Extracellular Vesicles in Viral Spread and Antiviral Response


1
Departamento de Biología Molecular, Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain
2
Centro de Biología Molecular Severo Ochoa, CSIC-UAM, Cantoblanco, 28049 Madrid, Spain
*
Author to whom correspondence should be addressed.
Viruses 2020, 12(6), 623; https://0-doi-org.brum.beds.ac.uk/10.3390/v12060623
Submission received: 30 April 2020
/
Revised: 1 June 2020
/
Accepted: 4 June 2020
/
Published: 8 June 2020
(This article belongs to the Special Issue Viruses and Extracellular Vesicles)
{kind=link}
Abstract
:Viral spread by both enveloped and non-enveloped viruses may be mediated by extracellular vesicles (EVs), including microvesicles (MVs) and exosomes. These secreted vesicles have been demonstrated to be an efficient mechanism that viruses can use to enter host cells, enhance spread or evade the host immune response. However, the complex interplay between viruses and EVs gives rise to antagonistic biological tasks—to benefit the viruses, enhancing infection and interfering with the immune system or to benefit the host, by mediating anti-viral responses. Exosomes from cells infected with herpes simplex type 1 (HSV-1) may transport viral and host transcripts, proteins and innate immune components. This virus may also use MVs to expand its tropism and evade the host immune response. This review aims to describe the current knowledge about EVs and their participation in viral infection, with a specific focus on the role of exosomes and MVs in herpesvirus infections, particularly that of HSV-1.
1. Introduction
Optimization of viral spread is crucial for viruses to maximize their biological success. Once virions have been assembled, matured and released from cells, they must disperse throughout the host and overcome barriers such as selective tropism and the host immune system. In this regard, viruses have evolved strategies to avoid neutralizing antibodies and facilitate dissemination within the host by wrapping themselves in membrane-wrapped clusters [1].
Viral spread by both enveloped and non-enveloped viruses may occur through extracellular vesicles (EVs) [2,3,4,5,6]. EVs have also proven to be an efficient mechanism for viruses to enter host cells or evade the host immune response [7,8,9]. EVs have key biological activities during viral infections, including the transport of viral genomes into target cells and interventions in cell physiology that support infection [10]. Their role as mediators of transplacental and sexually transmitted viral infections has also been proposed [11]. Other advantages ascribed to the use of EVs by viruses include several strategies for dispersing in groups, in so-called “collective infectious units.” One advantage of collective dispersal strategies is an increase in effective multiplicity of infection (MOI) [12]. It has been argued that a high MOI may enhance infectivity by allowing the virus to overcome different infection barriers and may reduce the probability of unsuccessful infections due to stochastic processes. In this regard, propagation as a pool of virions inside EVs can be considered a mode of collective dispersal strategy [12], presenting the concomitant advantages of that en bloc viral transmission [13]. Collective infectious units can consist not only of multiple virions inside a vesicle but also of multiple viral genomes within a single virion. The simultaneous delivery of multiple viral genomes to the same cell may have significant consequences for pathogenesis, antiviral resistance and social evolution [14]. In this way, EVs may support genetic cooperativity among viral quasispecies and increase the fitness of the whole viral population [15].
Numerous reports have highlighted the role of EVs in viral infection and their importance in viral entry, spread and immune evasion [2,5,8,9,15,16,17,18]. However, given the complex relationship between viruses and EVs, these vesicles can also trigger anti-viral host responses [6]. In this sense, it is widely accepted that EVs exert critical functions not only in the bolstering but also in the blockage of viral infections, modulating immune responses and serving as a vehicle of intercellular communication between infected and uninfected cells [4,6,19]. Indeed, because of their common biogenesis pathways, EVs and viruses may be considered to be close relatives and it has been argued that a deep understanding of the biology of EVs and the mechanisms by which they impact viral infections is necessary, especially for translation into therapy [20].
Herpes simplex type 1 (HSV-1) may spread by two main pathways—by cell-free virus; or by direct cell-to-cell spread, in which the viral transmission occurs through cell-to-cell contact [21], with cell adhesion proteins and their cytoskeletal connections used for lateral spread [22]. In cell-free virus infections, progeny viral particles must “escape” into the extracellular space and then infect another cell from the outside. Interactions of viral envelope proteins with the cell surface define cell-free viral spread and although it enhances dissemination by allowing diffusing virions to infect distant cells, there are some disadvantages, as free virions can be neutralized by antibodies or subjected to opsonization and phagocytosis [23]. Acquisition of an outer envelope may help shield HSV-1 from neutralizing antibodies while reinforcing viral dissemination [24]. Some strains of HSV-1 can also spread through syncytium formation, which occurs upon fusion of infected cells with neighboring uninfected ones [25].
This review describes current knowledge about the involvement of EVs in viral infections. We specifically focus on recent research on the role of EVs, both exosomes and microvesicles (MVs), during herpesvirus infections, particularly that of HSV-1.
2. EVs: Brief Overview
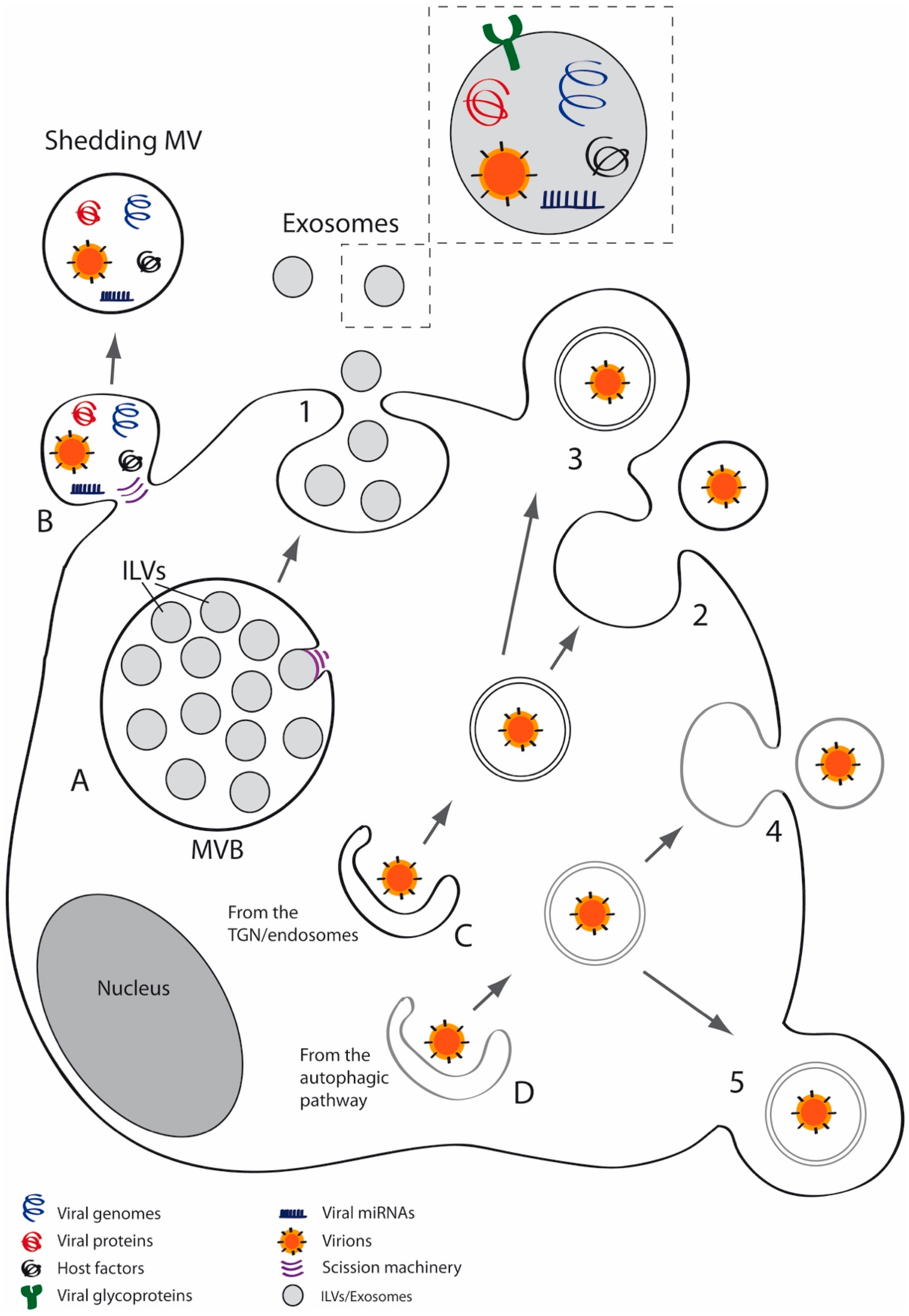
Extracellular vesicles (EVs) are membrane vesicles secreted by most cell types which have been isolated from several biological fluids such as blood, urine, cerebrospinal fluid and saliva [26,27,28,29]. Almost all cell types belonging to the three domains of life, Archaea, Bacteria and Eukarya, may secrete EVs [30,31]. Classification and nomenclature of EVs is complex but three major categories of EVs can be broadly established—apoptotic bodies, MVs and exosomes [26,32]. MVs derive from shedding of the plasma membrane [28,33], whereas exosomes are vesicles released to the extracellular space after fusion of multivesicular bodies (MVBs) with the cell membrane [26,32,34] (Figure 1). Exosomes have a typical diameter of 30–100 nm while MVs have a heterogeneous size, ranging from 100 nm to 1 μm in diameter [29,35]. MVs are enriched in lipid rafts and proteins such as flotillin-1 or integrins and expose phosphatidylserine (PS) on the outer plasma membrane leaflet [36,37,38], whereas exosomes are enriched in tetraspanins such as CD9, CD63 and CD81 and endosomal markers including ALIX and TSG101. The relative centrifugal force needed to isolate MVs is frequently between 10,000 and 20,000× g [39] and around 100,000× g is typically used to pellet exosomes [40,41,42,43].
EVs may be taken up by recipient cells by endocytosis or fusion with the plasma membrane and, given the diversity of EVs, these vesicles can enter cells using different routes. Endocytic pathways are heterogeneous and may include several mechanisms, such as endocytosis that is dependent or independent of clathrin, caveolin-dependent uptake, phagocytosis, macropinocytosis and lipid raft-mediated endocytosis [44]. EVs may be key mediators of several physiological and pathological processes [27,45,46] and are currently associated with cancer [47,48,49,50], infection [3,8,51], inflammation and immune responses [52] and myelination and neuron–glia communication [53,54,55,56,57]. Regarding virus-host interactions, EVs have emerged as a relevant object of attention because of their participation in the intercellular communication processes during viral infections.
3. EVs in Viral Infections
3.1. Non-Enveloped Viruses
EVs may modulate the infection of diverse viruses and non-enveloped viruses in particular exploit them to exit from cells non-lytically and to avoid and manipulate the immune system [15]. The canonical separation between enveloped and non-enveloped virus has been nuanced by the existence of non-enveloped viruses that, during a major part of their viral cycle, may spread while enclosed in vesicles, thus behaving as “quasi-enveloped” viruses. In this regard, several picornaviruses can be released from their host cells enclosed in EVs. This was first reported in hepatitis A virus (HAV), a hepatovirus which was found enclosed in host-derived membrane vesicles resembling exosomes which protected the virions from antibody-mediated neutralization [58]. The HAV structural protein pX was shown to interact with ALIX to promote the secretion of virions through exosome-like vesicles [59]. These quasi-enveloped viruses were infectious and circulated in the blood of infected humans and their biogenesis was dependent on host ESCRT machinery. Besides HAV, hepatitis E virus (HEV) can be also released through MVBs by the cellular exosomal pathway [60] and may circulate in the blood completely enclosed in membranes during infection, which is just as infectious as their naked counterparts [61]. Unlike enveloped viruses, quasi-enveloped viruses lack viral glycoproteins within the surrounding lipid bilayer but they may display internal proteins (such as VP1pX in the case of quasi-enveloped HEV) that are absent in canonical naked virions. The acquired membrane protects these viruses from neutralizing antibodies while facilitating spread within the host and, like enveloped viruses, they may hijack the host ESCRT machinery to exit infected cells non-cytolytically. In an alternative model, these viruses may also use autophagosome-mediated exit without lysis (AWOL), releasing virions enclosed in LC3-positive vesicles [61]. According to the AWOL model, which was first described for poliovirus release [62], viruses confined in double-membrane structures derived from the autophagic pathway can be released to the extracellular milieu via fusion with the plasma membrane. Another picornavirus, the aphthovirus responsible for foot-and-mouth disease (FMDV) was long ago observed to be released from cells by an exocytic mechanism involving membrane-limited vesicles [63]. More recently, a mechanism of exosome-mediated transmission of FMDV has been described in vivo and in vitro [64] and has also been considered as a potential immune evasion mechanism.
Cells infected by Coxsackie B virus (CBV) have demonstrated to release EVs. Thus, infected neural progenitor and stem cells (NPSCs) and C2C12 myoblast cells induced the release of abundant MVs containing viral proteins and infectious virus, a process that meant a novel route for virus dissemination [65]. Cells infected with CBV trigger fragmentation of mitochondria through a precursor of autophagic mitochondrial elimination (mitophagy) and virions may be released within MVs derived from these mitophagosomes [66]. The acquisition of this “cloak” may help the virus to evade the immune system, allowing an efficient non-lytic viral spread. CBV can also enhance replication efficiency by packaging microRNAs (miRNAs) into EVs from infected cells [67].
Enterovirus 71 (EV71) can be transported non-lytically between cells during early viral infection and exosomes containing this virus have been shown to establish a productive infection in human neuroblastoma cells [68]. EV71 virions were also found inside exosomes from infected human rhabdomyosarcoma cells and also have been detected in human samples [69]. Upon infection, another picornavirus, encephalomyocarditis virus (EMCV), triggers the release of multiple EV subpopulations that differ in their physical properties, composition and function [70].
Choroid plexus epithelial cells infected with the human polyomavirus 2 (JC virus) may produce EVs containing virions [71]. Those vesicles expressed exosomal markers such as CD9 and TSG101 and entered glial cells by macropinocytosis and clathrin-dependent endocytosis. The presence of virions in EVs may constitute a main pathway for its spread, since oligodendrocytes and astrocytes, the major targets of JC virus in the central nervous system (CNS), do not express the viral attachment receptors needed for direct viral fusion. EVs may also mediate the interaction between the human papilloma virus (HPV) and human immunodeficiency virus (HIV)-1. In this way, exosomes secreted from cells infected with HPV may increase HIV-1 replication in U1 cells via an oxidative stress pathway and treatment with antioxidants reduced EV-mediated enhancement of HIV-1 replication [72].
Gastroenteric non-enveloped pathogens such as noroviruses and rotaviruses may also spread enclosed in EVs, transferring a higher infectious dose to the next host cell and contributing to enhanced fecal-oral propagation [73]. Bluetongue virus (BTV) is a reovirus that may also be released from cells by a budding process. The majority of virions are released by cell lysis in mammalian cells but in insect cells, the release of BTV is nonlytic. Expression of NS3, a non-structural viral protein, in invertebrate cells infected with this arbovirus has been reported to correlate with nonlytic virus release. NS3 upholds virus release by recruiting ESCRT-I protein TSG101 [74] and it may act like the membrane protein of enveloped viruses, being responsible for intracellular trafficking and budding of virus particles [75]. The integrity of MVBs is also important for BTV assembly [76], as it occurs for other enveloped viruses.
3.2. Enveloped Viruses
Several enveloped viruses exploit EVs as a vehicle for enhancing their propagation. For example, EVs play a relevant role in hepatitis C virus (HCV) spread, as virions contained in exosomes can be transported to hepatocyte-like cells, establishing a productive infection [77]. Exosomes found in the serum of HCV infected patients can also mediate HCV transmission to hepatocytes [78].
Exosomes released by influenza virus-infected cells may modulate immune response through its transfer of different cargoes such as proteins, mRNAs or miRNAs. Upon infection, the expression of host miRNAs is altered and those miRNAs can regulate viral genes and both stimulate or suppress cell apoptosis and innate immune responses during infection. Therefore, secretion of exosomes and dysregulation of miRNAs are associated with immune regulation and pathogenicity during infection with this virus [79]. In mouse model, infection with influenza increases the levels of miRNAs in exosomes from bronchoalveolar lavage fluid. Moreover, serum exosomes from mice infected with influenza showed high levels of miR-483-3p (one of those miRNAs). The addition of those exosomes to MS1 murine cell line and their following uptake by those cells, potentiated the expression of proinflammatory cytokines in that vascular endothelial cell line [80]. A Y5 RNA-derived small RNA, Hsa-miR-1975, was secreted in exosomes and transferred to neighboring cells to induce interferon expression, which highlights the role of this Y-class small RNA in host’s defense against influenza as an antiviral mechanism based in exosomal traffic [81].
EVs may also modulate infections carried out by flaviviruses such as Dengue virus (DENV). Upon DENV infection, immune cells increase secretion of pro-inflammatory factors into the bloodstream, causing hyperpermeability. It has been hypothesized that exosomes and/or MVs might be hijacked to benefit viral spread and pathogenesis, perhaps triggering hyperpermeability and plasma leakage [82]. The versatility of the biological role of EVs is exemplified by virus-vector-host interactions that limit infection in mosquito cells. Host EVs may enter mosquito cells, inhibiting their infection through the restriction of virus-endosomal membrane fusion [83]. In addition, exosomes released from DENV-infected macrophages and added to the human EA.hy926 endothelial cells induced physiological changes in that cell line, leading to a protective effect during the early stages of infection that may help to maintain endothelial integrity [84]. Exosomes from C6/36 mosquito cells infected with Zika virus (ZIKV) may also modify host cells responses and contribute to the pathogenesis of ZIKV infection in humans. These exosomes induced a pro-inflammatory state and participated in endothelial vascular cell damage by inducing coagulation, inflammation and endothelial permeability [85].
EVs may also modulate retroviral pathogenesis [86,87]; HIV and simian immunodeficiency viruses modulate vesicle secretion through the Nef protein, which is secreted in exosomes and modifies intracellular trafficking pathways, enhancing viral infectivity and regulating host gene expression and immune responses [88,89,90,91]. In addition, TNFα release by cells upon incorporation of exosomes secreted by infected cells may reactivate latent HIV-1 [92]. The “Trojan exosome” hypothesis states that retroviruses use cellular exosome biogenesis pathways to form infectious particles and the exosome uptake pathway for receptor-independent, Env-independent routes of infection [93]. In other words, retroviruses coopt the cellular machinery for exosomal release; the strong concordance between the host exosome protein profile and that of HIV-1 supported this hypothesis [94], although other authors have questioned the existence of this type of specialized and shared release pathway for HIV-1 and exosomes [95]. Later reports supported the Trojan exosome hypothesis [96,97], proposing a model in which dendritic cells (DCs) internalize retroviruses by endocytosis and subsequently infect interacting CD4+ T cells, a mechanism known as trans-infection that may coexist with direct infection to a different extent depending on the maturation state of the DCs [98].
EVs are involved in the pathogenesis of other retroviruses, too. For instance, EVs produced by cells infected with human T-cell lymphotropic virus-1 (HTLV-1) may transport viral proteins and RNA, causing adverse effects on recipient uninfected cells and increasing viral spread via the upregulation of cell-to-cell contacts [99].
Exosomes may be key mediators of immune responses in patients with respiratory viral infections. Exosomes were detected in serum samples collected from lung transplant recipients with symptomatic respiratory infections caused by coronavirus, respiratory syncytial virus and the non-enveloped rhinovirus, as well as from non-symptomatic stable recipients. Those exosomes enclosed significantly higher levels of lung self-antigens, 20S proteasome and viral antigens when compared with controls. When mice were immunized with those exosomes obtained from transplant recipients with respiratory viral infections, they developed immune responses to self-antigens, small airway occlusion, fibrosis and cellular infiltration [100].
4. EVs in Herpesvirus Infections
4.1. Alphaherpesviruses
Alpha-, beta- and gammaherpesviruses may exploit EVs to enhance viral spread or may trigger EV-mediated host immune responses [101,102]. The relationship between EVs and HSV-1 is the most heavily investigated among the subfamily Alphaherpesvirinae. HSV-1 is a double-stranded DNA enveloped virus. It is a highly prevalent neurotropic human pathogen [103] that can establish latency in neurons [104]. Primary infection occurs in epithelial cells and then the virus spreads to neurons of the trigeminal ganglia, travelling in a retrograde direction toward the neuron cell bodies, where the virus establishes latent infections [105]. To establish and maintain latency, HSV-1 expresses miRNAs that can downregulate key viral immediate early proteins [106]. Herpesviruses, in general, use such miRNAs to induce and maintain latency [107].
Around 90% of people are seropositive for HSV-1 −which indicates a past exposure to the virus− and have latent HSV-1 genomes in the trigeminal ganglia. Although early studies discovered latent HSV-1 and HSV-2 in the trigeminal and sacral ganglia, respectively, both viruses may also spread to spinal ganglia [108]. Latent HSV-1 can reactivate periodically, either spontaneously or following different triggering events such as ultraviolet light exposure, immunosuppression, fever or x-ray irradiation [109]. Under certain circumstances, HSV-1 may cause severe pathologies such as keratoconjunctivitis or encephalitis [110], which is the major cause of sporadic fatal encephalitis worldwide. HSV-1 is also an increasing cause of genital herpes [111].
HSV-1 may infect many different hosts and cell types [112] using different receptors and different pathways—plasma membrane fusion at neutral pH; or endocytosis that can be dependent or independent of low pH [113,114,115]. In many cultured cell lines, such as HEp-2 and Vero, HSV-1 enters cells by a pH-neutral fusion with the cell membrane. However, it enters HeLa and CHO-K1 cells by endocytosis and subsequent exposure to a low pH [116]. Heparan sulfate glycosaminoglycans operate as attachment receptors for the viral glycoprotein gC [117]. Although this glycoprotein is not essential for viral entry, its absence decreases infectivity, due to the reduction of efficiency of viral binding to cells [116]. In the absence of gC, gB can mediate binding to heparan sulfate [118]. Regardless of the pathway, gD, fusion effector gB and fusion modulator complex gH/gL are essential for HSV-1 entry [119]. Capsid assembly and DNA packaging occur in the nucleus, whereas primary envelopment and de-envelopment take place at the nuclear envelope. Acquisition of tegument and secondary envelopment occurs in the cytoplasm, via budding into vesicles derived from the trans-Golgi network (TGN coated with viral glycoproteins and additional tegument proteins [120]. As with other viruses, MVBs may affect significantly the HSV-1 envelopment and egress, since MVBs modified by the virus constitute a site for envelopment of this virus [121,122].
Finally, viral particles may be released by exocytosis at the plasma membrane and/or transmitted by cell-to-cell at cell junctions [120]. In human tissues, the major mode of HSV-1 transmission is cell-to-cell spread, that is, the direct passage of progeny virus from an infected cell to an adjacent one; this mechanism may serve as an immune evasion strategy, since it protects the virus from immune surveillance [123]. The main entry receptors for gD glycoprotein are HVEM [9], nectin-1 [10] and 3-O-sulfated heparan sulfate [11]. Paired immunoglobulin-like type 2 receptor (PILR) alpha [124] and myelin-associated glycoprotein (MAG) [125] are receptors for gB. Many details about the process of viral dissemination are not completely understood yet and, therefore, clarifying the mechanisms of viral spread and subsequent entry into other cells is still required to fully understand the viral cycle. In this respect, deepen into the role of EVs during HSV-1 infection will shed light on this complex process.
The production of secreted vesicles by cells infected with HSV-1 has been known for a long time and is currently well described. The first to be discovered were the light particles (L-particles), which are similar to virions in appearance but lack the viral nucleocapsid and genome and therefore are not infectious [126,127]. L-particles are secreted after infection with all alphaherpesviruses tested to date, both in human and animal cells [128]. Although L-particles are not infectious, they may deliver viral proteins and cellular factors needed for virus replication and immune evasion such as ICP0, ICP4 and gB [129,130], thus facilitating HSV-1 infection. L-particles and virions show common features—vhs (virión host shutoff, a protein located in the tegument that selectively degrades mRNA early in infection) and α-TIF (a tegument protein that induces immediate early genes by interacting with two cellular proteins) exert the same function in both particles and both share similar assembly and egress pathways, suggesting that viral glycoproteins and tegument are sufficient to induce secondary envelopment [120,127]. There are another type of particles, pre-viral DNA replication enveloped particles (PREPs), which share morphological features with L-particles but differ in relative protein composition. For instance, gC and gD are reduced or absent in PREPs whereas VP22 or gE are increased [131].
Transfer of functional viral proteins to uninfected cells via L-particles may suggest a viral immune escape strategy. Incubation of mature dendritic cells with L particles reduced CD83 (a molecule with costimulatory properties) expression on uninfected bystander dendritic cells, showing that functional viral proteins may be transmitted via L particles from infected to uninfected bystander cells, thereby inducing CD83 downregulation [130]. Similarly, HSV-1 may also exploit the EV pathway to evade the immune system. In this regard, it has been demonstrated that this virus may manipulate the MHC class II processing pathway by modifying the endosomal sorting and trafficking of HLA-DR, hijacking these molecules from their typical cell membrane function and instead diverting them into the exosomal pathway [132]. Likewise, work in our laboratory showed that infection of Chinese hamster ovary (CHO) cells with virions enclosed in MVs was not completely neutralized by anti-HSV-1 antibodies, suggesting that these EVs may be shielding the viral particles from the immune system [133].
However, immune modulation by EVs is complex and multifaceted. Recent studies have described the release of EVs, ranging between 50 and 110 nm, from cells infected with HSV-1. Those vesicles carried viral and host transcripts −mRNAs, miRNAs and non-coding RNAs− and proteins, including the tetraspanins CD9, CD63 and CD81 and innate immune components such as stimulator of IFN genes (STING) [134]. STING activates transcription of type I IFNs, which induce an antiviral response. The use of exosomes to export STING to uninfected cells has been explained as a way to control dissemination of the virus, in such a way that HSV-1 might limit the spread of infection from cell-to-cell to control its virulence and facilitate the dissemination between individuals [129,134,135].
It has been reported that Rab27a, a small GTPase implicated in secretion of exosomes [136], may influence the assembly of viruses such as HIV-1 [137] and HCMV [138]. Likewise, studies carried out by our group showed that this GTPase plays a relevant role in HSV-1 infection of oligodendrocytes [139]. Our results showed a significant reduction in plaque size and viral production in Rab27a-silenced cells infected with HSV-1, suggesting that Rab27a depletion may affect viral egress. More recent studies have revealed the participation of MVs in HSV-1 spread. Our study [133] described the features of MVs released by different cell lines infected with HSV-1 and their participation in the infectious cycle, suggesting that MVs released by cells infected by HSV-1 contained viral particle and were endocytosed by naïve cells, leading to productive infection. These findings suggested that HSV-1 spread might use MVs to expand its tropism and evade the immune response [133,140]. The exact process of HSV-1 targeting to MVs is not known yet but our results suggest that autophagy may be involved in that process, since MVs isolated from infected cells were positive for LC3-II, an autophagy marker. Hence, those results point to a role for the autophagic pathway in the MVs-mediated HSV-1 spread [140].
Though HSV-1 is the best-studied alphaherpesvirus, other species belonging to this family have also been involved in secretion and/or modulation of EVs. Bovine herpesvirus-1 (BoHV-1) and pseudorabies virus (PRV) may also localize gB to EVs. This glycoprotein co-localized with CD63 and MHC II in late endosomes [141]. On the other hand, exosomes secreted by lymphocytes after infection with varicella-zoster virus (VZV) were found to contain selectively concentrated STING [142], suggesting that this virus may modulate the immune system via exosomes.
4.2. Betaherpesviruses
The exosome secretion pathway plays a significant role in the life cycle of human herpesvirus 6 (HHV-6), a betaherpesvirus that can modify the molecular transport machinery in infected cells. HHV-6 virions are released along with intraluminal vesicles via the exosomal pathway, by fusion of the limiting membrane of MVBs −in which virus particles and exosomes are enclosed− with the plasma membrane [143]. In addition, T cells infected with HHV-6 showed reduced surface and intracellular expression of MHC class I molecules but these molecules were redistributed to TGN- or post-TGN-derived vesicles and then incorporated into virions and exosomes. The reduction in the total expression of MHC class I in HHV-6-infected cells suggests that a fraction of these molecules may be transported to lysosomes and degraded through the same route that is used to transport particles to MVBs [144]. This mechanism may be important for the virus to evade immune surveillance [101].
A relevant role for MVBs in the release of human cytomegalovirus (HCMV) has been also reported. The association of HCMV proteins with MVBs has been demonstrated [145] and participation of MVBs in the final envelopment of HCMV has also been reported [146]. The presence of gB and gH (two envelope glycoproteins that are essential for HCMV infectivity) in exosomes secreted by HCMV-infected cells has also been recently observed [147]. The exosomes secreted by HCMV-infected cells might exert a relevant effect on the immune system. Thus, endothelial cells infected with HCMV generated viral antigens associated to ALIX-, TSG101- and CD63-positive exosomes that indirectly activated CD4+ T cells. This suggests that circulating exosomes from the HCMV-infected vascular endothelium might be a source of HCMV antigens in infected individuals, hypothetically contributing to the establishment of a HCMV-specific memory T cell population [148].
4.3. Gammaherpesviruses
The human gammaherpesviruses Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) have also been demonstrated to modulate the tumor microenvironment through exosomes. Thus, these oncogenic viruses may modify the protein content of exosomes to modulate the tumor microenvironment, enhance viral efficiency and promote tumorigenesis [149,150,151].
To contribute to the development and progression of malignancy, oncogenic viruses such as EBV promote a pro-tumoral environment, often by altering vesicle content and secretion. EBV-infected cells secrete exosomes carrying viral factors that can be internalized by recipient cells. For instance, exosomes released from latently-EBV-infected nasopharyngeal carcinoma cells (NPC) contained the latent membrane protein-1 (LMP1), signal transduction molecules and virus-encoded miRNAs [151]. LMP1 is the principal oncogene of EBV, being expressed in most EBV-related cancers and is critical for B cell immortalization and cellular transformation [152]. This viral oncoprotein, expressed by cells latently infected with EBV, critically contributes to pathogenesis by deregulation of cellular signal transduction pathways [153]. It has been recently shown that LMP1 promotes EVs secretion and may enhance cancer progression and metastasis by up-regulating syndecan-2 (SDC2) and synaptotagmin-like-4 (SYTL4) through nuclear factor (NF)-κB signaling [154].
LMP1 was initially detected by immunoelectron microscopy in budding plasma membrane protrusions compatible with MVs, although this oncoprotein was also found in the pellet obtained from the centrifugation of conditioned medium at 70,000× g [150]. A simultaneous report found that exosomes containing LMP1 inhibited the proliferation of peripheral blood mononuclear cells, suggesting that this viral oncogene may be involved in immune regulation helping the infected tumor cells to escape the immune system [155]. Inhibition of immune responses may not only promote viral spread but also its oncogenic transformation [17]. In addition, LMP1 associates with CD63 in endosomes and its secretion in exosomes reduces NF-κB activation, a relevant fact given that constitutive activation of NF-κB by LMP1 stimulates the proliferation of EBV-infected cells to establish viral persistence [156]. Therefore, since LMP1-modified exosomes enhance the growth, migration and invasion of malignant cells and thus enhance progression of EBV-associated tumors, the localization of LMP1 in exosomes is critical for tumor progression. The role of CD63 in this process is also crucial, since CD63 knockout resulted in a reduction of LMP1-induced particle secretion and LMP1 packaging impairment [157]. EBV-encoded LMP1 may also contribute to upregulation of intercellular adhesion molecule 1 (ICAM-1) [158].
On the other hand, exosomes can also transfer functional miRNAs from EBV-infected cells to subcellular sites of gene repression in uninfected recipient cells [159] and exosomes carrying miRNAs have been detected in blood samples from nasopharyngeal carcinoma patients [160]. The main functions of EBV-encoded miRNAs might be related to immune evasion, inhibition of apoptosis and cell transformation and proliferation, whereas the cellular miRNAs modulate their own biogenesis and latent/lytic infection [161]. A high expression of viral miRNAs encoded by human herpesviruses in diseased human tooth pulps has been recently demonstrated [162]. Examination of their influence on cellular miRNA of primary human oral keratinocytes showed high levels of viral miRNAs in exosomes derived from viral miRNA-transfected oral keratinocytes. In addition, those exosomes released their contents into macrophages, altering expression of their endogenous miRNAs, suggesting that herpesvirus-encoded miRNAs produced during oral infection might impact host defenses and exacerbate pathogenesis in oral inflammatory diseases generally considered to be of bacterial origin [162]. Herpesvirus-encoded miRNAs within exosomes have also been implicated in other diseases such as lichen planus, a chronic inflammatory disease with unclear etiology that has been associated with secretion of exosomes by cells infected with HCMV and other herpesviruses [163,164]. In addition, the miR-200 family of miRNAs acts as a cellular switch, regulating the shift from latency to the lytic cycle. In this context, it has been shown that miRNA-200 are packed into exosomes which create an epithelial microenvironment that promotes the EBV lytic cycle [165].
Exosomes may coat bystander lymphocytes with IL-35, an immunosuppressive cytokine composed of EBV-induced protein 3 (Ebi3) and IL-12α chain (p35) subunits. This EV-associated cytokine promotes infection tolerance in two ways—first, by inducing IL-35 production in non-Treg (regulatory T) cells; and second, by causing an immunosuppressive phenotype in EV-acquiring T and B cells, leading to secondary suppression of immune responses [166]. Another recent study has described a process due to a paracrine loop in which EBV M81 strain-infected B cells secreted exosomes, containing EBER2 RNA, that were endocytosed by neighboring cells. This RNA increased CXCL8 expression, a chemokine that enhanced spontaneous lytic replication levels in M81-infected B cells [167].
KSHV is the causative agent of Kaposi’s sarcoma, the most frequent cancer in untreated HIV patients suffering from acquired immunodeficiency syndrome (AIDS). Early studies showed that KSHV greatly alters the protein content of exosomes and that exosomes secreted from B cells infected with KSHV affect cellular metabolism and likely modulate cell death and survival [149]. It is widely accepted that the exosomal pathway is exploited by KSHV for viral spread and oncogenesis [168]. Exosomes obtained from patient primary effusion lymphoma pleural fluid produced an earlier and enhanced migration of endothelial cells, giving these patient-derived exosomes a functional biological role in cell migration Therefore, the exosomes derived from KSHV-associated malignancies were functional and in addition contained a distinct subset of miRNAs [169]. Later studies have demonstrated that exosomes isolated from the saliva of HIV patients and secreted by HIV-infected T-cell lines stimulated KSHV infectivity in epithelial cells, revealing that HIV-associated exosomes are a risk factor for KSHV infection in HIV-infected patients [170].
5. The Role of Lipid Rafts and the MAL Proteolipid
Myelin and lymphocyte protein (MAL) [171] is nonglycosylated integral membrane protein with four hydrophobic domains located in detergent-insoluble membrane fractions enriched in condensed membranes [172]. This proteolipid is the prototypical member of the MAL family, which reside in detergent-insoluble membranes enriched in cholesterol and glycolipids [173]. MAL is expressed in several cell types including epithelial and endothelial cells, hepatocytes and T cells that are found in several tissues [174]. In the nervous system, MAL is predominantly localized in compact myelin formed by oligodendrocytes and Schwann cells, playing an essential role in the stability of myelin [175] and regulating the distribution of PLP, the main myelin protein, into different microdomains [176]. This proteolipid plays a critical role in apical transport in epithelial cells [177,178], whereas in T lymphocytes, MAL is located at the immunological synapse and significantly affects exosome secretion [179,180].
The ESCRT machinery is typically involved in the budding and release of endosomal vesicles into MVBs. However many viruses, such as HIV or Ebola, utilize this machinery for budding and release. ESCRT proteins, which are localized in the neck region of membrane buds, mediate the membrane scission for viral release. Enveloped viruses acquire their lipid bilayers by budding through host membranes and many of them hijack the cellular ESCRT machinery to exit from the cells [181]. Some non-enveloped viruses such as BTV and HAV can also recruit the ESCRT pathway for release. However, the budding of other viruses, such as influenza, is ESCRT-independent. Lipid rafts substantially contribute to viral entry and assembly of enveloped and non-enveloped viruses [182] and they also contribute to the budding of several enveloped viruses via accumulation of viral components at the budding site, thus facilitating their interaction [183]. Some ESCRT proteins were identified in lipid rafts and it is probable that these proteins could associate to these lipid microdomains to promote membrane budding. Thus, the ESCRT proteins could bind to pre-existing rafts or induce lipid domain formation. Lipid rafts are a platform for assembly of many viruses and, therefore, the enrichment of ESCRT proteins in lipid rafts would facilitate their recruitment to assembling virions [184]. The role of TEMs in viral budding and release has also been demonstrated for several viruses [185].
The involvement of MAL in HSV-1 infection has been recently reported [186]. HSV-1 virions were transported in association with MAL-positive structures in oligodendrocytes to reach the end of cellular processes, which contact uninfected cells. In addition, as functional studies showed, the depletion of MAL led to a significant decrease in infection, with a drastic reduction in the number of lytic plaques in MAL-silenced cells. In this context, MAL proteolipid might be involved in viral spread via two main pathways—first, through its role in raft-mediated direct transport; and second, through its function on exosome secretion. In the absence of MAL, the formation and traffic of MVBs may be greatly impaired, because tetraspanin-enriched microdomains are not incorporated efficiently into intraluminal vesicles of MVBs. MAL accumulates on the limiting membrane of MVBs, triggering the rerouting of these aberrant MVBs to lysosomes for degradation. Therefore, MAL might exert a multifaceted role delivering virions either to cell-to-cell contacts or the plasma membrane or, otherwise allow their incorporation into MVBs. The detailed participation of the MAL proteolipid in viral spread is worth future investigation.
6. Conclusions
Both enveloped and non-enveloped viruses may exploit EVs to enhance their viral cycle but may also be the object of antiviral EV-mediated responses. The majority of viral families contain species that may hijack the EV-mediated endocytic machinery to enter cells and/or use secreted EVs for viral release. In addition, several viruses may interfere with the host immune system. In most cases, viruses use exosome secretion pathways, although viruses such as CBV, EBV or HSV-1 may use shedding MVs for viral spread and immune escape.
Alpha-, beta and gammaherpesviruses can use EVs to enhance viral spread or to modulate the immune response. Regarding HSV-1, exosomes secreted by infected cells may transport viral and host transcripts, proteins and innate immune components and this virus may use shedding MVs to expand its tropism and to evade the host immune response. A deep understanding of EVs and their involvement in host-viral interactions is essential for future use in diagnostics and anti-viral therapy.
Author Contributions
Conceptualization, R.B.-M. and J.A.L.-G.; writing—original draft preparation, R.B.-M.; writing—review and editing, R.B.-M. and J.A.L.-G.; infographics: R.B.-M. and I.R.: project administration, J.A.L.-G.; funding acquisition, J.A.L.-G. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support for the study was provided by Fundación Severo Ochoa-Aeromédica Canaria. The funders had no role in the decision to publish or preparation of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sanjuan, R.; Thoulouze, M.I. Why viruses sometimes disperse in groups. Virus Evol. 2019, 5, vez014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.R.; Kashanchi, F.; Jacobson, S. Exosomes in Viral Disease. Neurotherapeutics 2016, 13, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.W.; Lichty, B.D.; Bowdish, D.M. Microvesicles: Ubiquitous contributors to infection and immunity. J. Leukoc. Biol. 2015, 97, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr. Exosomal communication goes viral. J. Virol. 2015, 89, 5200–5203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alenquer, M.; Amorim, M.J. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses 2015, 7, 5066–5083. [Google Scholar] [CrossRef] [PubMed]
- Wurdinger, T.; Gatson, N.N.; Balaj, L.; Kaur, B.; Breakefield, X.O.; Pegtel, D.M. Extracellular vesicles and their convergence with viral pathways. Adv. Virol. 2012, 2012, 767694. [Google Scholar] [CrossRef]
- Schwab, A.; Meyering, S.S.; Lepene, B.; Iordanskiy, S.; van Hoek, M.L.; Hakami, R.M.; Kashanchi, F. Extracellular vesicles from infected cells: Potential for direct pathogenesis. Front. Microbiol. 2015, 6, 1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef] [Green Version]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, H.M.; Masoumi, N.; Witwer, K.W.; Pegtel, D.M. Extracellular Vesicles Exploit Viral Entry Routes for Cargo Delivery. Microbiol. Mol. Biol. Rev. Membr. 2016, 80, 369–386. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, V.L.; Ellwanger, J.H.; Chies, J.A.B. Extracellular vesicles in host-pathogen interactions and immune regulation—Exosomes as emerging actors in the immunological theater of pregnancy. Heliyon 2019, 5, e02355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreu-Moreno, I.; Sanjuan, R. Collective Viral Spread Mediated by Virion Aggregates Promotes the Evolution of Defective Interfering Particles. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan-Bonnet, N.; Perales, C.; Domingo, E. Extracellular vesicles: Vehicles of en bloc viral transmission. Virus Res. 2019, 265, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Leeks, A.; Sanjuan, R.; West, S.A. The evolution of collective infectious units in viruses. Virus Res. 2019, 265, 94–101. [Google Scholar] [CrossRef]
- Altan-Bonnet, N. Extracellular vesicles are the Trojan horses of viral infection. Curr. Opin. Microbiol. 2016, 32, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Kouwaki, T.; Okamoto, M.; Tsukamoto, H.; Fukushima, Y.; Oshiumi, H. Extracellular Vesicles Deliver Host and Virus RNA and Regulate Innate Immune Response. Int. J. Mol. Sci. 2017, 18, 666. [Google Scholar] [CrossRef] [Green Version]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The Role of Extracellular Vesicles in Viral Infection and Transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Crenshaw, B.J.; Gu, L.; Sims, B.; Matthews, Q.L. Exosome Biogenesis and Biological Function in Response to Viral Infections. Open Virol. J. 2018, 12, 134–148. [Google Scholar] [CrossRef] [Green Version]
- Raab-Traub, N.; Dittmer, D.P. Viral effects on the content and function of extracellular vesicles. Nat. Rev. Microbiol. 2017, 15, 559–572. [Google Scholar] [CrossRef]
- Nolte-’t Hoen, E.; Cremer, T.; Gallo, R.C.; Margolis, L.B. Extracellular vesicles and viruses: Are they close relatives? Proc. Natl. Acad. Sci. USA 2016, 113, 9155–9161. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.C.; Huber, M.T. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 2002, 76, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, M.; Generous, A.; Sinn, P.L.; Cattaneo, R. Connections matter—How viruses use cell-cell adhesion components. J. Cell Sci. 2015, 128, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graw, F.; Perelson, A.S. Modeling Viral Spread. Annu. Rev. Virol. 2016, 3, 555–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, J.; Mangale, V.; Thienphrapa, W.; Gottlieb, R.A.; Feuer, R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [Google Scholar] [CrossRef] [Green Version]
- Weed, D.J.; Nicola, A.V. Herpes simplex virus Membrane Fusion. Adv. Anat. Embryol. Cell Biol. 2017, 223, 29–47. [Google Scholar]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, A.E.; D’Souza-Schorey, C. The biology of extracellular microvesicles. Traffic 2018, 19, 319–327. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budnik, V.; Ruiz-Canada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef]
- Wei, X.; Liu, C.; Wang, H.; Wang, L.; Xiao, F.; Guo, Z.; Zhang, H. Surface Phosphatidylserine Is Responsible for the Internalization on Microvesicles Derived from Hypoxia-Induced Human Bone Marrow Mesenchymal Stem Cells into Human Endothelial Cells. PLoS ONE 2016, 11, e0147360. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.; Pendlebury, S.A.; Green, C. Lipid organization in erythrocyte membrane microvesicles. Biochem. J. 1984, 224, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Bello-Morales, R.; Lopez-Guerrero, J.A. Isolation/Analysis of Extracellular Microvesicles from HSV-1-Infected Cells. In Herpes Simplex Virus; Springer Nature: Basel, Switzerland, 2020; Volume 2060, pp. 305–317. [Google Scholar]
- Witwer, K.W.; Buzas, E.I.; Bemis, L.T.; Bora, A.; Lasser, C.; Lotvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2, 20389. [Google Scholar] [CrossRef]
- Lane, R.E.; Korbie, D.; Trau, M.; Hill, M.M. Purification Protocols for Extracellular Vesicles. Methods Mol. Biol. 2017, 1660, 111–130. [Google Scholar] [PubMed] [Green Version]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharm. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, F.M.; Carneiro, F.; Machado, J.C.; Melo, S.A. Exosomes and Immune Response in Cancer: Friends or Foes? Front. Immunol. 2018, 9, 730. [Google Scholar] [CrossRef] [PubMed]
- Nogues, L.; Benito-Martin, A.; Hergueta-Redondo, M.; Peinado, H. The influence of tumour-derived extracellular vesicles on local and distal metastatic dissemination. Mol. Asp. Med. 2018, 60, 15–26. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123 Pt 10, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.M.; Reiner, N.E. Exosomes and other microvesicles in infection biology: Organelles with unanticipated phenotypes. Cell. Microbiol. 2011, 13, 1–9. [Google Scholar] [CrossRef]
- Robbins, P.D.; Dorronsoro, A.; Booker, C.N. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J. Clin. Investig. 2016, 126, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Fruhbeis, C.; Frohlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Mobius, W.; Goebbels, S.; Nave, K.A.; et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusic, K.M.; Pusic, A.D.; Kraig, R.P. Environmental Enrichment Stimulates Immune Cell Secretion of Exosomes that Promote CNS Myelination and May Regulate Inflammation. Cell Mol. Neurobiol. 2016, 36, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, M.M.; Kaiser, J.; Schwab, M.E. Extracellular Vesicles: Multimodal Envoys in Neural Maintenance and Repair. Trends Neurosci. 2018, 41, 360–372. [Google Scholar] [CrossRef]
- Lopez-Leal, R.; Court, F.A. Schwann Cell Exosomes Mediate Neuron-Glia Communication and Enhance Axonal Regeneration. Cell. Mol. Neurobiol. 2016, 36, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Basso, M.; Bonetto, V. Extracellular Vesicles and a Novel Form of Communication in the Brain. Front. Neurosci. 2016, 10, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Ma, P.; Deng, L.; Liu, Z.; Wang, X.; Liu, X.; Long, G. Hepatitis A virus structural protein pX interacts with ALIX and promotes the secretion of virions and foreign proteins through exosome-like vesicles. J. Extracell. Vesicles 2020, 9, 1716513. [Google Scholar] [CrossRef]
- Nagashima, S.; Jirintai, S.; Takahashi, M.; Kobayashi, T.; Tanggis; Nishizawa, T.; Kouki, T.; Yashiro, T.; Okamoto, H. Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J. Gen. Virol. 2014, 95 Pt 10, 2166–2175. [Google Scholar] [CrossRef]
- Feng, Z.; Hirai-Yuki, A.; McKnight, K.L.; Lemon, S.M. Naked Viruses That Aren’t Always Naked: Quasi-Enveloped Agents of Acute Hepatitis. Annu. Rev. Virol. 2014, 1, 539–560. [Google Scholar] [CrossRef]
- Taylor, M.P.; Burgon, T.B.; Kirkegaard, K.; Jackson, W.T. Role of microtubules in extracellular release of poliovirus. J. Virol. 2009, 83, 6599–6609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwell, J.H.; Wool, S.; Kosikowski, F.V. Vesicular exocytosis of foot- and -mouth disease virus from mammary gland secretory epithelium of infected cows. J. Gen. Virol. 1981, 56 Pt 1, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Xu, S.; Shi, X.; Xu, G.; Shen, C.; Liu, X.; Zheng, H. Exosomes-mediated transmission of foot-and-mouth disease virus in vivo and in vitro. Vet. Microbiol. 2019, 233, 164–173. [Google Scholar] [CrossRef]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Sin, J.; McIntyre, L.; Stotland, A.; Feuer, R.; Gottlieb, R.A. Coxsackievirus B Escapes the Infected Cell in Ejected Mitophagosomes. J. Virol. 2017, 91, e01347-17. [Google Scholar] [CrossRef] [Green Version]
- Germano, J.F.; Sawaged, S.; Saadaeijahromi, H.; Andres, A.M.; Feuer, R.; Gottlieb, R.A.; Sin, J. Coxsackievirus B infection induces the extracellular release of miR-590-5p, a proviral microRNA. Virology 2019, 529, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Wu, J.; Shen, L.; Yang, J.; Chen, J.; Xu, H. Enterovirus 71 transmission by exosomes establishes a productive infection in human neuroblastoma cells. Virus Genes 2016, 52, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Wu, J.; Fang, D.; Qiu, Y.; Zou, X.; Jia, X.; Yin, Y.; Shen, L.; Mao, L. Exosomes cloak the virion to transmit Enterovirus 71 non-lytically. Virulence 2020, 11, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Van der Grein, S.G.; Defourny, K.A.Y.; Rabouw, H.H.; Galiveti, C.R.; Langereis, M.A.; Wauben, M.H.M.; Arkesteijn, G.J.A.; van Kuppeveld, F.J.M.; Nolte-’t Hoen, E.N.M. Picornavirus infection induces temporal release of multiple extracellular vesicle subsets that differ in molecular composition and infectious potential. Plos Pathog. 2019, 15, e1007594. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, B.A.; Morris-Love, J.; Gee, G.V.; Haley, S.A.; Atwood, W.J. JC Virus infected choroid plexus epithelial cells produce extracellular vesicles that infect glial cells independently of the virus attachment receptor. PLoS Pathog. 2020, 16, e1008371. [Google Scholar] [CrossRef]
- Ranjit, S.; Kodidela, S.; Sinha, N.; Chauhan, S.; Kumar, S. Extracellular Vesicles from Human Papilloma Virus-Infected Cervical Cancer Cells Enhance HIV-1 Replication in Differentiated U1 Cell Line. Viruses 2020, 12, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiana, M.; Ghosh, S.; Ho, B.A.; Rajasekaran, V.; Du, W.L.; Mutsafi, Y.; De Jesus-Diaz, D.A.; Sosnovtsev, S.V.; Levenson, E.A.; Parra, G.I.; et al. Vesicle-Cloaked Virus Clusters Are Optimal Units for Inter-organismal Viral Transmission. Cell Host Microbe 2018, 24, 208–220 e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirblich, C.; Bhattacharya, B.; Roy, P. Nonstructural protein 3 of bluetongue virus assists virus release by recruiting ESCRT-I protein Tsg101. J. Virol. 2006, 80, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celma, C.C.; Roy, P. A viral nonstructural protein regulates bluetongue virus trafficking and release. J. Virol. 2009, 83, 6806–6816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, B.; Celma, C.C.; Roy, P. Influence of cellular trafficking pathway on bluetongue virus infection in ovine cells. Viruses 2015, 7, 2378–2403. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [Green Version]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Zhou, J.; Wang, H. Host microRNAs and exosomes that modulate influenza virus infection. Virus Res. 2020, 279, 197885. [Google Scholar] [CrossRef]
- Maemura, T.; Fukuyama, S.; Kawaoka, Y. High Levels of miR-483-3p Are Present in Serum Exosomes Upon Infection of Mice With Highly Pathogenic Avian Influenza Virus. Front. Microbiol. 2020, 11, 144. [Google Scholar] [CrossRef]
- Liu, Y.M.; Tseng, C.H.; Chen, Y.C.; Yu, W.Y.; Ho, M.Y.; Ho, C.Y.; Lai, M.M.C.; Su, W.C. Exosome-delivered and Y RNA-derived small RNA suppresses influenza virus replication. J. Biomed. Sci. 2019, 26, 58. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Lata, S.; Ali, A.; Banerjea, A.C. Dengue haemorrhagic fever: A job done via exosomes? Emerg. Microbes Infect. 2019, 8, 1626–1635. [Google Scholar] [CrossRef] [Green Version]
- Freitas, M.N.; Marten, A.D.; Moore, G.A.; Tree, M.O.; McBrayer, S.P.; Conway, M.J. Extracellular vesicles restrict dengue virus fusion in Aedes aegypti cells. Virology 2020, 541, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Velandia-Romero, M.L.; Calderon-Pelaez, M.A.; Balbas-Tepedino, A.; Marquez-Ortiz, R.A.; Madronero, L.J.; Barreto Prieto, A.; Castellanos, J.E. Extracellular vesicles of U937 macrophage cell line infected with DENV-2 induce activation in endothelial cells EA.hy926. PLoS ONE 2020, 15, e0227030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Rojas, P.P.; Quiroz-Garcia, E.; Monroy-Martinez, V.; Agredano-Moreno, L.T.; Jimenez-Garcia, L.F.; Ruiz-Ordaz, B.H. Participation of Extracellular Vesicles from Zika-Virus-Infected Mosquito Cells in the Modification of Naive Cells’ Behavior by Mediating Cell-to-Cell Transmission of Viral Elements. Cells 2020, 9, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madison, M.N.; Okeoma, C.M. Exosomes: Implications in HIV-1 Pathogenesis. Viruses 2015, 7, 4093–4118. [Google Scholar] [CrossRef] [Green Version]
- Ellwanger, J.H.; Veit, T.D.; Chies, J.A.B. Exosomes in HIV infection: A review and critical look. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2017, 53, 146–154. [Google Scholar] [CrossRef]
- Felli, C.; Vincentini, O.; Silano, M.; Masotti, A. HIV-1 Nef Signaling in Intestinal Mucosa Epithelium Suggests the Existence of an Active Inter-kingdom Crosstalk Mediated by Exosomes. Front. Microbiol. 2017, 8, 1022. [Google Scholar] [CrossRef]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef]
- Pereira, E.A.; daSilva, L.L. HIV-1 Nef: Taking Control of Protein Trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef] [Green Version]
- McNamara, R.P.; Costantini, L.M.; Myers, T.A.; Schouest, B.; Maness, N.J.; Griffith, J.D.; Damania, B.A.; MacLean, A.G.; Dittmer, D.P. Nef Secretion into Extracellular Vesicles or Exosomes Is Conserved across Human and Simian Immunodeficiency Viruses. mBio 2018, 9, e02344-17. [Google Scholar] [CrossRef] [Green Version]
- Arenaccio, C.; Anticoli, S.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Federico, M. Latent HIV-1 is activated by exosomes from cells infected with either replication-competent or defective HIV-1. Retrovirology 2015, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, S.J.; Booth, A.M.; Hildreth, J.E. The Trojan exosome hypothesis. Proc. Natl. Acad. Sci. USA 2003, 100, 10592–10597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.G.; Booth, A.; Gould, S.J.; Hildreth, J.E. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J. Biol. Chem. 2003, 278, 52347–52354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coren, L.V.; Shatzer, T.; Ott, D.E. CD45 immunoaffinity depletion of vesicles from Jurkat T cells demonstrates that exosomes contain CD45: No evidence for a distinct exosome/HIV-1 budding pathway. Retrovirology 2008, 5, 64. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-Useros, N.; Naranjo-Gomez, M.; Archer, J.; Hatch, S.C.; Erkizia, I.; Blanco, J.; Borras, F.E.; Puertas, M.C.; Connor, J.H.; Fernandez-Figueras, M.T.; et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 2009, 113, 2732–2741. [Google Scholar] [CrossRef] [Green Version]
- Hildreth, J.E.K. HIV As Trojan Exosome: Immunological Paradox Explained? Front. Immunol. 2017, 8, 1715. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-Useros, N.; Naranjo-Gomez, M.; Erkizia, I.; Puertas, M.C.; Borras, F.E.; Blanco, J.; Martinez-Picado, J. HIV and mature dendritic cells: Trojan exosomes riding the Trojan horse? Plos Pathog. 2010, 6, e1000740. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.O.; DeMarino, C.; Pleet, M.L.; Cowen, M.; Branscome, H.; Al Sharif, S.; Jones, J.; Dutartre, H.; Lepene, B.; Liotta, L.A.; et al. HTLV-1 Extracellular Vesicles Promote Cell-to-Cell Contact. Front. Microbiol. 2019, 10, 2147. [Google Scholar] [CrossRef]
- Gunasekaran, M.; Bansal, S.; Ravichandran, R.; Sharma, M.; Perincheri, S.; Rodriguez, F.; Hachem, R.; Fisher, C.E.; Limaye, A.P.; Omar, A.; et al. Respiratory viral infection in lung transplantation induces exosomes that trigger chronic rejection. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2020, 39, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Sadeghipour, S.; Mathias, R.A. Herpesviruses hijack host exosomes for viral pathogenesis. Semin. Cell Dev. Biol. 2017, 67, 91–100. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, Q.; Xie, Y.; Zuo, L.; Zhu, F.; Lu, J. Extracellular vesicles: Novel vehicles in herpesvirus infection. Virol. Sin. 2017, 32, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Corey, L. Persistence in the population: Epidemiology, transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Roizman, B.; Zhou, G.; Du, T. Checkpoints in productive and latent infections with herpes simplex virus 1: Conceptualization of the issues. J. Neurovirol. 2011, 17, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Derfuss, T. Control of HSV-1 latency in human trigeminal ganglia—Current overview. J. Neurovirol. 2011, 17, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Vanicek, J.; Robins, H.; Shenk, T.; Levine, A.J. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: Implications for latency. Proc. Natl. Acad. Sci. USA 2008, 105, 5453–5458. [Google Scholar] [CrossRef] [Green Version]
- Obara, Y.; Furuta, Y.; Takasu, T.; Suzuki, S.; Suzuki, H.; Matsukawa, S.; Fujioka, Y.; Takahashi, H.; Kurata, T.; Nagashima, K. Distribution of herpes simplex virus types 1 and 2 genomes in human spinal ganglia studied by PCR and in situ hybridization. J. Med. Virol. 1997, 52, 136–142. [Google Scholar] [CrossRef]
- Kennedy, P.G.; Chaudhuri, A. Herpes simplex encephalitis. J. Neurol. Neurosurg. Psychiatry 2002, 73, 237–238. [Google Scholar] [CrossRef]
- Whitley, R.J. Herpes simplex encephalitis: Adolescents and adults. Antivir. Res. 2006, 71, 141–148. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Bellamy, A.R.; Hook, E.W., 3rd; Levin, M.J.; Wald, A.; Ewell, M.G.; Wolff, P.A.; Deal, C.D.; Heineman, T.C.; Dubin, G.; et al. Epidemiology, clinical presentation, and antibody response to primary infection with herpes simplex virus type 1 and type 2 in young women. Clin. Infect. Dis. 2013, 56, 344–351. [Google Scholar] [CrossRef]
- Karasneh, G.A.; Shukla, D. Herpes simplex virus infects most cell types in vitro: Clues to its success. Virol. J. 2011, 8, 481. [Google Scholar] [CrossRef] [Green Version]
- Agelidis, A.M.; Shukla, D. Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell Mol. Life Sci 2008, 65, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Reske, A.; Pollara, G.; Krummenacher, C.; Chain, B.M.; Katz, D.R. Understanding HSV-1 entry glycoproteins. Rev. Med. Virol. 2007, 17, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. Febs J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510. [Google Scholar] [CrossRef]
- Schweighardt, B.; Atwood, W.J. Virus receptors in the human central nervous system. J. Neurovirol. 2001, 7, 187–195. [Google Scholar]
- Hilterbrand, A.T.; Heldwein, E.E. Go go gadget glycoprotein!: HSV-1 draws on its sizeable glycoprotein tool kit to customize its diverse entry routes. PLoS Pathog. 2019, 15, e1007660. [Google Scholar] [CrossRef]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [Green Version]
- Calistri, A.; Sette, P.; Salata, C.; Cancellotti, E.; Forghieri, C.; Comin, A.; Gottlinger, H.; Campadelli-Fiume, G.; Palu, G.; Parolin, C. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 2007, 81, 11468–11478. [Google Scholar] [CrossRef] [Green Version]
- Crump, C.M.; Yates, C.; Minson, T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 2007, 81, 7380–7387. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G. The egress of alphaherpesviruses from the cell. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suenaga, T.; Satoh, T.; Somboonthum, P.; Kawaguchi, Y.; Mori, Y.; Arase, H. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. USA 2010, 107, 866–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szilagyi, J.F.; Cunningham, C. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 1991, 72 Pt 3, 661–668. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J.; Addison, C.; Craigie, M.C.; Rixon, F.J. Noninfectious L-particles supply functions which can facilitate infection by HSV-1. Virology 1992, 190, 682–688. [Google Scholar] [CrossRef]
- Heilingloh, C.S.; Krawczyk, A. Role of L-Particles during Herpes Simplex Virus Infection. Front. Microbiol. 2017, 8, 2565. [Google Scholar] [CrossRef] [Green Version]
- Kalamvoki, M.; Deschamps, T. Extracellular vesicles during Herpes Simplex Virus type 1 infection: An inquire. Virol. J. 2016, 13, 63. [Google Scholar] [CrossRef] [Green Version]
- Heilingloh, C.S.; Kummer, M.; Muhl-Zurbes, P.; Drassner, C.; Daniel, C.; Klewer, M.; Steinkasserer, A. L Particles Transmit Viral Proteins from Herpes Simplex Virus 1-Infected Mature Dendritic Cells to Uninfected Bystander Cells, Inducing CD83 Downmodulation. J. Virol. 2015, 89, 11046–11055. [Google Scholar] [CrossRef] [Green Version]
- Dargan, D.J.; Patel, A.H.; Subak-Sharpe, J.H. PREPs: Herpes simplex virus type 1-specific particles produced by infected cells when viral DNA replication is blocked. J. Virol. 1995, 69, 4924–4932. [Google Scholar] [CrossRef] [Green Version]
- Temme, S.; Eis-Hubinger, A.M.; McLellan, A.D.; Koch, N. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J. Immunol. 2010, 184, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Bello-Morales, R.; Praena, B.; de la Nuez, C.; Rejas, M.T.; Guerra, M.; Galan-Ganga, M.; Izquierdo, M.; Calvo, V.; Krummenacher, C.; Lopez-Guerrero, J.A. Role of Microvesicles in the Spread of Herpes Simplex Virus 1 in Oligodendrocytic Cells. J. Virol. 2018, 92, e00088-18. [Google Scholar] [CrossRef] [Green Version]
- Kalamvoki, M.; Du, T.; Roizman, B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc. Natl. Acad. Sci. USA 2014, 111, E4991–E4996. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, T.; Kalamvoki, M. Extracellular Vesicles Released by Herpes Simplex Virus 1-Infected Cells Block Virus Replication in Recipient Cells in a STING-Dependent Manner. J. Virol. 2018, 92, e01102-18. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12 (Suppl. 1–13), 19–30. [Google Scholar] [CrossRef] [Green Version]
- Gerber, P.P.; Cabrini, M.; Jancic, C.; Paoletti, L.; Banchio, C.; von Bilderling, C.; Sigaut, L.; Pietrasanta, L.I.; Duette, G.; Freed, E.O.; et al. Rab27a controls HIV-1 assembly by regulating plasma membrane levels of phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 2015, 209, 435–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraile-Ramos, A.; Cepeda, V.; Elstak, E.; van der Sluijs, P. Rab27a is required for human cytomegalovirus assembly. PLoS ONE 2010, 5, e15318. [Google Scholar] [CrossRef] [PubMed]
- Bello-Morales, R.; Crespillo, A.J.; Fraile-Ramos, A.; Tabares, E.; Alcina, A.; Lopez-Guerrero, J.A. Role of the small GTPase Rab27a during herpes simplex virus infection of oligodendrocytic cells. BMC Microbiol. 2012, 12, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello-Morales, R.; Lopez-Guerrero, J.A. Extracellular Vesicles in Herpes Viral Spread and Immune Evasion. Front. Microbiol. 2018, 9, 2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowska, K.; Wachalska, M.; Graul, M.; Rychlowski, M.; Bienkowska-Szewczyk, K.; Lipinska, A.D. Alphaherpesvirus gB Homologs Are Targeted to Extracellular Vesicles, but They Differentially Affect MHC Class II Molecules. Viruses 2020, 12, 429. [Google Scholar] [CrossRef] [Green Version]
- Gershon, M.; Gershon, A. Varicella-Zoster Virus and the Enteric Nervous System. J. Infect. Dis. 2018, 218 (Suppl. 2), S113–S119. [Google Scholar] [CrossRef]
- Mori, Y.; Koike, M.; Moriishi, E.; Kawabata, A.; Tang, H.; Oyaizu, H.; Uchiyama, Y.; Yamanishi, K. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic 2008, 9, 1728–1742. [Google Scholar] [CrossRef] [Green Version]
- Ota, M.; Serada, S.; Naka, T.; Mori, Y. MHC class I molecules are incorporated into human herpesvirus-6 viral particles and released into the extracellular environment. Microbiol. Immunol. 2014, 58, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraile-Ramos, A.; Pelchen-Matthews, A.; Risco, C.; Rejas, M.T.; Emery, V.C.; Hassan-Walker, A.F.; Esteban, M.; Marsh, M. The ESCRT machinery is not required for human cytomegalovirus envelopment. Cell. Microbiol. 2007, 9, 2955–2967. [Google Scholar] [CrossRef] [PubMed]
- Schauflinger, M.; Fischer, D.; Schreiber, A.; Chevillotte, M.; Walther, P.; Mertens, T.; von Einem, J. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J. Virol. 2011, 85, 3821–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zicari, S.; Arakelyan, A.; Palomino, R.A.N.; Fitzgerald, W.; Vanpouille, C.; Lebedeva, A.; Schmitt, A.; Bomsel, M.; Britt, W.; Margolis, L. Human cytomegalovirus-infected cells release extracellular vesicles that carry viral surface proteins. Virology 2018, 524, 97–105. [Google Scholar] [CrossRef]
- Walker, J.D.; Maier, C.L.; Pober, J.S. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J. Immunol. 2009, 182, 1548–1559. [Google Scholar] [CrossRef] [Green Version]
- Meckes, D.G., Jr.; Gunawardena, H.P.; Dekroon, R.M.; Heaton, P.R.; Edwards, R.H.; Ozgur, S.; Griffith, J.D.; Damania, B.; Raab-Traub, N. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc. Natl. Acad. Sci. USA 2013, 110, E2925–E2933. [Google Scholar] [CrossRef] [Green Version]
- Vazirabadi, G.; Geiger, T.R.; Coffin, W.F., 3rd; Martin, J.M. Epstein-Barr virus latent membrane protein-1 (LMP-1) and lytic LMP-1 localization in plasma membrane-derived extracellular vesicles and intracellular virions. J. Gen. Virol. 2003, 84 Pt 8, 1997–2008. [Google Scholar] [CrossRef]
- Meckes, D.G., Jr.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [Green Version]
- Cone, A.S.; York, S.B.; Meckes, D.G., Jr. Extracellular Vesicles in Epstein-Barr Virus Pathogenesis. Curr. Clin. Microbiol. Rep. 2019, 6, 121–131. [Google Scholar] [CrossRef]
- Kieser, A.; Sterz, K.R. The Latent Membrane Protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar]
- Liao, C.; Zhou, Q.; Zhang, Z.; Wu, X.; Zhou, Z.; Li, B.; Peng, J.; Shen, L.; Li, D.; Luo, X.; et al. Epstein-Barr virus-encoded latent membrane protein 1 promotes extracellular vesicle secretion through syndecan-2 and synaptotagmin-like-4 in nasopharyngeal carcinoma cells. Cancer Sci. 2020, 111, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.; Middeldorp, J.; Sculley, T. Localization of the Epstein-Barr virus protein LMP 1 to exosomes. J. Gen. Virol. 2003, 84 Pt 7, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Verweij, F.J.; van Eijndhoven, M.A.; Hopmans, E.S.; Vendrig, T.; Wurdinger, T.; Cahir-McFarland, E.; Kieff, E.; Geerts, D.; van der Kant, R.; Neefjes, J.; et al. LMP1 association with CD63 in endosomes and secretion via exosomes limits constitutive NF-kappaB activation. EMBO J. 2011, 30, 2115–2129. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.N.; Nkosi, D.; Conlon, M.M.; York, S.B.; Liu, X.; Tremblay, D.C.; Meckes, D.G., Jr. CD63 Regulates Epstein-Barr Virus LMP1 Exosomal Packaging, Enhancement of Vesicle Production, and Noncanonical NF-kappaB Signaling. J. Virol. 2017, 91, e02251-16. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Wurdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [Green Version]
- Gourzones, C.; Gelin, A.; Bombik, I.; Klibi, J.; Verillaud, B.; Guigay, J.; Lang, P.; Temam, S.; Schneider, V.; Amiel, C.; et al. Extra-cellular release and blood diffusion of BART viral micro-RNAs produced by EBV-infected nasopharyngeal carcinoma cells. Virol. J. 2010, 7, 271. [Google Scholar] [CrossRef] [Green Version]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.R.; Shango, J.; Seal, A.; Shukla, D.; Nares, S. Viral miRNAs Alter Host Cell miRNA Profiles and Modulate Innate Immune Responses. Front. Immunol. 2018, 9, 433. [Google Scholar] [CrossRef]
- Li, C.; He, H.; Wang, J.; Xia, X.; Zhang, M.; Wu, Q. Possible roles of exosomal miRNAs in the pathogenesis of oral lichen planus. Am. J. Transl. Res. 2019, 11, 5313–5323. [Google Scholar]
- Ding, M.; Wang, X.; Wang, C.; Liu, X.; Zen, K.; Wang, W.; Zhang, C.Y.; Zhang, C. Distinct expression profile of HCMV encoded miRNAs in plasma from oral lichen planus patients. J. Transl. Med. 2017, 15, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Nanbo, A.; Sun, L.; Lin, Z. Extracellular Vesicles in Epstein-Barr Virus’ Life Cycle and Pathogenesis. Microorganisms 2019, 7, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, J.A.; Tomita, Y.; Jankowska-Gan, E.; Lema, D.A.; Arvedson, M.P.; Nair, A.; Bracamonte-Baran, W.; Zhou, Y.; Meyer, K.K.; Zhong, W.; et al. Treg-Cell-Derived IL-35-Coated Extracellular Vesicles Promote Infectious Tolerance. Cell Rep. 2020, 30, 1039–1051.e5. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tsai, M.H.; Shumilov, A.; Baccianti, F.; Tsao, S.W.; Poirey, R.; Delecluse, H.J. Epstein-Barr virus ncRNA from a nasopharyngeal carcinoma induces an inflammatory response that promotes virus production. Nat. Microbiol. 2019, 4, 2475–2486. [Google Scholar] [CrossRef]
- Zheng, J.; Shi, Y.; Feng, Z.; Zheng, Y.; Li, Z.; Zhao, Y.; Wang, Y. Oncogenic effects of exosomes in gamma-herpesvirus-associated neoplasms. J. Cell. Physiol. 2019, 234, 19167–19179. [Google Scholar] [CrossRef] [PubMed]
- Chugh, P.E.; Sin, S.H.; Ozgur, S.; Henry, D.H.; Menezes, P.; Griffith, J.; Eron, J.J.; Damania, B.; Dittmer, D.P. Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog. 2013, 9, e1003484. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Feng, Z.; Yuan, G.; Emerson, C.C.; Stewart, P.L.; Ye, F.; Jin, G. Human Immunodeficiency Virus-Associated Exosomes Promote Kaposi’s Sarcoma-Associated Herpesvirus Infection via the Epidermal Growth Factor Receptor. J. Virol. 2020, 94, e01782-19. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.A.; Weissman, S.M. cDNA cloning and sequence of MAL, a hydrophobic protein associated with human T-cell differentiation. Proc. Natl. Acad. Sci. USA 1987, 84, 1997–2001. [Google Scholar] [CrossRef] [Green Version]
- Millan, J.; Puertollano, R.; Fan, L.; Rancano, C.; Alonso, M.A. The MAL proteolipid is a component of the detergent-insoluble membrane subdomains of human T-lymphocytes. Biochem. J. 1997, 321 Pt 1, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, P.; Puertollano, R.; Alonso, M.A. Structural and biochemical similarities reveal a family of proteins related to the MAL proteolipid, a component of detergent-insoluble membrane microdomains. Biochem. Biophys. Res. Commun. 1997, 232, 618–621. [Google Scholar] [CrossRef]
- Marazuela, M.; Acevedo, A.; Adrados, M.; Garcia-Lopez, M.A.; Alonso, M.A. Expression of MAL, an integral protein component of the machinery for raft-mediated pical transport, in human epithelia. J. Histochem. Cytochem. 2003, 51, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, M. MAL, a proteolipid in glycosphingolipid enriched domains: Functional implications in myelin and beyond. Prog. Neurobiol. 2000, 60, 531–544. [Google Scholar] [CrossRef]
- Bijlard, M.; de Jonge, J.C.; Klunder, B.; Nomden, A.; Hoekstra, D.; Baron, W. MAL Is a Regulator of the Recruitment of Myelin Protein PLP to Membrane Microdomains. PLoS ONE 2016, 11, e0155317. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R.; Martin-Belmonte, F.; Millan, J.; de Marco, M.C.; Albar, J.P.; Kremer, L.; Alonso, M.A. The MAL proteolipid is necessary for normal apical transport and accurate sorting of the influenza virus hemagglutinin in Madin-Darby canine kidney cells. J. Cell Biol. 1999, 145, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Martin-Belmonte, F.; Arvan, P.; Alonso, M.A. MAL mediates apical transport of secretory proteins in polarized epithelial Madin-Darby canine kidney cells. J. Biol. Chem. 2001, 276, 49337–49342. [Google Scholar] [CrossRef] [Green Version]
- Ventimiglia, L.N.; Fernandez-Martin, L.; Martinez-Alonso, E.; Anton, O.M.; Guerra, M.; Martinez-Menarguez, J.A.; Andres, G.; Alonso, M.A. Cutting Edge: Regulation of Exosome Secretion by the Integral MAL Protein in T Cells. J. Immunol. 2015, 195, 810–814. [Google Scholar] [CrossRef] [Green Version]
- Ventimiglia, L.N.; Alonso, M.A. Biogenesis and Function of T Cell-Derived Exosomes. Front. Cell Dev. Biol. 2016, 4, 84. [Google Scholar] [CrossRef] [Green Version]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Chazal, N.; Gerlier, D. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. Mmbr 2003, 67, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Nayak, D.P.; Barman, S. Role of lipid rafts in virus assembly and budding. Adv. Virus Res. 2002, 58, 1–28. [Google Scholar]
- Bukrinsky, M.I.; Mukhamedova, N.; Sviridov, D. Lipid rafts and pathogens: The art of deception and exploitation. J. Lipid Res. 2020, 61, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hantak, M.P.; Qing, E.; Earnest, J.T.; Gallagher, T. Tetraspanins: Architects of Viral Entry and Exit Platforms. J. Virol. 2019, 93, e01429-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Guerrero, J.A.; de la Nuez, C.; Praena, B.; Sanchez-Leon, E.; Krummenacher, C.; Bello-Morales, R. Herpes Simplex Virus 1 Spread in Oligodendrocytic Cells Is Highly Dependent on MAL Proteolipid. J. Virol. 2020, 94, e01739-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic models of viral spread via extracellular vesicles (EVs). The EV-mediated release of virions, viral components and induced/altered host factors by infected cells, may be performed using different pathways and molecular machineries. Several viral components such as DNAs, RNAs, miRNAs, and/or proteins, may be packaged into EVs. Infected cells may also release EVs containing host factors such as DNAs, RNAs, miRNAs and/ or proteins, induced or altered by the infection. All these components may trigger both pro- or anti-viral effects. A. Viruses may use the multivesicular bodies (MVBs) for viral spread, packaging virions or viral factors into intraluminal vesicles (ILVs) that will be released to the extracellular medium (exosomes) after fusion of the MVB with the plasma membrane (1). B. Viruses may also exit cells enclosed in shedding microvesicles (MVs), which might also contain viral and/or host factors. Other feasible routes for viral spread involve the secretory (C) and the autophagic (D) pathways. C. Several viruses use the TGN or endosomes for envelopment. In the canonical pathway for HSV-1, the viral egress involves the fusion of a double-membrane vesicle with the plasma membrane (2), giving rise to free enveloped virions. Hypothetically, this structure might exit the cell after shedding of the plasma membrane (3), giving rise to a three-membraned EV, which would correspond to an enveloped virion enclosed within a shedding MV. D. Viruses can also be wrapped into vesicles/tubules belonging to the autophagic pathway. These vesicles containing virions could also be released by fusion (4) or by membrane shedding (5). For routes A-D, different molecular machineries, such as the endosomal sorting complexes required for transport (ESCRT) complex, tetraspanin-enriched microdomains (TEMs) and/or lipid rafts, may operate and collaborate for vesicle scission.
Figure 1.
Schematic models of viral spread via extracellular vesicles (EVs). The EV-mediated release of virions, viral components and induced/altered host factors by infected cells, may be performed using different pathways and molecular machineries. Several viral components such as DNAs, RNAs, miRNAs, and/or proteins, may be packaged into EVs. Infected cells may also release EVs containing host factors such as DNAs, RNAs, miRNAs and/ or proteins, induced or altered by the infection. All these components may trigger both pro- or anti-viral effects. A. Viruses may use the multivesicular bodies (MVBs) for viral spread, packaging virions or viral factors into intraluminal vesicles (ILVs) that will be released to the extracellular medium (exosomes) after fusion of the MVB with the plasma membrane (1). B. Viruses may also exit cells enclosed in shedding microvesicles (MVs), which might also contain viral and/or host factors. Other feasible routes for viral spread involve the secretory (C) and the autophagic (D) pathways. C. Several viruses use the TGN or endosomes for envelopment. In the canonical pathway for HSV-1, the viral egress involves the fusion of a double-membrane vesicle with the plasma membrane (2), giving rise to free enveloped virions. Hypothetically, this structure might exit the cell after shedding of the plasma membrane (3), giving rise to a three-membraned EV, which would correspond to an enveloped virion enclosed within a shedding MV. D. Viruses can also be wrapped into vesicles/tubules belonging to the autophagic pathway. These vesicles containing virions could also be released by fusion (4) or by membrane shedding (5). For routes A-D, different molecular machineries, such as the endosomal sorting complexes required for transport (ESCRT) complex, tetraspanin-enriched microdomains (TEMs) and/or lipid rafts, may operate and collaborate for vesicle scission.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bello-Morales, R.; Ripa, I.; López-Guerrero, J.A. Extracellular Vesicles in Viral Spread and Antiviral Response. Viruses 2020, 12, 623. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060623
AMA Style
Bello-Morales R, Ripa I, López-Guerrero JA. Extracellular Vesicles in Viral Spread and Antiviral Response. Viruses. 2020; 12(6):623. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060623
Chicago/Turabian StyleBello-Morales, Raquel, Inés Ripa, and José Antonio López-Guerrero. 2020. "Extracellular Vesicles in Viral Spread and Antiviral Response" Viruses 12, no. 6: 623. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060623
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.