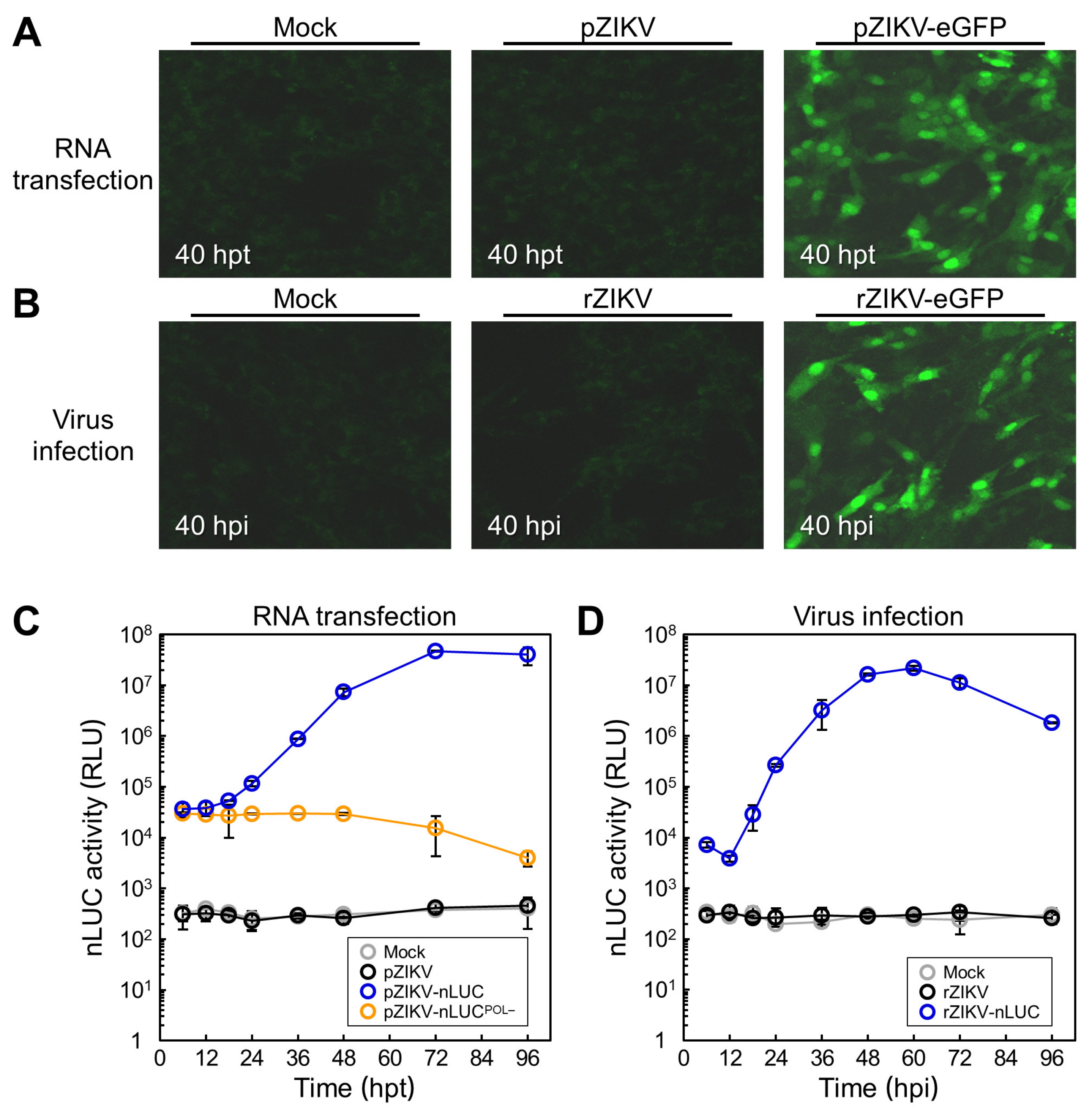
3.1. Construction of Two Reporter-Encoding Full-Length ZIKV cDNA Clones
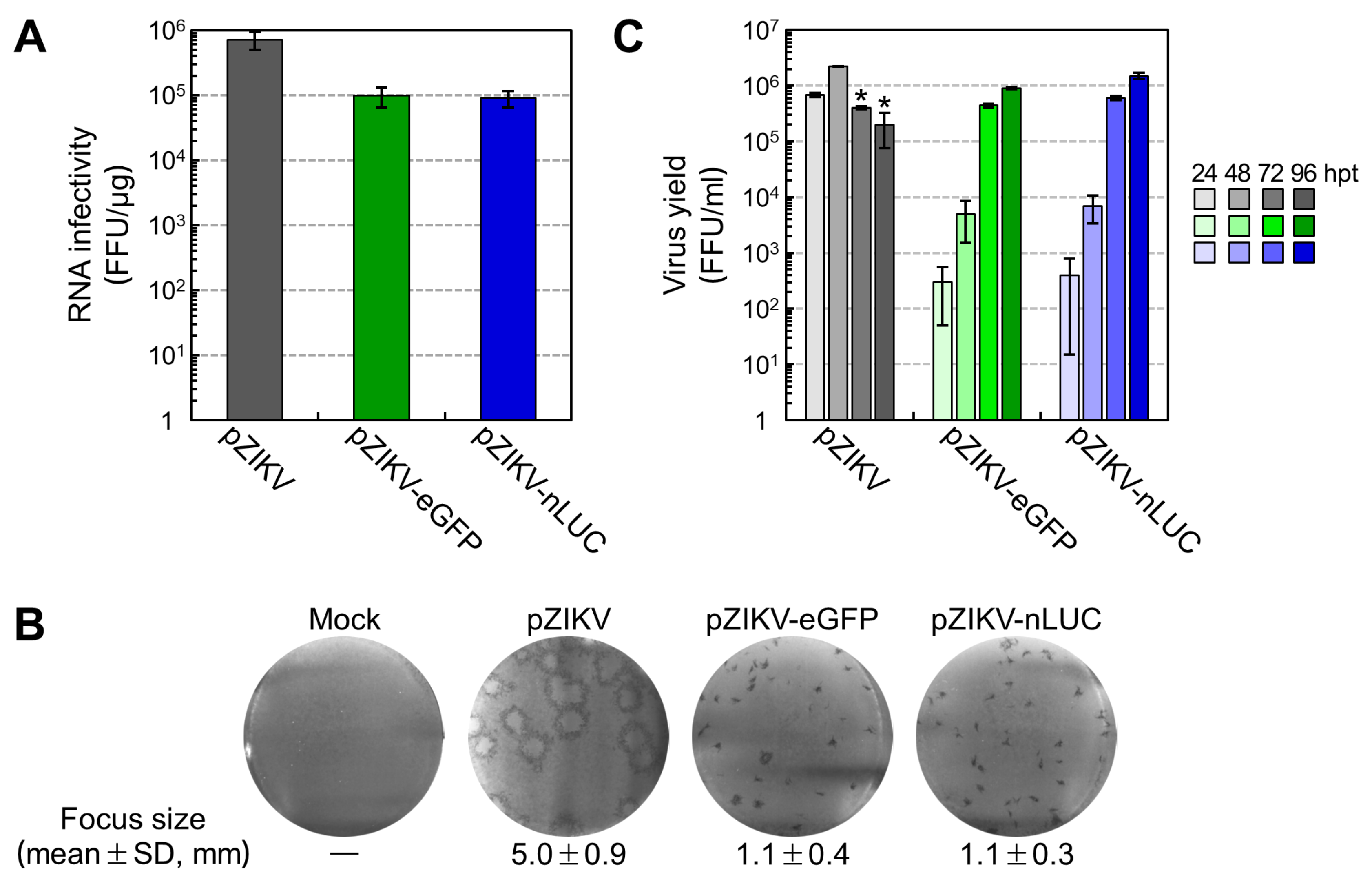
3.2. Production of Infectious RNA Transcripts from Two Reporter-Encoding Full-Length ZIKV cDNA Clones
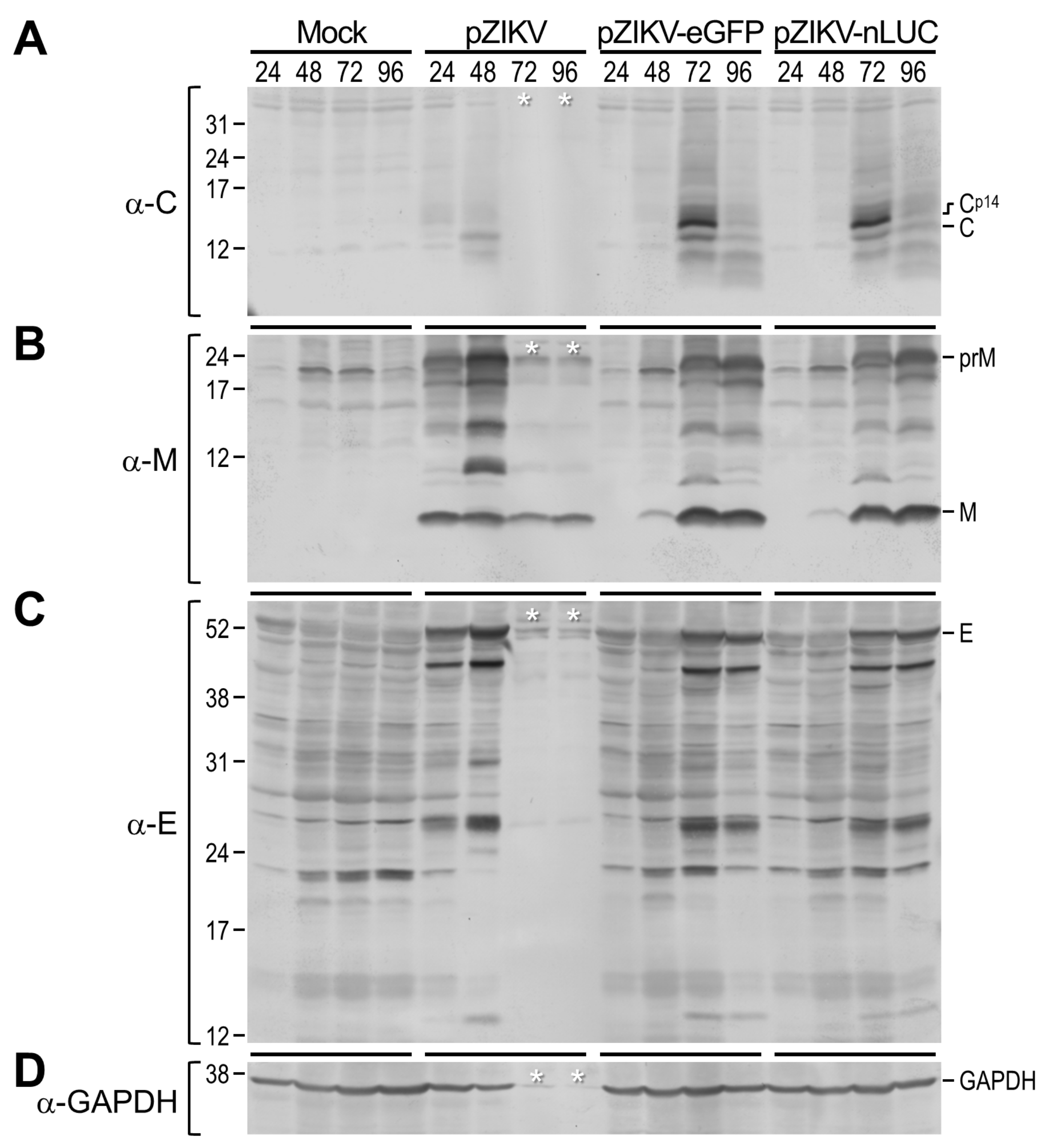
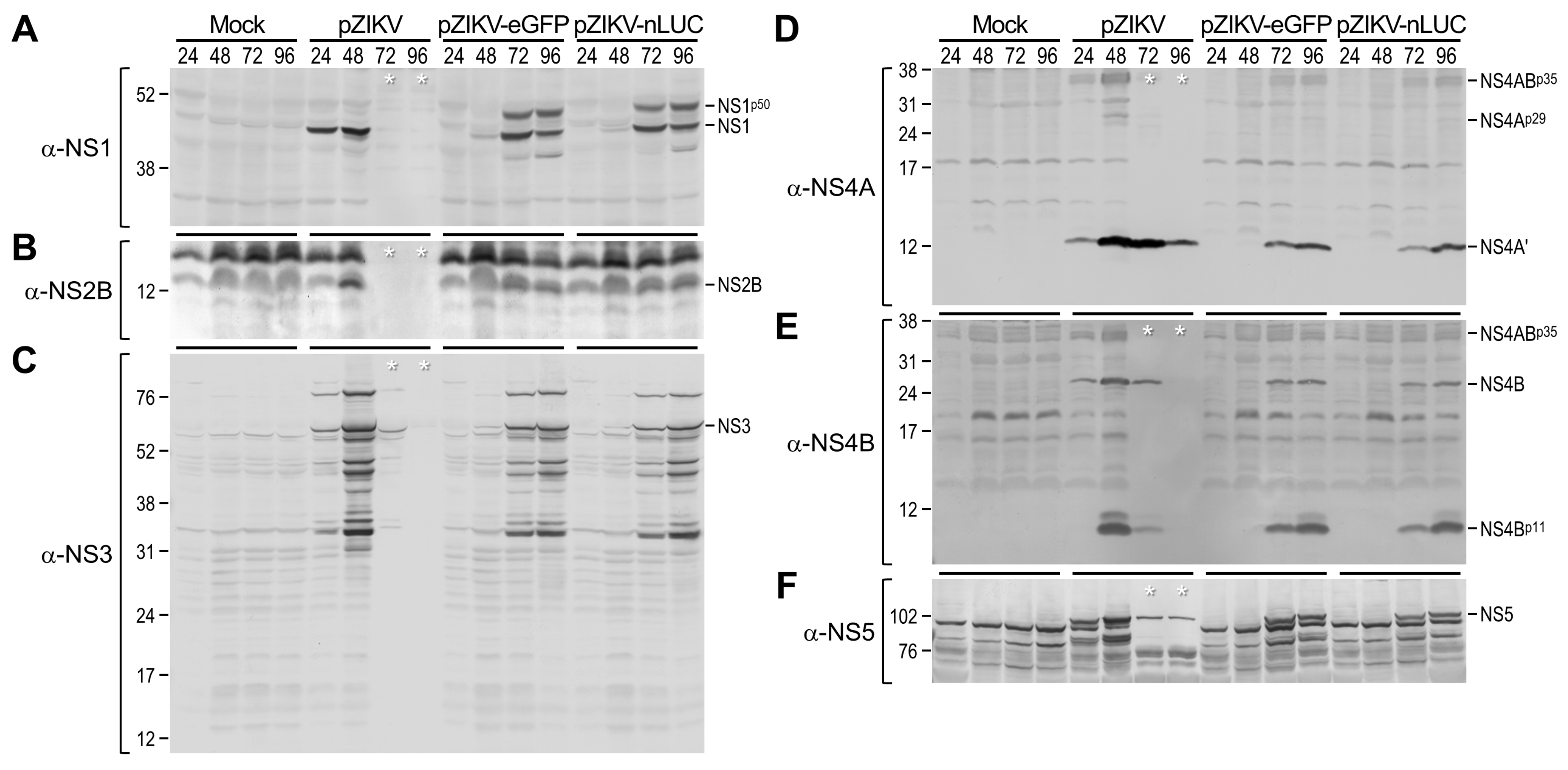
3.3. Probing of Viral Protein Accumulation During ZIKV Genomic RNA Replication
3.4. Characterization of Reporter Gene Expression During ZIKV Genomic RNA Replication
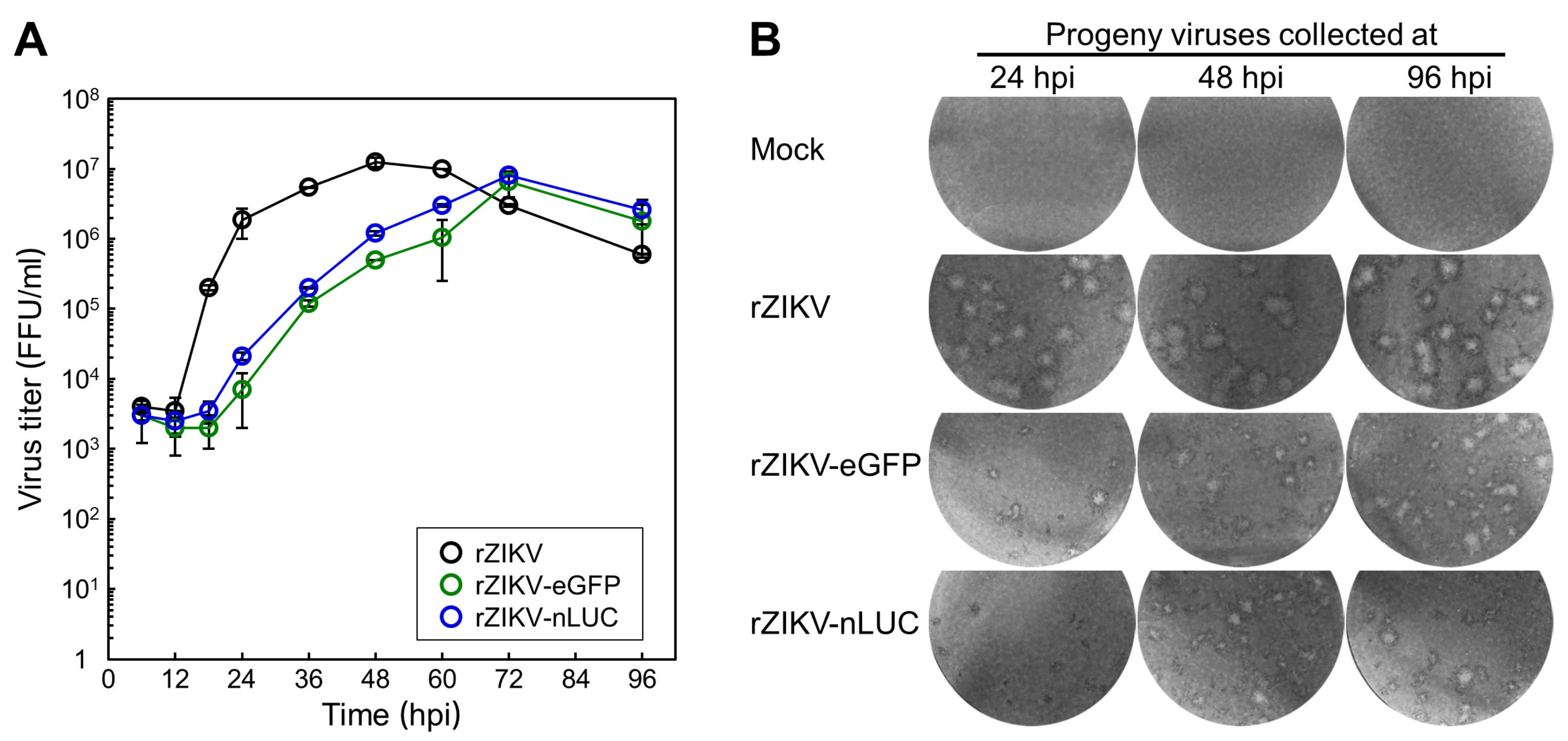
3.5. Examination of the Viral Growth and Stability of Two Reporter-Expressing ZIKVs
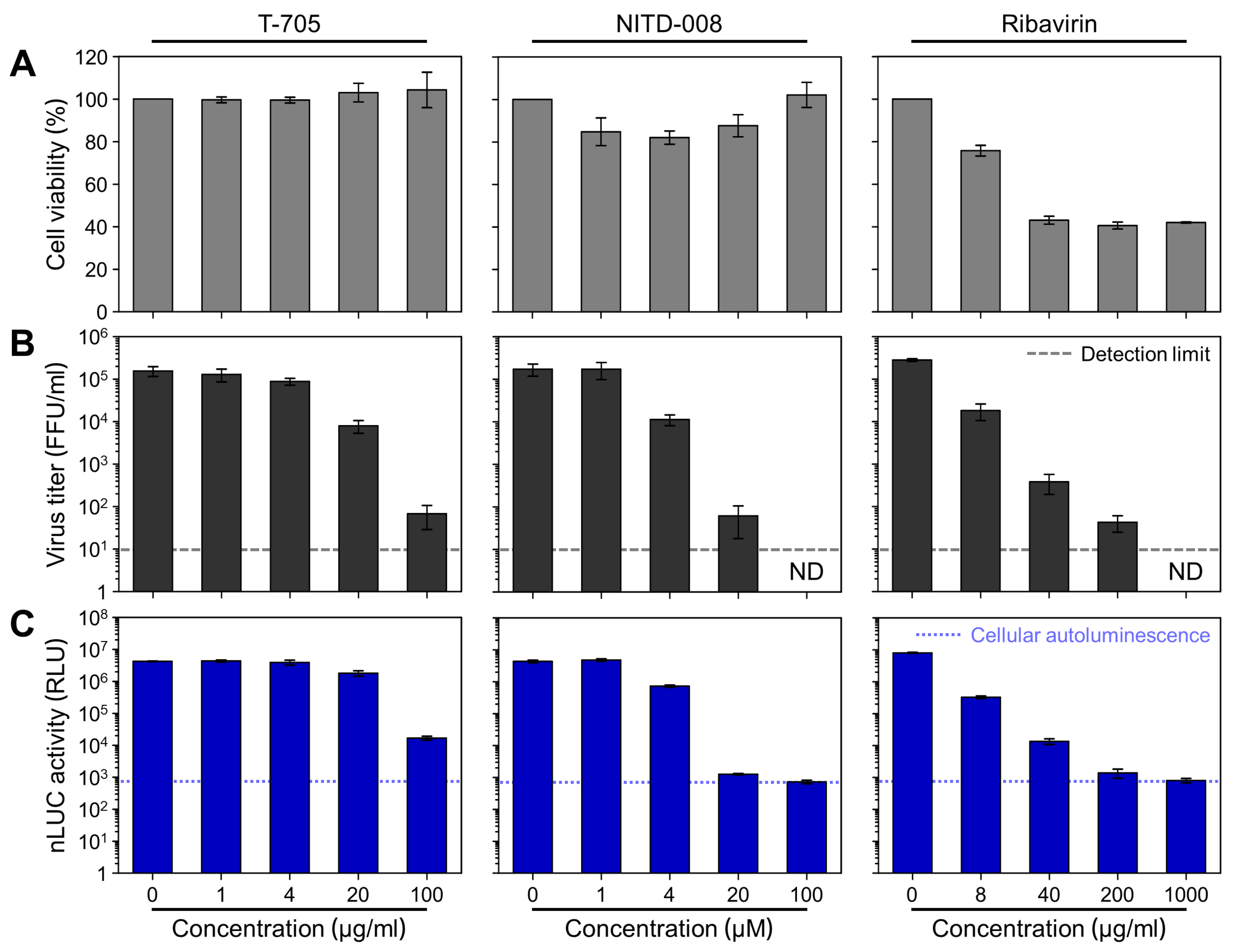
3.6. Evaluation of the Antiviral Activity of Three Compounds Using rZIKV-nLUC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yun, S.I.; Lee, Y.M. Zika virus: An emerging flavivirus. J. Microbiol. 2017, 55, 204–219. [Google Scholar] [CrossRef]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Diamond, M.S.; Mysorekar, I.U. Maternal-fetal transmission of Zika virus: Routes and signals for infection. J. Interferon Cytokine Res. 2017, 37, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, N.; Mayorquin Galvan, E.E.; Zavala Trujillo, I.G.; Zavala-Cerna, M.G. Congenital Zika syndrome: Pitfalls in the placental barrier. Rev. Med. Virol. 2018, 28, e1985. [Google Scholar] [CrossRef]
- Sakkas, H.; Bozidis, P.; Giannakopoulos, X.; Sofikitis, N.; Papadopoulou, C. An update on sexual transmission of Zika virus. Pathogens 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epelboin, Y.; Talaga, S.; Epelboin, L.; Dusfour, I. Zika virus: An updated review of competent or naturally infected mosquitoes. PLoS Negl. Trop. Dis. 2017, 11, e0005933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, B.H.; Yun, S.I.; Woolley, M.; Lee, Y.M. Zika virus: History, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 2017, 308, 50–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhaes, T.; Foy, B.D.; Marques, E.T.A.; Ebel, G.D.; Weger-Lucarelli, J. Mosquito-borne and sexual transmission of Zika virus: Recent developments and future directions. Virus Res. 2018, 254, 1–9. [Google Scholar] [CrossRef]
- Colt, S.; Garcia-Casal, M.N.; Pena-Rosas, J.P.; Finkelstein, J.L.; Rayco-Solon, P.; Weise Prinzo, Z.C.; Mehta, S. Transmission of Zika virus through breast milk and other breastfeeding-related bodily-fluids: A systematic review. PLoS Negl. Trop. Dis. 2017, 11, e0005528. [Google Scholar] [CrossRef] [Green Version]
- Musso, D.; Nhan, T.; Robin, E.; Roche, C.; Bierlaire, D.; Zisou, K.; Shan Yan, A.; Cao-Lormeau, V.M.; Broult, J. Potential for Zika virus transmission through blood transfusion demonstrated during an outbreak in French Polynesia, November 2013 to February 2014. Eurosurveillance 2014, 19, 20761. [Google Scholar] [CrossRef] [Green Version]
- Haby, M.M.; Pinart, M.; Elias, V.; Reveiz, L. Prevalence of asymptomatic Zika virus infection: A systematic review. Bull. World Health Organ. 2018, 96, 402D–413D. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Song, B.H.; Frank, J.C.; Julander, J.G.; Olsen, A.L.; Polejaeva, I.A.; Davies, C.J.; White, K.L.; Lee, Y.M. Functional genomics and immunologic tools: The impact of viral and host genetic variations on the outcome of Zika virus infection. Viruses 2018, 10, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaver, J.T.; Lelutiu, N.; Habib, R.; Skountzou, I. Evolution of two major Zika virus lineages: Implications for pathology, immune response, and vaccine development. Front. Immunol. 2018, 9, 1640. [Google Scholar] [CrossRef] [PubMed]
- Fuller, T.L.; Calvet, G.; Genaro Estevam, C.; Rafael Angelo, J.; Abiodun, G.J.; Halai, U.A.; De Santis, B.; Carvalho Sequeira, P.; Machado Araujo, E.; Alves Sampaio, S.; et al. Behavioral, climatic, and environmental risk factors for Zika and Chikungunya virus infections in Rio de Janeiro, Brazil, 2015–2016. PLoS ONE 2017, 12, e0188002. [Google Scholar] [CrossRef] [Green Version]
- Musso, D.; Gubler, D.J. Zika virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [Green Version]
- Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Rodriguez, Y.; Ramirez-Santana, C.; Anaya, J.M. Zika virus and autoimmunity. One-step forward. Autoimmun. Rev. 2017, 16, 1237–1245. [Google Scholar] [CrossRef]
- Nascimento, O.J.M.; da Silva, I.R.F. Guillain-Barre syndrome and Zika virus outbreaks. Curr. Opin. Neurol. 2017, 30, 500–507. [Google Scholar] [CrossRef]
- Wen, Z.; Song, H.; Ming, G.L. How does Zika virus cause microcephaly? Genes Dev. 2017, 31, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.A.; Staples, J.E.; Dobyns, W.B.; Pessoa, A.; Ventura, C.V.; Fonseca, E.B.; Ribeiro, E.M.; Ventura, L.O.; Neto, N.N.; Arena, J.F.; et al. Characterizing the pattern of anomalies in congenital Zika syndrome for pediatric clinicians. JAMA Pediatr. 2017, 171, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Platt, D.J.; Miner, J.J. Consequences of congenital Zika virus infection. Curr. Opin. Virol. 2017, 27, 1–7. [Google Scholar] [CrossRef]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 A resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostyuchenko, V.A.; Lim, E.X.; Zhang, S.; Fibriansah, G.; Ng, T.S.; Ooi, J.S.; Shi, J.; Lok, S.M. Structure of the thermally stable Zika virus. Nature 2016, 533, 425–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Z.; Song, H.; Shi, Y.; Qi, J.; Gao, G.F. Crystal structure of the capsid protein from Zika virus. J. Mol. Biol. 2018, 430, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Song, B.H.; Frank, J.C.; Julander, J.G.; Polejaeva, I.A.; Davies, C.J.; White, K.L.; Lee, Y.M. Complete genome sequences of three historically important, spatiotemporally distinct, and genetically divergent strains of Zika virus: MR-766, P6-740, and PRVABC-59. Genome Announc. 2016, 4, e00800-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Fink, K.; Zust, R.; Lim, S.P.; Qin, C.F.; Shi, P.Y. Flavivirus RNA methylation. J. Gen. Virol. 2014, 95, 763–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.Y.; Yu, J.Y.; Huang, X.Y.; Fan, H.; Li, X.F.; Deng, Y.Q.; Ji, X.; Cheng, M.L.; Ye, Q.; Zhao, H.; et al. Characterization of cis-acting RNA elements of Zika virus by using a self-splicing ribozyme-dependent infectious clone. J. Virol. 2017, 91, e00484-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donald, C.L.; Brennan, B.; Cumberworth, S.L.; Rezelj, V.V.; Clark, J.J.; Cordeiro, M.T.; Freitas de Oliveira Franca, R.; Pena, L.J.; Wilkie, G.S.; Da Silva Filipe, A.; et al. Full genome sequence and sfRNA interferon antagonist activity of Zika virus from Recife, Brazil. PLoS Negl. Trop. Dis. 2016, 10, e0005048. [Google Scholar] [CrossRef]
- Akiyama, B.M.; Laurence, H.M.; Massey, A.R.; Costantino, D.A.; Xie, X.; Yang, Y.; Shi, P.Y.; Nix, J.C.; Beckham, J.D.; Kieft, J.S. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 2016, 354, 1148–1152. [Google Scholar] [CrossRef] [Green Version]
- Goertz, G.P.; Abbo, S.R.; Fros, J.J.; Pijlman, G.P. Functional RNA during Zika virus infection. Virus Res. 2018, 254, 41–53. [Google Scholar] [CrossRef]
- Sirohi, D.; Kuhn, R.J. Zika virus structure, maturation, and receptors. J. Infect. Dis. 2017, 216, S935–S944. [Google Scholar] [CrossRef] [Green Version]
- Rey, F.A.; Stiasny, K.; Heinz, F.X. Flavivirus structural heterogeneity: Implications for cell entry. Curr. Opin. Virol. 2017, 24, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Lee, Y.M. Early events in Japanese encephalitis virus infection: Viral entry. Pathogens 2018, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural characterization of Zika virus replication factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badshah, S.L.; Naeem, A.; Mabkhot, Y. The new high resolution crystal structure of NS2B-NS3 protease of Zika virus. Viruses 2017, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phoo, W.W.; Li, Y.; Zhang, Z.; Lee, M.Y.; Loh, Y.R.; Tan, Y.B.; Ng, E.Y.; Lescar, J.; Kang, C.; Luo, D. Structure of the NS2B-NS3 protease from Zika virus after self-cleavage. Nat. Commun. 2016, 7, 13410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Y.; Loh, Y.R.; Phoo, W.W.; Hung, A.W.; Kang, C.; Luo, D. Crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science 2016, 354, 1597–1600. [Google Scholar] [CrossRef]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.; Coloma, J.; Garcia-Sastre, A.; Aggarwal, A.K. Structure of the NS3 helicase from Zika virus. Nat. Struct. Mol. Biol. 2016, 23, 752–754. [Google Scholar] [CrossRef] [Green Version]
- Coloma, J.; Jain, R.; Rajashankar, K.R.; Garcia-Sastre, A.; Aggarwal, A.K. Structures of NS5 methyltransferase from Zika virus. Cell Rep. 2016, 16, 3097–3102. [Google Scholar] [CrossRef] [Green Version]
- Coutard, B.; Barral, K.; Lichiere, J.; Selisko, B.; Martin, B.; Aouadi, W.; Lombardia, M.O.; Debart, F.; Vasseur, J.J.; Guillemot, J.C.; et al. Zika virus methyltransferase: Structure and functions for drug design perspectives. J. Virol. 2017, 91, e02202-16. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Song, H.; Wang, H.; Chai, Y.; Su, C.; Qi, J.; Shi, Y.; Gao, G.F. The crystal structure of Zika virus NS5 reveals conserved drug targets. EMBO J. 2017, 36, 919–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tan, X.F.; Thurmond, S.; Zhang, Z.M.; Lin, A.; Hai, R.; Song, J. The structure of Zika virus NS5 reveals a conserved domain conformation. Nat. Commun. 2017, 8, 14763. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Yi, G.; Du, F.; Chuang, Y.C.; Vaughan, R.C.; Sankaran, B.; Kao, C.C.; Li, P. Structure and function of the Zika virus full-length NS5 protein. Nat. Commun. 2017, 8, 14762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy, A.S.; Lima, G.M.; Oliveira, K.I.; Torres, N.U.; Maluf, F.V.; Guido, R.V.; Oliva, G. Crystal structure of Zika virus NS5 RNA-dependent RNA polymerase. Nat. Commun. 2017, 8, 14764. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.M.; Miller, A.S.; Klose, T.; Sirohi, D.; Buda, G.; Jiang, W.; Kuhn, R.J.; Rossmann, M.G. Structure of the immature Zika virus at 9 A resolution. Nat. Struct. Mol. Biol. 2017, 24, 184–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sager, G.; Gabaglio, S.; Sztul, E.; Belov, G.A. Role of host cell secretory machinery in Zika virus life cycle. Viruses 2018, 10, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, S.S.; Sevvana, M.; Kuhn, R.J.; Rossmann, M.G. Structural biology of Zika virus and other flaviviruses. Nat. Struct. Mol. Biol. 2018, 25, 13–20. [Google Scholar] [CrossRef]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Gubler, D.J.; Vasilakis, N.; Musso, D. History and emergence of Zika virus. J. Infect. Dis. 2017, 216, S860–S867. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Bossin, H.; Mallet, H.P.; Besnard, M.; Broult, J.; Baudouin, L.; Levi, J.E.; Sabino, E.C.; Ghawche, F.; Lanteri, M.C.; et al. Zika virus in French Polynesia 2013–14: Anatomy of a completed outbreak. Lancet Infect. Dis. 2018, 18, e172–e182. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.M.; Musso, D. Emerging arboviruses in the Pacific. Lancet 2014, 384, 1571–1572. [Google Scholar] [CrossRef]
- Wikan, N.; Smith, D.R. Zika virus: History of a newly emerging arbovirus. Lancet Infect. Dis. 2016, 16, e119–e126. [Google Scholar] [CrossRef] [Green Version]
- Zanluca, C.; Melo, V.C.; Mosimann, A.L.; Santos, G.I.; Santos, C.N.; Luz, K. First report of autochthonous transmission of Zika virus in Brazil. Mem. Inst. Oswaldo Cruz 2015, 110, 569–572. [Google Scholar] [CrossRef]
- Campos, G.S.; Bandeira, A.C.; Sardi, S.I. Zika virus outbreak, Bahia, Brazil. Emerg. Infect. Dis. 2015, 21, 1885–1886. [Google Scholar] [CrossRef]
- Avelino-Silva, V.I.; Kallas, E.G. Untold stories of the Zika virus epidemic in Brazil. Rev. Med. Virol. 2018, 28, e2000. [Google Scholar] [CrossRef] [Green Version]
- Baud, D.; Gubler, D.J.; Schaub, B.; Lanteri, M.C.; Musso, D. An update on Zika virus infection. Lancet 2017, 390, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Lessler, J.; Chaisson, L.H.; Kucirka, L.M.; Bi, Q.; Grantz, K.; Salje, H.; Carcelen, A.C.; Ott, C.T.; Sheffield, J.S.; Ferguson, N.M.; et al. Assessing the global threat from Zika virus. Science 2016, 353, aaf8160. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.I.; Kim, S.Y.; Rice, C.M.; Lee, Y.M. Development and application of a reverse genetics system for Japanese encephalitis virus. J. Virol. 2003, 77, 6450–6465. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.I.; Choi, Y.J.; Song, B.H.; Lee, Y.M. 3′ cis-acting elements that contribute to the competence and efficiency of Japanese encephalitis virus genome replication: Functional importance of sequence duplications, deletions, and substitutions. J. Virol. 2009, 83, 7909–7930. [Google Scholar] [CrossRef] [Green Version]
- Song, B.H.; Yun, S.I.; Choi, Y.J.; Kim, J.M.; Lee, C.H.; Lee, Y.M. A complex RNA motif defined by three discontinuous 5-nucleotide-long strands is essential for flavivirus RNA replication. RNA 2008, 14, 1791–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.K.; Kim, J.M.; Song, B.H.; Yun, S.I.; Yun, G.N.; Byun, S.J.; Lee, Y.M. Profiling of viral proteins expressed from the genomic RNA of Japanese encephalitis virus using a panel of 15 region-specific polyclonal rabbit antisera: Implications for viral gene expression. PLoS ONE 2015, 10, e0124318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef]
- Munster, M.; Plaszczyca, A.; Cortese, M.; Neufeldt, C.J.; Goellner, S.; Long, G.; Bartenschlager, R. A reverse genetics system for Zika virus based on a simple molecular cloning strategy. Viruses 2018, 10, 368. [Google Scholar] [CrossRef] [Green Version]
- Zmurko, J.; Marques, R.E.; Schols, D.; Verbeken, E.; Kaptein, S.J.; Neyts, J. The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl. Trop. Dis. 2016, 10, e0004695. [Google Scholar] [CrossRef]
- Lanko, K.; Eggermont, K.; Patel, A.; Kaptein, S.; Delang, L.; Verfaillie, C.M.; Neyts, J. Replication of the Zika virus in different iPSC-derived neuronal cells and implications to assess efficacy of antivirals. Antivir. Res. 2017, 145, 82–86. [Google Scholar] [CrossRef]
- Pires de Mello, C.P.; Tao, X.; Kim, T.H.; Vicchiarelli, M.; Bulitta, J.B.; Kaushik, A.; Brown, A.N. Clinical regimens of favipiravir inhibit Zika virus replication in the hollow-fiber infection model. Antimicrob. Agents Chemother. 2018, 62, e00967-18. [Google Scholar] [CrossRef] [Green Version]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.A.; Seong, R.K.; Kumar, M.; Shin, O.S. Favipiravir and ribavirin inhibit replication of Asian and African strains of Zika virus in different cell models. Viruses 2018, 10, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.Q.; Zhang, N.N.; Li, C.F.; Tian, M.; Hao, J.N.; Xie, X.P.; Shi, P.Y.; Qin, C.F. Adenosine analog NITD008 is a potent inhibitor of Zika virus. Open Forum Infect. Dis. 2016, 3, ofw175. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.K.; Shi, P.Y.; Chen, Y.L.; Flint, M.; Spiropoulou, C.F. In vitro antiviral activity of adenosine analog NITD008 against tick-borne flaviviruses. Antivir. Res. 2016, 130, 46–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Chen, Y.L.; Schul, W.; Wang, Q.Y.; Gu, F.; Duraiswamy, J.; Kondreddi, R.R.; Niyomrattanakit, P.; Lakshminarayana, S.B.; Goh, A.; et al. An adenosine nucleoside inhibitor of dengue virus. Proc. Natl. Acad. Sci. USA 2009, 106, 20435–20439. [Google Scholar] [CrossRef] [Green Version]
- Kamiyama, N.; Soma, R.; Hidano, S.; Watanabe, K.; Umekita, H.; Fukuda, C.; Noguchi, K.; Gendo, Y.; Ozaki, T.; Sonoda, A.; et al. Ribavirin inhibits Zika virus (ZIKV) replication in vitro and suppresses viremia in ZIKV-infected STAT1-deficient mice. Antivir. Res. 2017, 146, 1–11. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.F.; Yu, S.; Wang, X.; Zhang, L.; Yu, J.; Xie, L.; Li, W.; Ali, R.; Qiu, H.J. Applications of replicating-competent reporter-expressing viruses in diagnostic and molecular virology. Viruses 2016, 8, 127. [Google Scholar] [CrossRef] [Green Version]
- Avila-Perez, G.; Nogales, A.; Martin, V.; Almazan, F.; Martinez-Sobrido, L. Reverse genetic approaches for the generation of recombinant Zika virus. Viruses 2018, 10, 597. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.I.; Song, B.H.; Kim, J.K.; Lee, Y.M. Bacterial artificial chromosomes: A functional genomics tool for the study of positive-strand RNA viruses. J. Vis. Exp. 2015, 106, e53164. [Google Scholar] [CrossRef] [Green Version]
- Aubry, F.; Nougairede, A.; Gould, E.A.; de Lamballerie, X. Flavivirus reverse genetic systems, construction techniques and applications: A historical perspective. Antivir. Res. 2015, 114, 67–85. [Google Scholar] [CrossRef] [Green Version]
- Gorman, M.J.; Caine, E.A.; Zaitsev, K.; Begley, M.C.; Weger-Lucarelli, J.; Uccellini, M.B.; Tripathi, S.; Morrison, J.; Yount, B.L.; Dinnon, K.H., 3rd; et al. An immunocompetent mouse model of Zika virus infection. Cell Host Microbe 2018, 23, 672–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, C.L.; Zhang, Q.Y.; Chen, D.D.; Liu, S.Q.; Qin, C.F.; Zhang, B.; Ye, H.Q. Recovery of the Zika virus through an in vitro ligation approach. J. Gen. Virol. 2017, 98, 1739–1743. [Google Scholar] [CrossRef] [PubMed]
- Weger-Lucarelli, J.; Duggal, N.K.; Bullard-Feibelman, K.; Veselinovic, M.; Romo, H.; Nguyen, C.; Ruckert, C.; Brault, A.C.; Bowen, R.A.; Stenglein, M.; et al. Development and characterization of recombinant virus generated from a New World Zika virus infectious clone. J. Virol. 2017, 91, e01765-16. [Google Scholar] [CrossRef] [Green Version]
- Pu, S.Y.; Wu, R.H.; Yang, C.C.; Jao, T.M.; Tsai, M.H.; Wang, J.C.; Lin, H.M.; Chao, Y.S.; Yueh, A. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. J. Virol. 2011, 85, 2927–2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widman, D.G.; Young, E.; Yount, B.L.; Plante, K.S.; Gallichotte, E.N.; Carbaugh, D.L.; Peck, K.M.; Plante, J.; Swanstrom, J.; Heise, M.T.; et al. A reverse genetics platform that spans the Zika virus family tree. mBio 2017, 8, e02014-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, C.; Xie, X.; Muruato, A.E.; Rossi, S.L.; Roundy, C.M.; Azar, S.R.; Yang, Y.; Tesh, R.B.; Bourne, N.; Barrett, A.D.; et al. An infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe 2016, 19, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Mutso, M.; Saul, S.; Rausalu, K.; Susova, O.; Zusinaite, E.; Mahalingam, S.; Merits, A. Reverse genetic system, genetically stable reporter viruses and packaged subgenomic replicon based on a Brazilian Zika virus isolate. J. Gen. Virol. 2017, 98, 2712–2724. [Google Scholar] [CrossRef]
- Annamalai, A.S.; Pattnaik, A.; Sahoo, B.R.; Muthukrishnan, E.; Natarajan, S.K.; Steffen, D.; Vu, H.L.X.; Delhon, G.; Osorio, F.A.; Petro, T.M.; et al. Zika virus encoding nonglycosylated envelope protein is attenuated and defective in neuroinvasion. J. Virol. 2017, 91, e01348-17. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Xu, Y.; Lavillette, D.; Zhong, J.; Zou, G.; Long, G. Negligible contribution of M2634V substitution to ZIKV pathogenesis in AG6 mice revealed by a bacterial promoter activity reduced infectious clone. Sci. Rep. 2018, 8, 10491. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Zou, J.; Shan, C.; Yang, Y.; Kum, D.B.; Dallmeier, K.; Neyts, J.; Shi, P.Y. Zika virus replicons for drug discovery. EBioMedicine 2016, 12, 156–160. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Q.; Deng, C.L.; Gu, D.; Li, X.; Shi, L.; He, J.; Zhang, Q.Y.; Zhang, B.; Ye, H.Q. Development of a replicon cell line-based high throughput antiviral assay for screening inhibitors of Zika virus. Antivir. Res. 2018, 150, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Kenney, H.; Chen, R.; Liu, G.; Manukyan, H.; Whitehead, S.S.; Laassri, M.; Chumakov, K.; Pletnev, A.G. A full-length infectious cDNA clone of Zika virus from the 2015 epidemic in Brazil as a genetic platform for studies of virus-host interactions and vaccine development. mBio 2016, 7, e01114-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, M.C.; Sourisseau, M.; Espino, M.M.; Gray, E.S.; Chambers, M.T.; Tortorella, D.; Evans, M.J. Rescue of the 1947 Zika virus prototype strain with a cytomegalovirus promoter-driven cDNA clone. mSphere 2016, 1, e00246-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez-Jurado, S.; Nogales, A.; Avila-Perez, G.; Iborra, F.J.; Martinez-Sobrido, L.; Almazan, F. An alanine-to-valine substitution in the residue 175 of Zika virus NS2A protein affects viral RNA synthesis and attenuates the virus in vivo. Viruses 2018, 10, 547. [Google Scholar] [CrossRef] [Green Version]
- Setoh, Y.X.; Prow, N.A.; Peng, N.; Hugo, L.E.; Devine, G.; Hazlewood, J.E.; Suhrbier, A.; Khromykh, A.A. De novo generation and characterization of new Zika virus isolate using sequence data from a microcephaly case. mSphere 2017, 2, e00190-17. [Google Scholar] [CrossRef] [Green Version]
- Atieh, T.; Baronti, C.; de Lamballerie, X.; Nougairede, A. Simple reverse genetics systems for Asian and African Zika viruses. Sci. Rep. 2016, 6, 39384. [Google Scholar] [CrossRef] [Green Version]
- Gadea, G.; Bos, S.; Krejbich-Trotot, P.; Clain, E.; Viranaicken, W.; El-Kalamouni, C.; Mavingui, P.; Despres, P. A robust method for the rapid generation of recombinant Zika virus expressing the GFP reporter gene. Virology 2016, 497, 157–162. [Google Scholar] [CrossRef]
- Frumence, E.; Viranaicken, W.; Gadea, G.; Despres, P. A GFP reporter MR766-based flow cytometry neutralization test for rapid detection of Zika virus-neutralizing antibodies in serum specimens. Vaccines 2019, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Zou, G.; Xu, H.Y.; Qing, M.; Wang, Q.Y.; Shi, P.Y. Development and characterization of a stable luciferase dengue virus for high-throughput screening. Antivir. Res. 2011, 91, 11–19. [Google Scholar] [CrossRef]
- Fischl, W.; Bartenschlager, R. High-throughput screening using dengue virus reporter genomes. Methods Mol. Biol. 2013, 1030, 205–219. [Google Scholar] [CrossRef]
- Kato, F.; Hishiki, T. Dengue virus reporter replicon is a valuable tool for antiviral drug discovery and analysis of virus replication mechanisms. Viruses 2016, 8, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig-Basagoiti, F.; Deas, T.S.; Ren, P.; Tilgner, M.; Ferguson, D.M.; Shi, P.Y. High-throughput assays using a luciferase-expressing replicon, virus-like particles, and full-length virus for West Nile virus drug discovery. Antimicrob. Agents Chemother. 2005, 49, 4980–4988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehle, A. Fiat Luc: Bioluminescence imaging reveals in vivo viral replication dynamics. PLoS Pathog. 2015, 11, e1005081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.I.; Choi, Y.J.; Yu, X.F.; Song, J.Y.; Shin, Y.H.; Ju, Y.R.; Kim, S.Y.; Lee, Y.M. Engineering the Japanese encephalitis virus RNA genome for the expression of foreign genes of various sizes: Implications for packaging capacity and RNA replication efficiency. J. Neurovirol. 2007, 13, 522–535. [Google Scholar] [CrossRef]
- Deas, T.S.; Binduga-Gajewska, I.; Tilgner, M.; Ren, P.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kauffman, E.B.; Kramer, L.D.; Shi, P.Y. Inhibition of flavivirus infections by antisense oligomers specifically suppressing viral translation and RNA replication. J. Virol. 2005, 79, 4599–4609. [Google Scholar] [CrossRef] [Green Version]
- Pierson, T.C.; Diamond, M.S.; Ahmed, A.A.; Valentine, L.E.; Davis, C.W.; Samuel, M.A.; Hanna, S.L.; Puffer, B.A.; Doms, R.W. An infectious West Nile virus that expresses a GFP reporter gene. Virology 2005, 334, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Kaptein, S.J.; De Burghgraeve, T.; Froeyen, M.; Pastorino, B.; Alen, M.M.; Mondotte, J.A.; Herdewijn, P.; Jacobs, M.; de Lamballerie, X.; Schols, D.; et al. A derivate of the antibiotic doxorubicin is a selective inhibitor of dengue and yellow fever virus replication in vitro. Antimicrob. Agents Chemother. 2010, 54, 5269–5280. [Google Scholar] [CrossRef] [Green Version]
- Mondotte, J.A.; Lozach, P.Y.; Amara, A.; Gamarnik, A.V. Essential role of dengue virus envelope protein N glycosylation at asparagine-67 during viral propagation. J. Virol. 2007, 81, 7136–7148. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.T.; Shan, C.; Li, X.D.; Liu, S.Q.; Deng, C.L.; Ye, H.Q.; Shang, B.D.; Shi, P.Y.; Lv, M.; Shen, B.F.; et al. Generation of a recombinant West Nile virus stably expressing the Gaussia luciferase for neutralization assay. Virus Res. 2016, 211, 17–24. [Google Scholar] [CrossRef]
- Suphatrakul, A.; Duangchinda, T.; Jupatanakul, N.; Prasittisa, K.; Onnome, S.; Pengon, J.; Siridechadilok, B. Multi-color fluorescent reporter dengue viruses with improved stability for analysis of a multi-virus infection. PLoS ONE 2018, 13, e0194399. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; Dorner, M.; Feulner, M.; Imanaka, N.; Murphy, M.Y.; Ploss, A.; Rice, C.M. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proc. Natl. Acad. Sci. USA 2012, 109, 14610–14615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaturro, P.; Trist, I.M.; Paul, D.; Kumar, A.; Acosta, E.G.; Byrd, C.M.; Jordan, R.; Brancale, A.; Bartenschlager, R. Characterization of the mode of action of a potent dengue virus capsid inhibitor. J. Virol. 2014, 88, 11540–11555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaldo, M.C.; Mello, S.M.; Trindade, G.F.; Rangel, A.A.; Duarte, A.S.; Oliveira, P.J.; Freire, M.S.; Kubelka, C.F.; Galler, R. Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virol. J. 2007, 4, 115. [Google Scholar] [CrossRef] [Green Version]
- Vandergaast, R.; Hoover, L.I.; Zheng, K.; Fredericksen, B.L. Generation of West Nile virus infectious clones containing amino acid insertions between capsid and capsid anchor. Viruses 2014, 6, 1637–1653. [Google Scholar] [CrossRef] [Green Version]
- Tamura, T.; Fukuhara, T.; Uchida, T.; Ono, C.; Mori, H.; Sato, A.; Fauzyah, Y.; Okamoto, T.; Kurosu, T.; Setoh, Y.X.; et al. Characterization of recombinant Flaviviridae viruses possessing a small reporter tag. J. Virol. 2018, 92, e01582-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usme-Ciro, J.A.; Lopera, J.A.; Enjuanes, L.; Almazan, F.; Gallego-Gomez, J.C. Development of a novel DNA-launched dengue virus type 2 infectious clone assembled in a bacterial artificial chromosome. Virus Res. 2014, 180, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Borman, A.M.; Le Mercier, P.; Girard, M.; Kean, K.M. Comparison of picornaviral IRES-driven internal initiation of translation in cultured cells of different origins. Nucleic Acids Res. 1997, 25, 925–932. [Google Scholar] [CrossRef] [Green Version]
- Borman, A.M.; Bailly, J.L.; Girard, M.; Kean, K.M. Picornavirus internal ribosome entry segments: Comparison of translation efficiency and the requirements for optimal internal initiation of translation in vitro. Nucleic Acids Res. 1995, 23, 3656–3663. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, Z. IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell. Biol. 2019, 11, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Salas, E.; Francisco-Velilla, R.; Fernandez-Chamorro, J.; Embarek, A.M. Insights into structural and mechanistic features of viral IRES elements. Front. Microbiol. 2017, 8, 2629. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Mugavero, J.; Stauft, C.B.; Wimmer, E. Dengue and Zika virus 5′ untranslated regions harbor internal ribosomal entry site functions. mBio 2019, 10, e00459-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, A.J.; Gerdes, B.C.; Kaja, S.; Koulen, P. Insert sequence length determines transfection efficiency and gene expression levels in bicistronic mammalian expression vectors. Int. J. Biochem. Mol. Biol. 2013, 4, 201–208. [Google Scholar] [PubMed]
- Firth, A.E.; Atkins, J.F. A conserved predicted pseudoknot in the NS2A-encoding sequence of West Nile and Japanese encephalitis flaviviruses suggests NS1’ may derive from ribosomal frameshifting. Virol. J. 2009, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melian, E.B.; Hinzman, E.; Nagasaki, T.; Firth, A.E.; Wills, N.M.; Nouwens, A.S.; Blitvich, B.J.; Leung, J.; Funk, A.; Atkins, J.F.; et al. NS1’ of flaviviruses in the Japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J. Virol. 2010, 84, 1641–1647. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Li, X.F.; Zhao, H.; Li, S.H.; Deng, Y.Q.; Cao, R.Y.; Song, K.Y.; Wang, H.J.; Hua, R.H.; Yu, Y.X.; et al. A single nucleotide mutation in NS2A of Japanese encephalitis-live vaccine virus (SA14-14-2) ablates NS1’ formation and contributes to attenuation. J. Gen. Virol. 2012, 93, 1959–1964. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence (5′→3′) | Polarity |
|---|---|---|
| ORF3f | AAACGGACCGGGATCCCTTATAGGGCACAGAC | Sense |
| ORF3r | TTTGAATTCACACTAAAATTGGTGCTTACAA | Antisense |
| 3UTRf | AAAATGCATGCACCAATTTTAGTGTTGTCAGG | Sense |
| 3UTRr | TTTCCGCGGAGGGCGGCCGCGTATGTCGCGTT | Antisense |
| FORf | GTGGAAGGAGCTGGGGAAACGCAAGCGGCC | Sense |
| POLr | TGGCTTCACAACGCAGGCAGCTCCACTGACCGCCA | Antisense |
| POLf | TGGCGGTCAGTGGAGCTGCCTGCGTTGTGAAGCCA | Sense |
| REVr | CCAAGTGGTGCGGGGTCTGTGCCCTATAAG | Antisense |
| EGFPf | ACAACCATGGTGAGCAAGGGCGAGGAGCTGTT | Sense |
| EGFPr | TGCATGCATTCATTAACCGTCGACTGCAGAATT | Antisense |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, S.-I.; Song, B.-H.; Woolley, M.E.; Frank, J.C.; Julander, J.G.; Lee, Y.-M. Development, Characterization, and Application of Two Reporter-Expressing Recombinant Zika Viruses. Viruses 2020, 12, 572. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050572
Yun S-I, Song B-H, Woolley ME, Frank JC, Julander JG, Lee Y-M. Development, Characterization, and Application of Two Reporter-Expressing Recombinant Zika Viruses. Viruses. 2020; 12(5):572. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050572
Chicago/Turabian StyleYun, Sang-Im, Byung-Hak Song, Michael E. Woolley, Jordan C. Frank, Justin G. Julander, and Young-Min Lee. 2020. "Development, Characterization, and Application of Two Reporter-Expressing Recombinant Zika Viruses" Viruses 12, no. 5: 572. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050572