Differential Effects of Human Adenovirus E1A Protein Isoforms on Aerobic Glycolysis in A549 Human Lung Epithelial Cells

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Protein Extraction and Western Blot
2.3. Seahorse Glycolytic Stress Test
2.4. Seahorse Mitochondrial Stress Test
2.5. RNA Extraction and qPCR
2.6. RNA Sequencing Analysis
2.7. Statistics
3. Results
3.1. A549-13S Cells Increased Baseline Glycolysis and Decreased Maximum Respiration
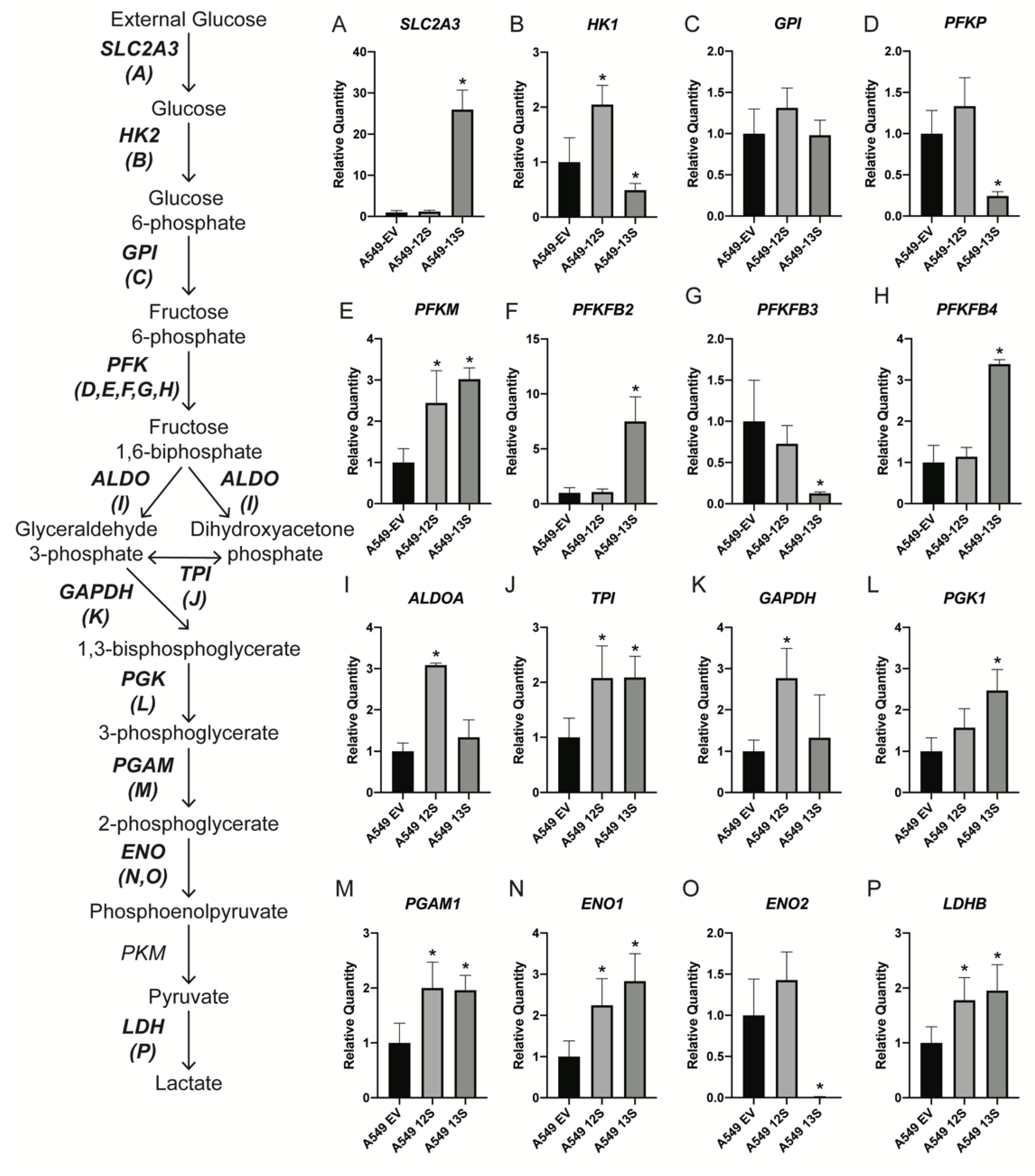
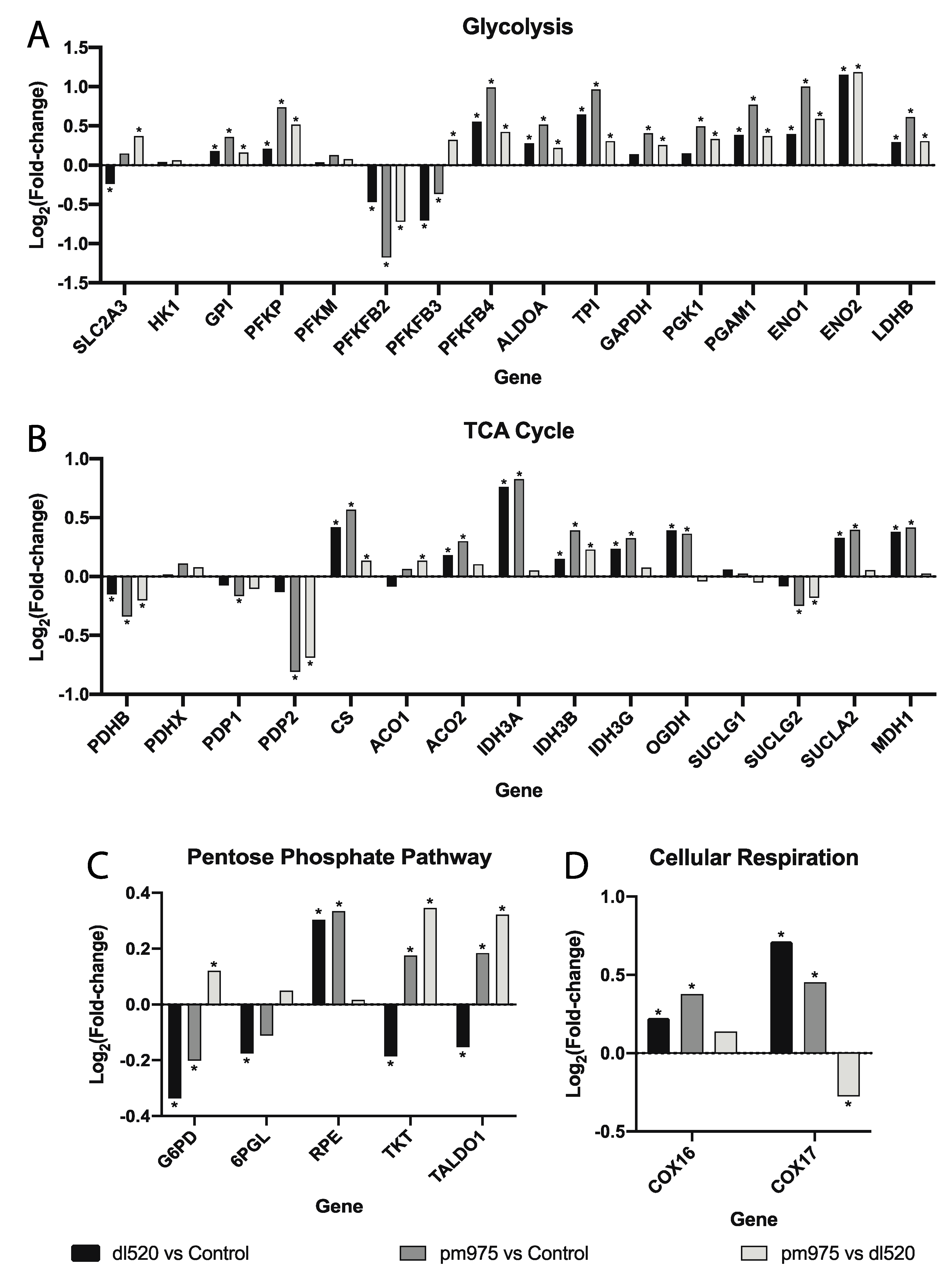
3.2. Glycolytic Genes Are Upregulated in A59-13S Cells
3.3. Pentose Phosphate Pathway Genes Are Differentially Regulated in A549-13S Cells
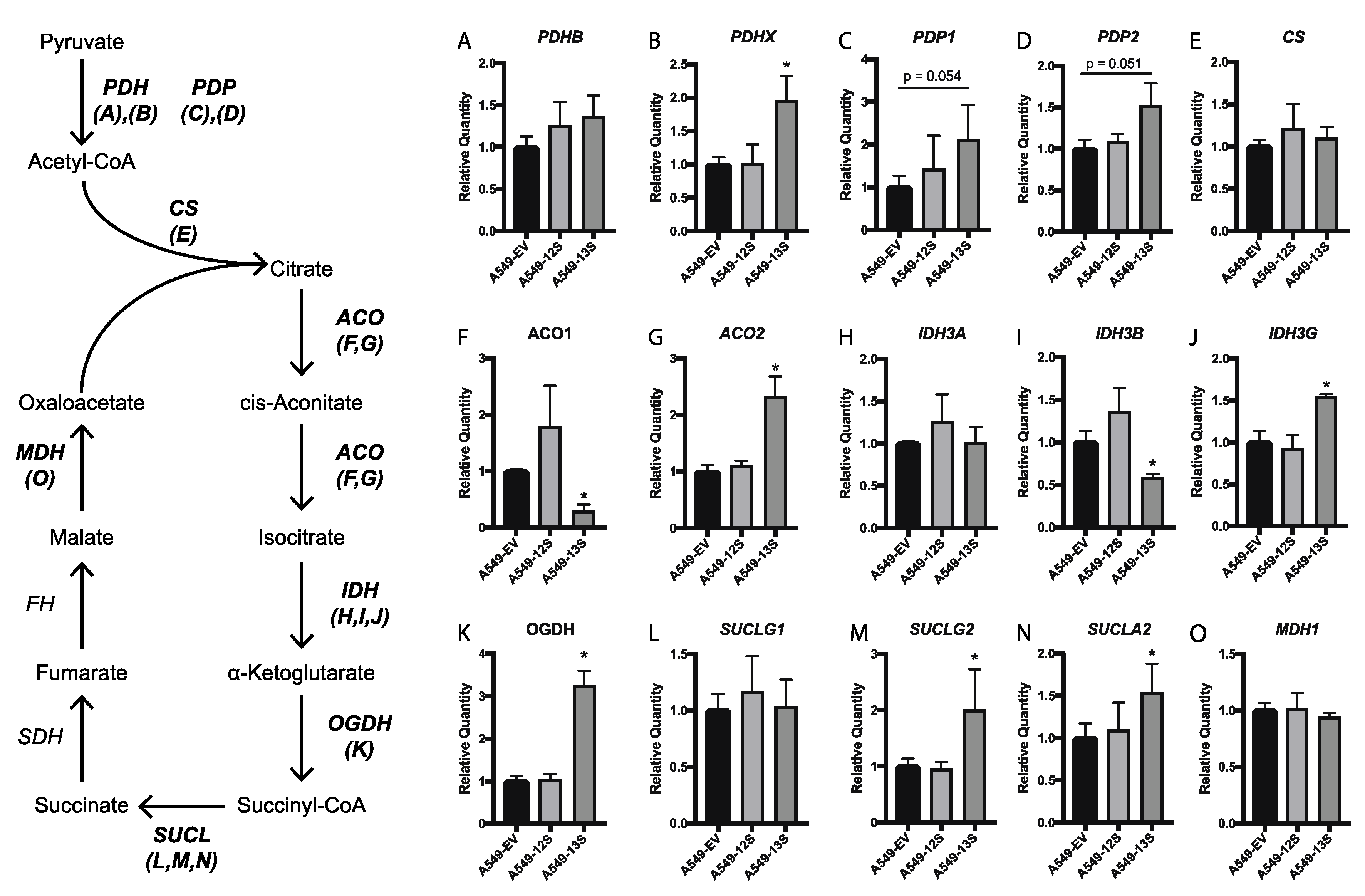
3.4. Tricarboxylic Acid Cycle Genes Are Upregulated in A549-13S Cells
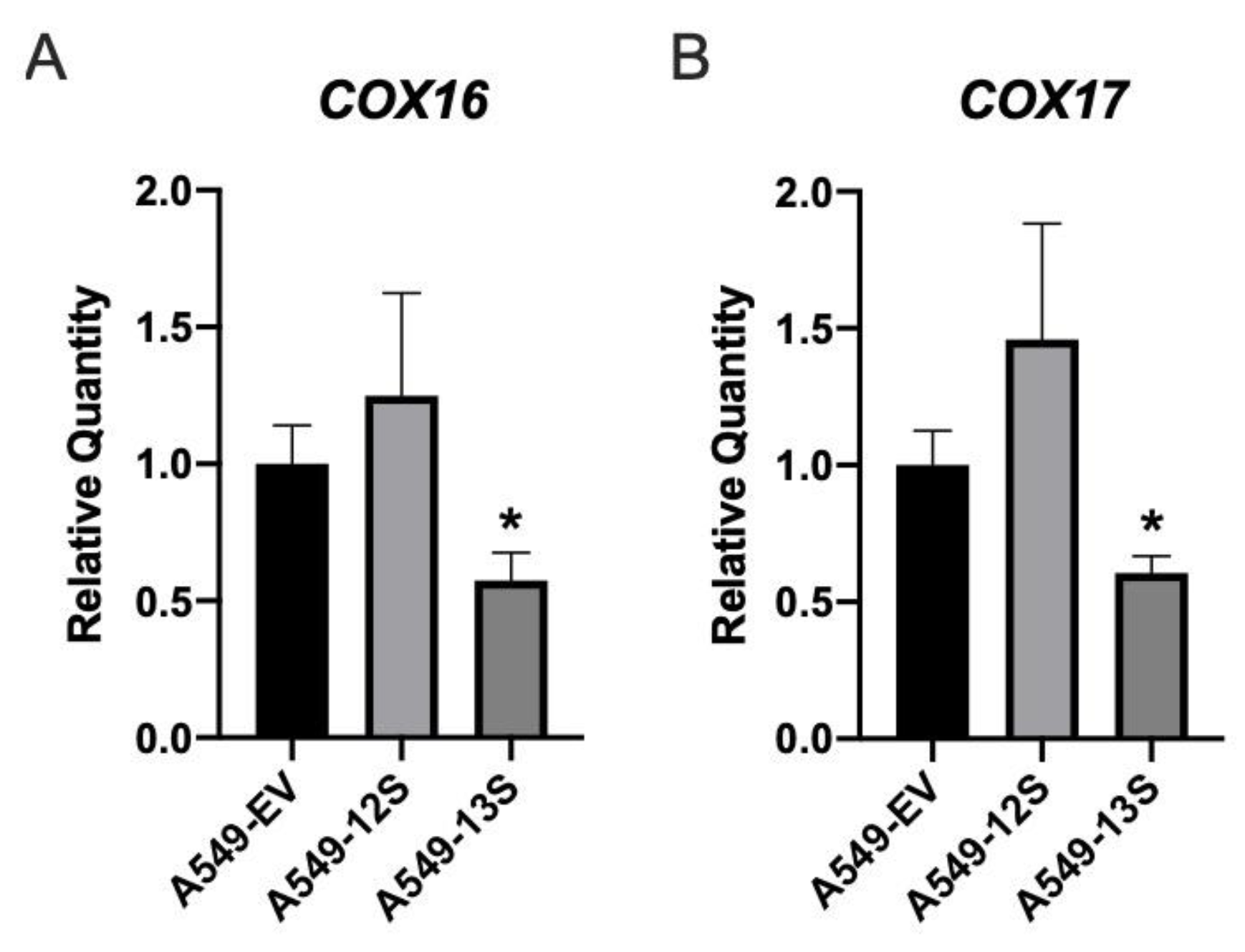
3.5. Oxidative Phosphorylation Genes Are Downregulated in A549-13S Cells
3.6. 13S E1A Influences Metabolism to a Greater Extent than 12S E1A in HAdV-5 Infected Primary IMR-90 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goodwin, C.M.; Xu, S.; Munger, J. Stealing the keys to the kitchen: Viral manipulation of the host cell metabolic network. Trends Microbiol. 2015, 23, 789–798. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Thaker, S.K.; Ch’ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef]
- Prusinkiewicz, M.A.; Mymryk, J.S. Metabolic reprogramming of the host cell by human adenovirus infection. Viruses 2019, 11, 141. [Google Scholar] [CrossRef] [Green Version]
- Ou, H.D.; May, A.P.; O’Shea, C.C. The critical protein interactions and structures that elicit growth deregulation in cancer and viral replication. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 48–73. [Google Scholar] [CrossRef] [Green Version]
- Thai, M.; Thaker, S.K.; Feng, J.; Du, Y.; Hu, H.; Ting Wu, T.; Graeber, T.G.; Braas, D.; Christofk, H.R. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 2015, 6, 8873. [Google Scholar] [CrossRef] [PubMed]
- Thai, M.; Graham, N.A.; Braas, D.; Nehil, M.; Komisopoulou, E.; Kurdistani, S.K.; McCormick, F.; Graeber, T.G.; Christofk, H.R. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 2014, 19, 694–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frese, K.K.; Lee, S.S.; Thomas, D.L.; Latorre, I.J.; Weiss, R.S.; Glaunsinger, B.A.; Javier, R.T. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-ORF1 oncoprotein. Oncogene 2003, 22, 710–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.S.; Weiss, R.S.; Javier, R.T. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6670–6675. [Google Scholar] [CrossRef] [Green Version]
- Dix, I.; Leppard, K.N. Regulated splicing of adenovirus type 5 E4 transcripts and regulated cytoplasmic accumulation of E4 mRNA. J. Virol. 1993, 67, 3226–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelka, P.; Ablack, J.N.G.; Fonseca, G.J.; Yousef, A.F.; Mymryk, J.S. Intrinsic structural disorder in adenovirus E1A: A viral molecular hub linking multiple diverse processes. J. Virol. 2008, 82, 7252–7263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, C.R.; Zhang, A.; Tessier, T.M.; Gameiro, S.F.; Mymryk, J.S. Hacking the cell: Network intrusion and exploitation by adenovirus E1A. MBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Crisostomo, L.; Soriano, A.M.; Mendez, M.; Graves, D.; Pelka, P. Temporal dynamics of adenovirus 5 gene expression in normal human cells. PLoS ONE 2019, 14, e0211192. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Granberg, F.; Pettersson, U. How adenovirus strives to control cellular gene expression. Virology 2007, 363, 357–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevins, J.R. Regulation of early adenovirus gene expression. Microbiol. Rev. 1987, 51, 419–430. [Google Scholar] [CrossRef]
- Radko, S.; Jung, R.; Olanubi, O.; Pelka, P. Effects of adenovirus type 5 E1A isoforms on viral replication in arrested human cells. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Lillie, J.W.; Loewenstein, P.M.; Green, M.R.; Green, M. Functional domains of adenovirus type 5 E1a proteins. Cell 1987, 50, 1091–1100. [Google Scholar] [CrossRef]
- Stevens, J.L.; Cantin, G.T.; Wang, G.; Shevchenko, A.; Shevchenko, A.; Berk, A.J. Transcription control by E1A and MAP kinase pathway via Sur2 Mediator subunit. Science 2002, 296, 755–758. [Google Scholar] [CrossRef]
- Pelka, P.; Ablack, J.N.G.; Torchia, J.; Turnell, A.S.; Grand, R.J.A.; Mymryk, J.S. Transcriptional control by adenovirus E1A conserved region 3 via p300/CBP. Nucleic Acids Res. 2009, 37, 1095–1106. [Google Scholar] [CrossRef]
- Madison, D.L.; Yaciuk, P.; Kwok, R.P.S.; Lundblad, J.R. Acetylation of the adenovirus-transforming protein E1A determines nuclear localization by disrupting association with importin-α. J. Biol. Chem. 2002, 277, 38755–38763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, E.; Mathews, M.B. Multiple functional domains in the adenovirus E1A gene. Cell 1987, 48, 177–178. [Google Scholar] [CrossRef]
- Soriano, A.M.; Crisostomo, L.; Mendez, M.; Graves, D.; Frost, J.R.; Olanubi, O.; Whyte, P.F.; Hearing, P.; Pelka, P. Adenovirus 5 E1A interacts with E4orf3 to regulate viral chromatin organization. J. Virol. 2019, 93, e00157-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.D.; Rosman, G.J. Improved retroviral vectors for gene transfer and expression. Biotechniques 1989, 7, 980–990. [Google Scholar]
- Zhang, A.; Tessier, T.M.; Galpin, K.J.C.; King, C.R.; Gameiro, S.F.; Anderson, W.W.; Yousef, A.F.; Qin, W.T.; Li, S.S.C.; Mymryk, J.S. The transcriptional repressor bs69 is a conserved target of the e1a proteins from several human adenovirus species. Viruses 2018, 10, 662. [Google Scholar] [CrossRef] [Green Version]
- Harlow, E.; Franza, B.R.; Schley, C. Monoclonal antibodies specific for adenovirus early region 1A proteins: Extensive heterogeneity in early region 1A products. J. Virol. 1985, 55, 533–546. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Haley, K.P.; Overhauser, J.; Babiss, L.E.; Ginsberg, H.S.; Jones, N.C. Transformation properties of type 5 adenovirus mutants that differentially express the E1A gene products. Proc. Natl. Acad. Sci. USA 1984, 81, 5734–5738. [Google Scholar] [CrossRef] [Green Version]
- Montell, C.; Fisher, E.F.; Caruthers, M.H.; Berk, A.J. Resolving the functions of overlapping viral genes by site-specific mutagenesis at a mRNA splice site. Nature 1982, 295, 380–384. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carinhas, N.; Koshkin, A.; Pais, D.A.M.; Alves, P.M.; Teixeira, A.P. 13C-metabolic flux analysis of human adenovirus infection: Implications for viral vector production. Biotechnol. Bioeng. 2017, 114, 195–207. [Google Scholar] [CrossRef]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamrahian, A.H.; Zhang, J.Z.; Elkhairi, F.S.; Prasad, R.; Ismail-Beigi, F. Activation of Glut1 glucose transporter in response to inhibition of oxidative phosphorylation. Role of sites of mitochondrial inhibition and mechanism of Glut1 activation. Arch. Biochem. Biophys. 1999, 368, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Thompson, C.B. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 270–276. [Google Scholar] [CrossRef]
- Watson, E.; Yilmaz, L.S.; Walhout, A.J.M. Understanding metabolic regulation at a systems level: Metabolite sensing, mathematical predictions, and model organisms. Annu. Rev. Genet. 2015, 49, 553–575. [Google Scholar] [CrossRef] [Green Version]
- Wegner, A.; Meiser, J.; Weindl, D.; Hiller, K. How metabolites modulate metabolic flux. Curr. Opin. Biotechnol. 2015, 34, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Valdés, A.; Zhao, H.; Pettersson, U.; Lind, S.B. Time-resolved proteomics of adenovirus infected cells. PLoS ONE 2018, 13, 1–23. [Google Scholar] [CrossRef]
- Allen, E.L.; Ulanet, D.B.; Pirman, D.; Mahoney, C.E.; Coco, J.; Si, Y.; Chen, Y.; Huang, L.; Ren, J.; Choe, S.; et al. Differential aspartate usage identifies a subset of cancer cells particularly dependent on OGDH. Cell Rep. 2016, 17, 876–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; Deberardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, R.; Pasquel, F.; Turcu, A.; Pongratz, R.L.; Roden, M.; Cline, G.W.; Shulman, G.I.; Kibbey, R.G. Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J. Biol. Chem. 2009, 284, 26578–26590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, W.C.; Cheng, A.L.; Brotto, M.; Chuang, C.Y. Visual gene-network analysis reveals the cancer gene co-expression in human endometrial cancer. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smallwood, H.S.; Duan, S.; Morfouace, M.; Rezinciuc, S.; Shulkin, B.L.; Shelat, A.; Zink, E.E.; Milasta, S.; Bajracharya, R.; Oluwaseum, A.J.; et al. Targeting Metabolic reprogramming by influenza infection for therapeutic intervention. Cell Rep. 2017, 19, 1640–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, E.; Grodzicker, T.; Roberts, R.J.; Mathews, M.B.; Zerler, B. Lytic and transforming functions of individual products of the adenovirus E1A gene. J. Virol. 1986, 57, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Hošek, T.; Calçada, E.O.; Nogueira, M.O.; Salvi, M.; Pagani, T.D.; Felli, I.C.; Pierattelli, R. Structural and dynamic characterization of the molecular hub early region 1A (E1A) from human adenovirus. Chem. A Eur. J. 2016, 22, 13010–13013. [Google Scholar] [CrossRef]
- Ablack, J.N.G.; Pelka, P.; Yousef, A.F.; Turnell, A.S.; Grand, R.J.A.; Mymryk, J.S. Comparison of E1A CR3-dependent transcriptional activation across six different human adenovirus subgroups. J. Virol. 2010, 84, 12771–12781. [Google Scholar] [CrossRef] [Green Version]
- Gallimore, P.H.; Turnell, A.S. Adenovirus E1A: Remodelling the host cell, a life or death experience. Oncogene 2001, 20, 7824–7835. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, P.A.; Blair, G.E. Expression and interactions of human adenovirus oncoproteins. Biochem. J. 1991, 275, 281–299. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prusinkiewicz, M.A.; Tu, J.; Dodge, M.J.; MacNeil, K.M.; Radko-Juettner, S.; Fonseca, G.J.; Pelka, P.; Mymryk, J.S. Differential Effects of Human Adenovirus E1A Protein Isoforms on Aerobic Glycolysis in A549 Human Lung Epithelial Cells. Viruses 2020, 12, 610. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060610
Prusinkiewicz MA, Tu J, Dodge MJ, MacNeil KM, Radko-Juettner S, Fonseca GJ, Pelka P, Mymryk JS. Differential Effects of Human Adenovirus E1A Protein Isoforms on Aerobic Glycolysis in A549 Human Lung Epithelial Cells. Viruses. 2020; 12(6):610. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060610
Chicago/Turabian StylePrusinkiewicz, Martin A., Jessie Tu, Mackenzie J. Dodge, Katelyn M. MacNeil, Sandi Radko-Juettner, Gregory J. Fonseca, Peter Pelka, and Joe S. Mymryk. 2020. "Differential Effects of Human Adenovirus E1A Protein Isoforms on Aerobic Glycolysis in A549 Human Lung Epithelial Cells" Viruses 12, no. 6: 610. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060610