Cyclin-Dependent Kinases 8 and 19 Regulate Host Cell Metabolism during Dengue Virus Serotype 2 Infection

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and DENV2 Infection
2.2. CDK8/19 Small-Molecule Inhibition and Cell Viability Assay
2.3. qRT-PCR Analysis
2.4. Plaque-Forming Unit and Extracellular Genome Equivalent Analysis
2.5. Lentivirus-Mediated shRNA Gene Knockdown
2.6. Subcellular Extracts and Protein Analysis
2.7. Mitochondrial and Glycolytic Stress Tests
2.8. Statistics
3. Results
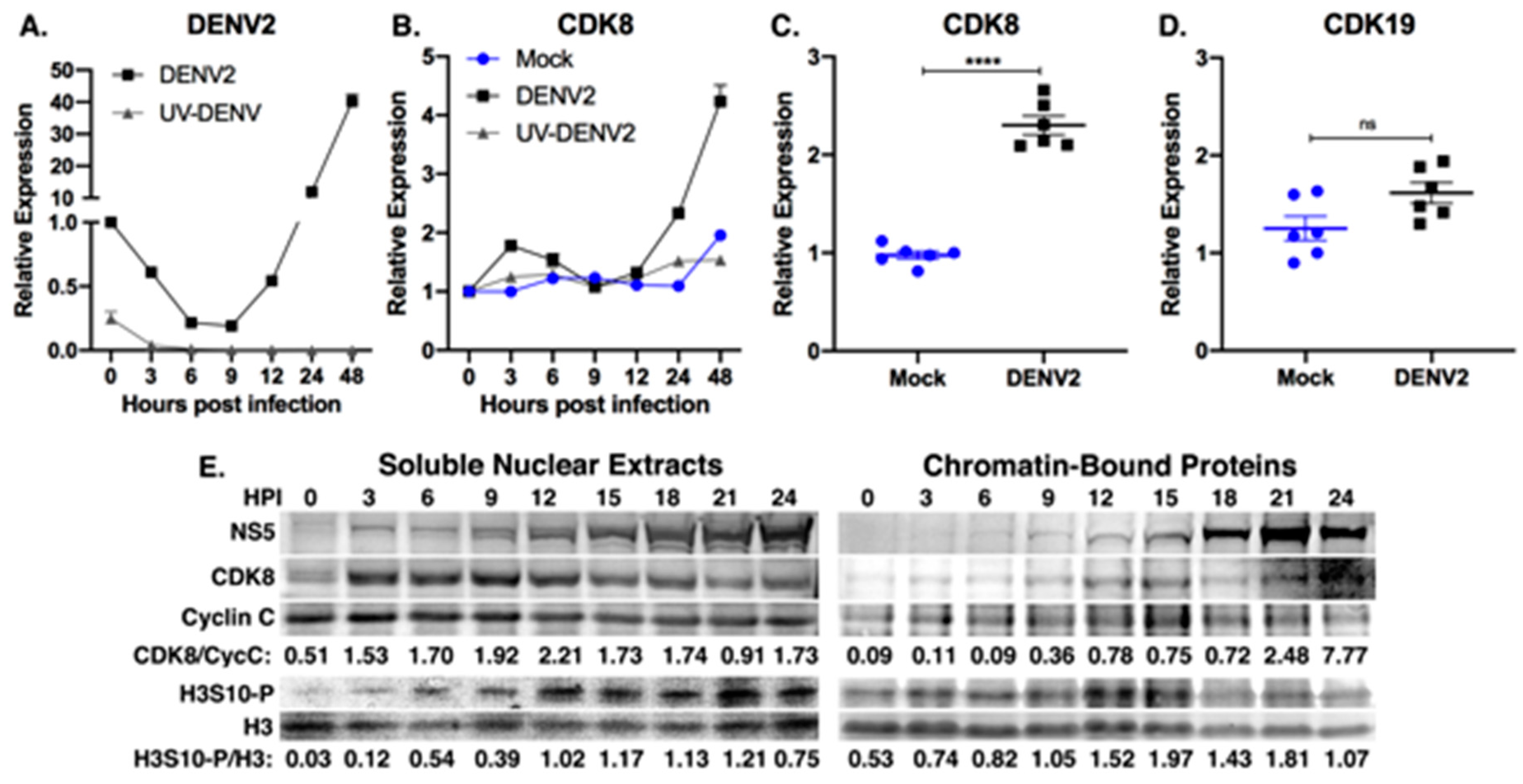
3.1. Cyclin-Dependent Kinase 8 Is Upregulated during DENV2 Infection
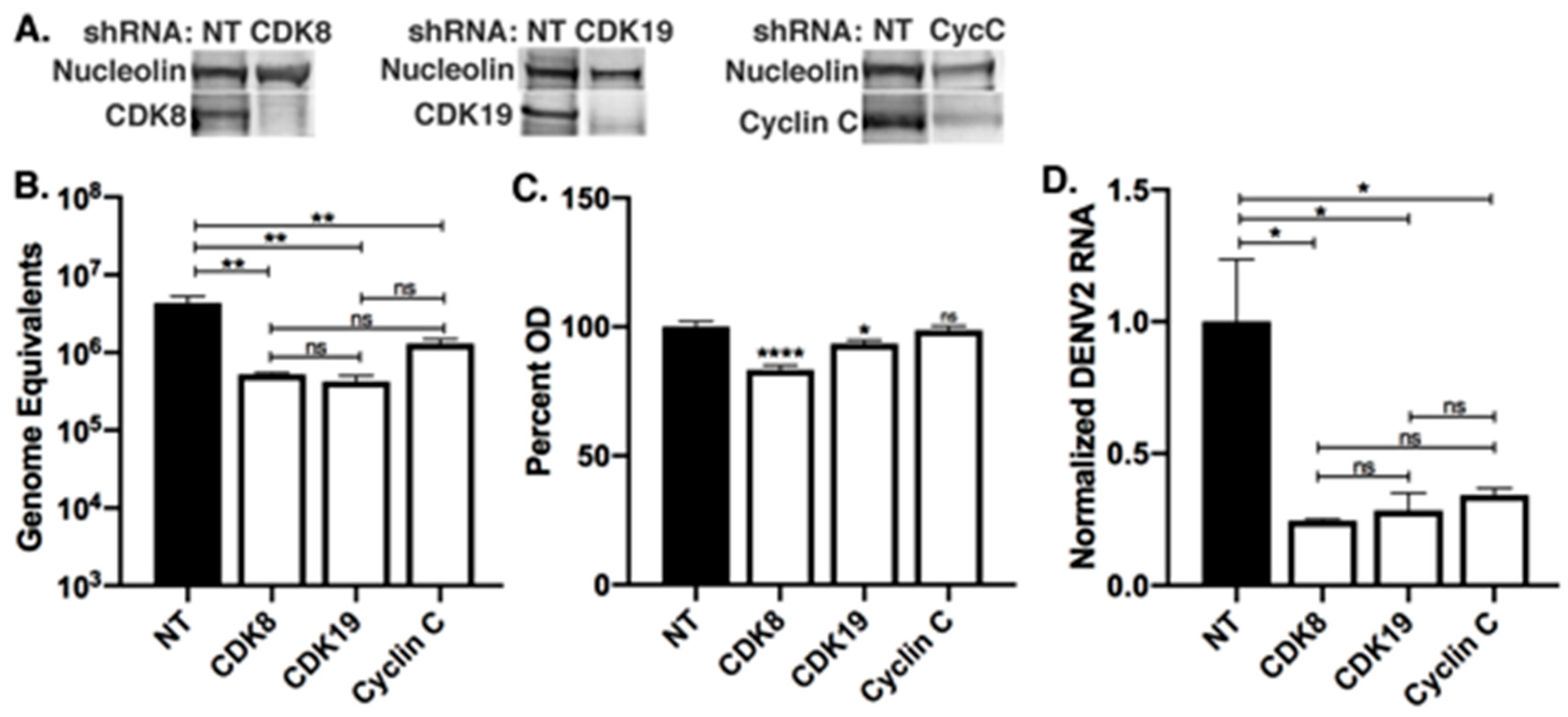
3.2. Knockdowns of CDK8 and CDK19 Reduce DENV2 Replication
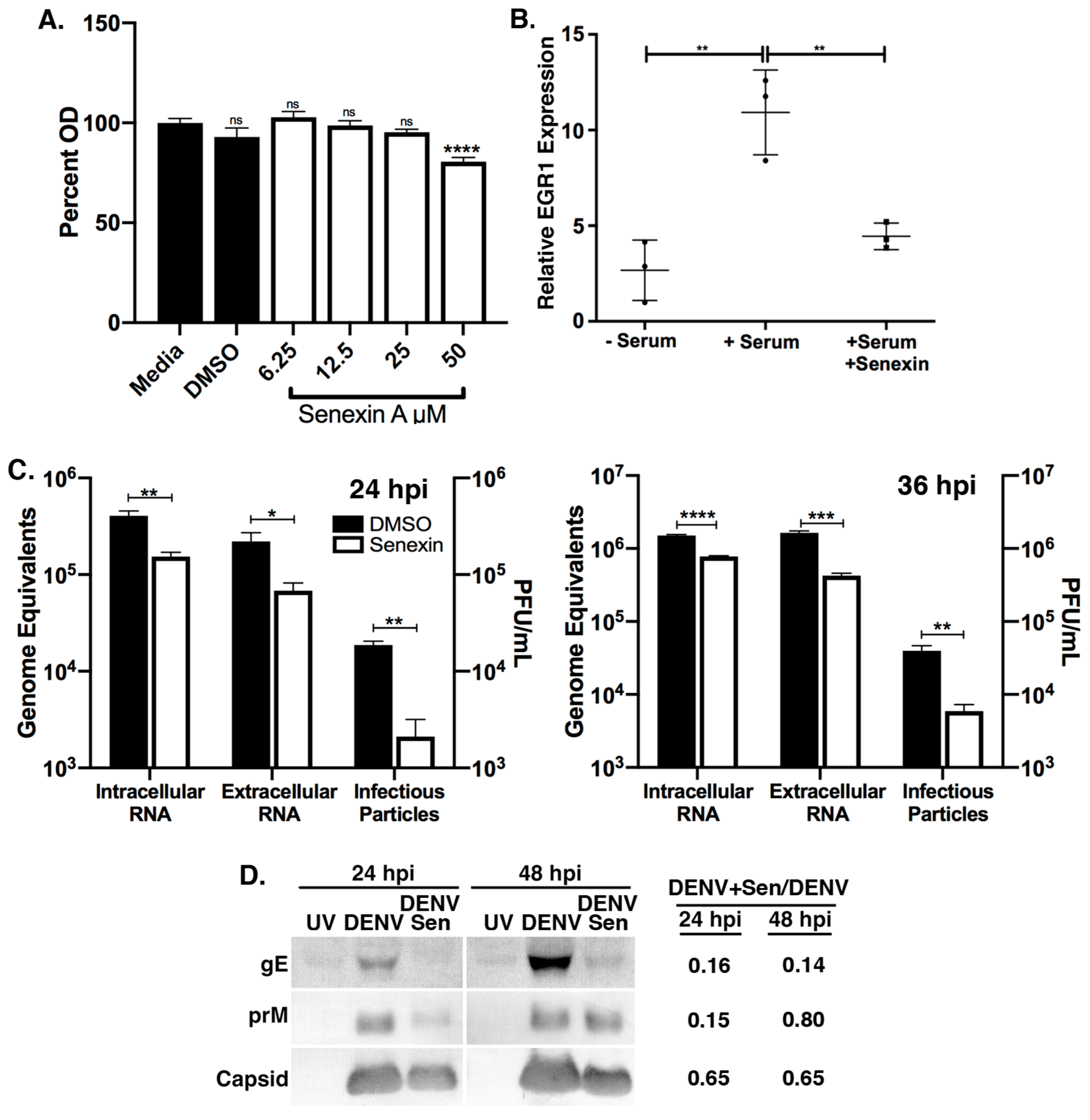
3.3. CDK8/19 Chemical Inhibition Reduces DENV2 Replication
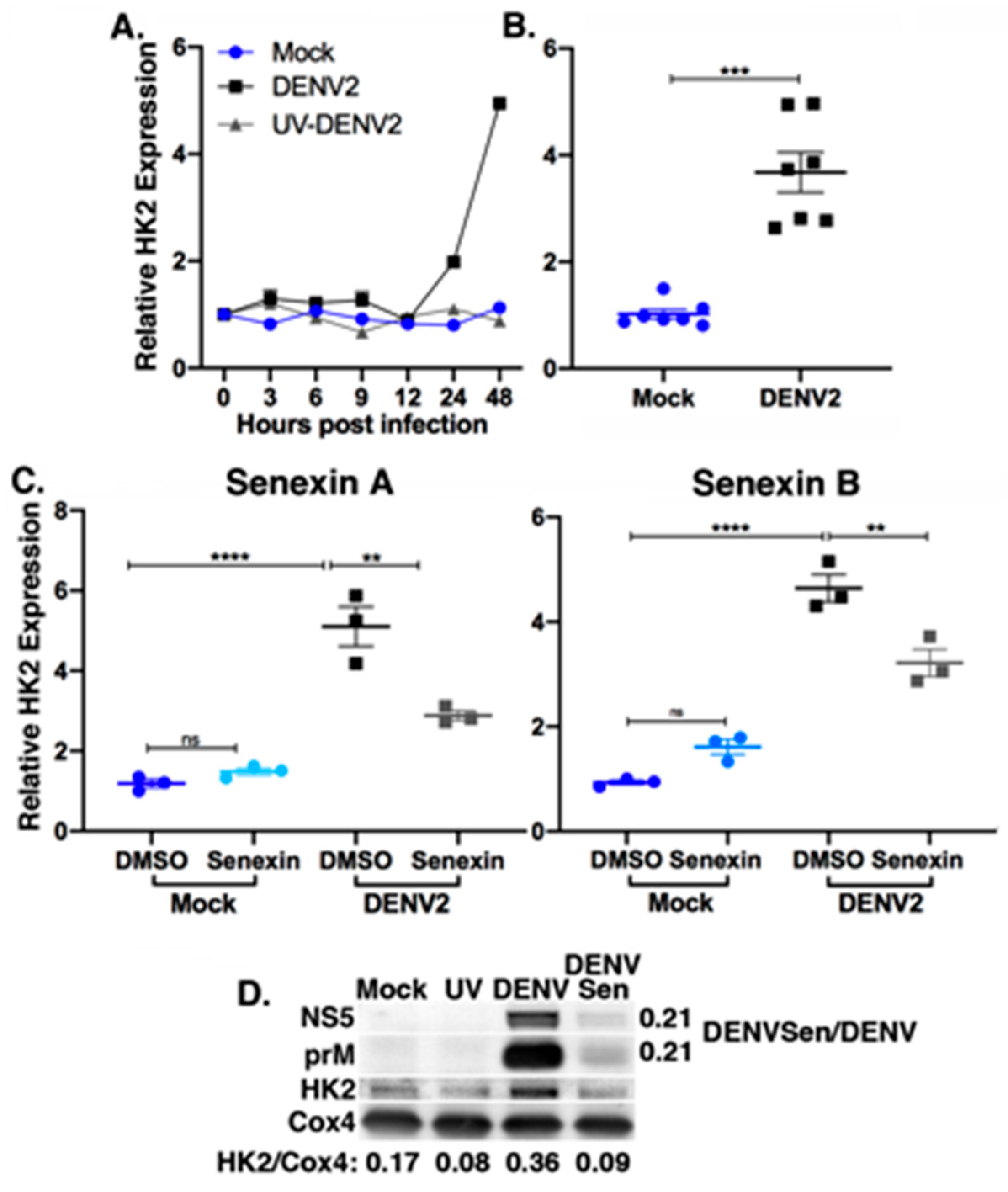
3.4. Metabolic Gene Expression Is Dependent on CDK8/19 Kinase Activity
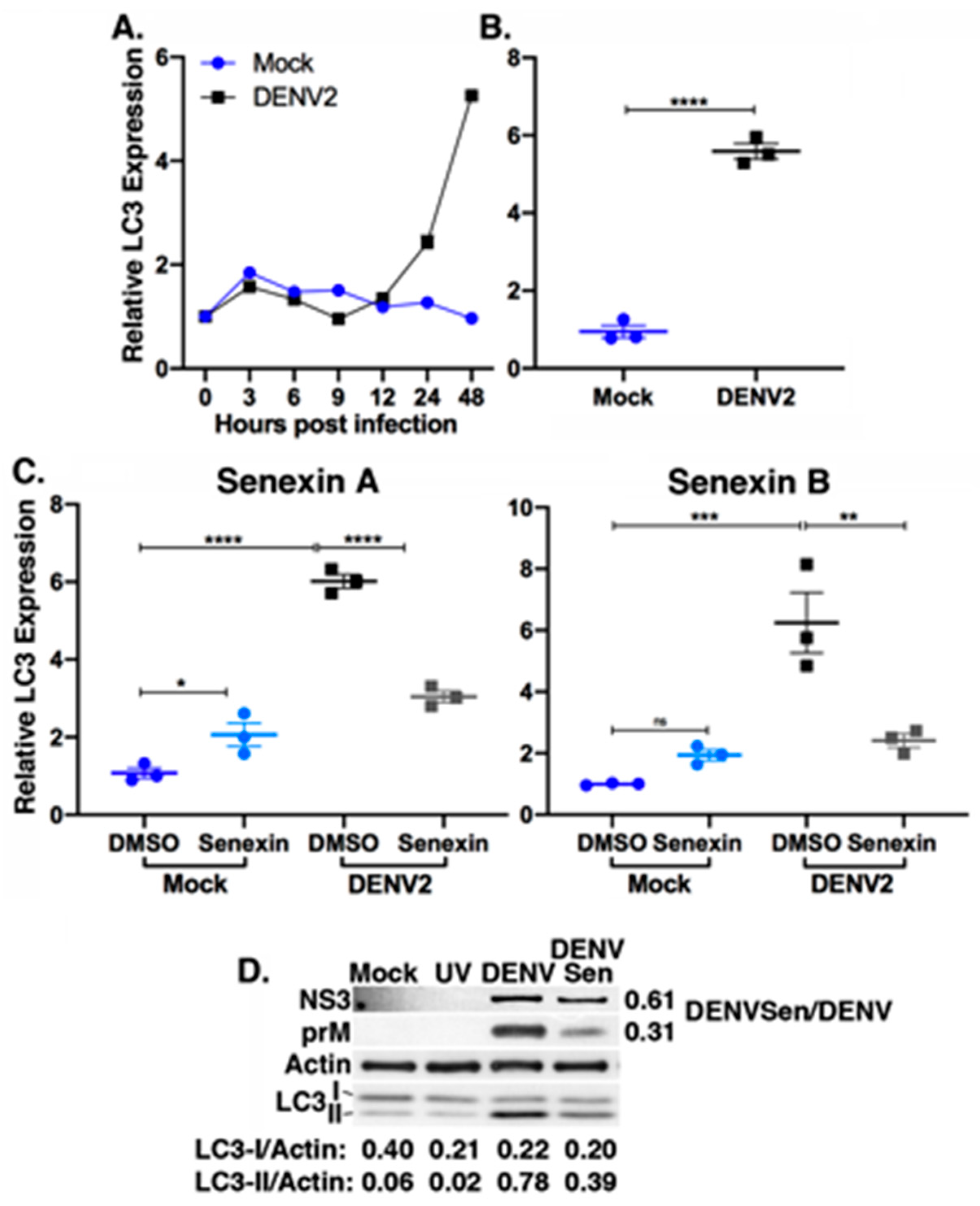
3.5. Senexin A Reduces Induction of Lipophagic Gene Expression
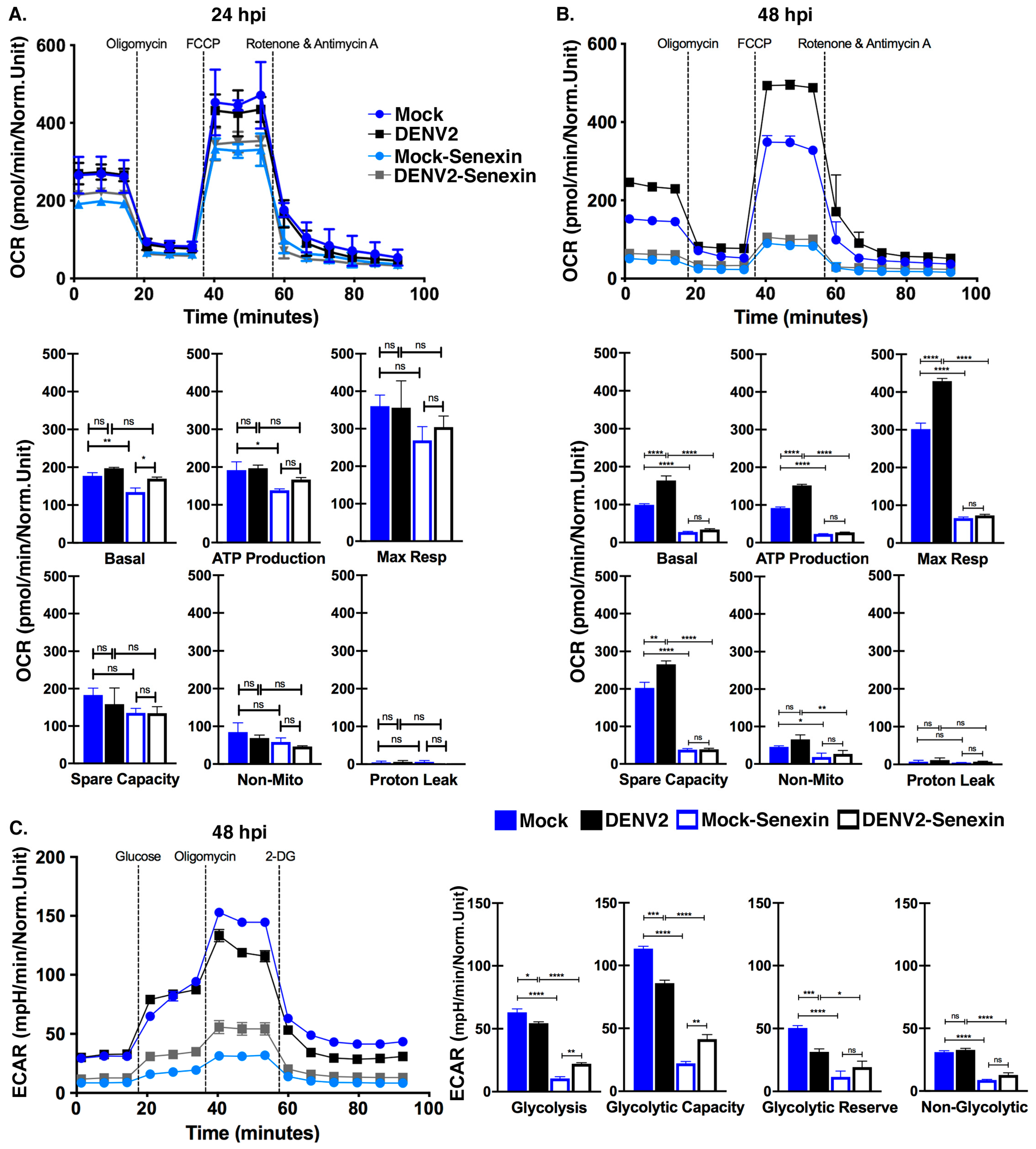
3.6. Senexin A Inhibits Mitochondrial Respiration
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Dengue and Severe Dengue. Available online: https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (accessed on 20 January 2020).
- World Health Organization; Pan American Health Organization. Epidemiological Update: Dengue. Available online: http://www.paho.org (accessed on 13 November 2019).
- Mayer, K.A.; Stockl, J.; Zlabinger, G.J.; Gualdoni, G.A. Hijacking the supplies: Metabolism as a novel facet of virus-host interaction. Front. Immunol. 2019, 10, 1533. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Altamirano, M.M.B.; Kolstoe, S.E.; Sanchez-Garcia, F.J. Virus control of cell metabolism for replication and evasion of host immune responses. Front. Cell. Infect. Microbiol. 2019, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Thaker, S.K.; Ch’ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Chotiwan, N.; Andre, B.G.; Sanchez-Vargas, I.; Islam, M.N.; Grabowski, J.M.; Hopf-Jannasch, A.; Gough, E.; Nakayasu, E.; Blair, C.D.; Belisle, J.T.; et al. Dynamic remodeling of lipids coincides with dengue virus replication in the midgut of Aedes aegypti mosquitoes. PLoS Pathog. 2018, 14, e1006853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes-Siqueira, L.O.; Zeidler, J.D.; Sousa, B.G.; Ferreira, T.; Da Poian, A.T. Anaplerotic role of glucose in the oxidation of endogenous fatty acids during dengue virus infection. mSphere 2018, 3, e00458-17. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue virus induces and requires glycolysis for optimal replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [Green Version]
- Gullberg, R.C.; Steel, J.J.; Pujari, V.; Rovnak, J.; Crick, D.C.; Perera, R. Stearoly-CoA desaturase 1 differentiates early and advanced dengue virus infections and determines virus particle infectivity. PLoS Pathog. 2018, 14, e1007261. [Google Scholar] [CrossRef] [Green Version]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [Green Version]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Melo, C.; Delafiori, J.; Dabaja, M.Z.; de Oliveira, D.N.; Guerreiro, T.M.; Colombo, T.E.; Nogueira, M.L.; Proenca-Modena, J.L.; Catharino, R.R. The role of lipids in the inception, maintenance and complications of dengue virus infection. Sci. Rep. 2018, 8, 11826. [Google Scholar] [CrossRef]
- Perera, R.; Riley, C.; Isaac, G.; Hopf-Jannasch, A.S.; Moore, R.J.; Weitz, K.W.; Pasa-Tolic, L.; Metz, T.O.; Adamec, J.; Kuhn, R.J. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 2012, 8, e1002584. [Google Scholar] [CrossRef] [PubMed]
- Pleet, M.L.; Branscome, H.; DeMarino, C.; Pinto, D.O.; Zadeh, M.A.; Rodriguez, M.; Sariyer, I.K.; El-Hage, N.; Kashanchi, F. Autophagy, EVs, and Infections: A perfect question for a perfect time. Front. Cell. Infect. Microbiol. 2018, 8, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Mak, T.K.; Gubler, D.J. Prevention and control of dengue-the light at the end of the tunnel. Lancet Infect. Dis. 2017, 17, e79–e87. [Google Scholar] [CrossRef]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385, 453–465. [Google Scholar] [CrossRef]
- Halstead, S.B. Dengue. Lancet 2007, 370, 1644–1652. [Google Scholar] [CrossRef]
- Silva, E.M.; Conde, J.N.; Allonso, D.; Ventura, G.T.; Coelho, D.R.; Carneiro, P.H.; Silva, M.L.; Paes, M.V.; Rabelo, K.; Weissmuller, G.; et al. Dengue virus nonstructural 3 protein interacts directly with human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and reduces its glycolytic activity. Sci. Rep. 2019, 9, 2651. [Google Scholar] [CrossRef]
- Abernathy, E.; Mateo, R.; Majzoub, K.; van Buuren, N.; Bird, S.W.; Carette, J.E.; Kirkegaard, K. Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PLoS Biol. 2019, 17, e2006926. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.R.; Chuang, Y.C.; Lin, Y.S.; Liu, H.S.; Liu, C.C.; Perng, G.C.; Yeh, T.M. Dengue virus nonstructural protein 1 induces vascular leakage through macrophage migration inhibitory factor and autophagy. PLoS Negl. Trop. Dis. 2016, 10, e0004828. [Google Scholar] [CrossRef]
- Jordan, T.X.; Randall, G. Dengue virus activates the AMP kinase-mTOR axis to stimulate a proviral lipophagy. J. Virol. 2017, 91, e02020-16. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses exploit the lipid droplet protein AUP1 to trigger lipophagy and drive virus production. Cell Host Microbe 2018, 23, 819–831 e815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allonso, D.; Andrade, I.S.; Conde, J.N.; Coelho, D.R.; Rocha, D.C.; da Silva, M.L.; Ventura, G.T.; Silva, E.M.; Mohana-Borges, R. Dengue virus NS1 protein modulates cellular energy metabolism by increasing glyceraldehyde-3-phosphate dehydrogenase activity. J. Virol. 2015, 89, 11871–11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dolken, L.; Strobl, B.; Muller, M.; Taatjes, D.J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donner, A.J.; Ebmeier, C.C.; Taatjes, D.J.; Espinosa, J.M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010, 17, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.D.; Donner, A.J.; Knuesel, M.T.; York, A.G.; Espinosa, J.M.; Taatjes, D.J. Cooperative activity of cdk8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. EMBO J. 2008, 27, 1447–1457. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Feng, D.; Wang, Q.; Abdulla, A.; Xie, X.J.; Zhou, J.; Sun, Y.; Yang, E.S.; Liu, L.P.; Vaitheesvaran, B.; et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J. Clin. Investig. 2012, 122, 2417–2427. [Google Scholar] [CrossRef]
- Birkenheuer, C.H.; Brewster, C.D.; Quackenbush, S.L.; Rovnak, J. Retroviral cyclin controls cyclin-dependent kinase 8-mediated transcription elongation and reinitiation. J. Virol. 2015, 89, 5450–5461. [Google Scholar] [CrossRef] [Green Version]
- Rovnak, J.; Brewster, C.D.; Quackenbush, S.L. Retroviral cyclin enhances cyclin-dependent kinase 8 activity. J. Virol. 2012, 86, 5742–5751. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, M.D.; Andrysik, Z.; Pandey, A.; Hoh, M.; Bonner, E.A.; Hill, A.A.; Sullivan, K.D.; Espinosa, J.M. CDK8 kinase activity promotes glycolysis. Cell Rep. 2017, 21, 1495–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.W.; Hu, Y.; Chen, C.L.; Xia, B.; Zirin, J.; Yuan, M.; Asara, J.M.; Rabinow, L.; Perrimon, N. The TORC1-regulated CPA complex rewires an RNA processing network to drive autophagy and metabolic reprogramming. Cell Metab. 2018, 27, 1040–1054 e1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewster, C.; Birkenheuer, C.; Vogt, M.; Quackenbush, S.; Rovnak, J. The retroviral cyclin of walleye dermal sarcoma virus binds cyclin-dependent kinases 3 and 8. Virology 2011, 409, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovnak, J.; Quackenbush, S.L. Walleye dermal sarcoma virus cyclin interacts with components of the Mediator complex and the RNA polymerase II holoenzyme. J. Virol. 2002, 76, 8031–8039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.S.; Chumanevich, A.A.; Lim, C.U.; Liang, J.; Chen, M.; Altilia, S.; Oliver, D.; Rae, J.M.; Shtutman, M.; Kiaris, H.; et al. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget 2017, 8, 12558–12575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.C.; Farmaki, E.; Altilia, S.; Schools, G.P.; West, D.K.; Chen, M.; Chang, B.D.; Puzyrev, A.T.; Lim, C.U.; Rokow-Kittell, R.; et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc. Natl. Acad. Sci. USA 2012, 109, 13799–13804. [Google Scholar] [CrossRef] [Green Version]
- Kinney, R.M.; Butrapet, S.; Chang, G.J.; Tsuchiya, K.R.; Roehrig, J.T.; Bhamarapravati, N.; Gubler, D.J. Construction of infectious cDNA clones for dengue 2 virus: Strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology 1997, 230, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Watson, S.; Mercier, S.; Bye, C.; Wilkinson, J.; Cunningham, A.L.; Harman, A.N. Determination of suitable housekeeping genes for normalisation of quantitative real time PCR analysis of cells infected with human immunodeficiency virus and herpes viruses. Virol. J. 2007, 4, 130. [Google Scholar] [CrossRef] [Green Version]
- Laue, T.; Emmerich, P.; Schmitz, H. Detection of dengue virus RNA in patients after primary or secondary dengue infection by using the TaqMan automated amplification system. J. Clin. Microbiol. 1999, 37, 2543–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.A.; Singh, B.K.; Zhou, J.; Wu, Y.; Farah, B.L.; Ohba, K.; Lesmana, R.; Gooding, J.; Bay, B.H.; Yen, P.M. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015, 11, 1341–1357. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Liu, R.; Li, J.; Zhang, C.; Wang, Y.; Cai, Q.; Qian, X.; Xia, Y.; Zheng, Y.; Piao, Y.; et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 2017, 8, 949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A.; Shadel, G.S. Isolation of mitochondria from cells and tissues. Cold Spring Harb. Protoc. 2014, 2014, pdb top074542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fant, C.B.; Taatjes, D.J. Regulatory functions of the Mediator kinases CDK8 and CDK19. Transcription 2019, 10, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Steinparzer, I.; Sedlyarov, V.; Rubin, J.D.; Eislmayr, K.; Galbraith, M.D.; Levandowski, C.B.; Vcelkova, T.; Sneezum, L.; Wascher, F.; Amman, F.; et al. Transcriptional responses to IFN-gamma require mediator kinase-dependent pause release and mechanistically distinct CDK8 and CDK19 functions. Mol. Cell 2019, 76, 485–499 e488. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, M.; Donner, A.; Espinosa, J. CDK8: A positive regulator of transcription. Transcription 2010, 1, 4–12. [Google Scholar] [CrossRef] [Green Version]
- De Maio, F.A.; Risso, G.; Iglesias, N.G.; Shah, P.; Pozzi, B.; Gebhard, L.G.; Mammi, P.; Mancini, E.; Yanovsky, M.J.; Andino, R.; et al. The dengue virus NS5 protein intrudes in the cellular spliceosome and modulates splicing. PLoS Pathog. 2016, 12, e1005841. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, H.; Sung, P.Y.; Chiu, H.C.; Yousuf, A.; Bird, J.; Lim, S.P.; Davidson, A.D. Serotype-specific differences in dengue virus non-structural protein 5 nuclear localization. J. Biol. Chem. 2013, 288, 22621–22635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Buhler, S.; Selisko, B.; Davidson, A.; Mulder, K.; Canard, B.; Miller, S.; Bartenschlager, R. Nuclear localization of dengue virus nonstructural protein 5 does not strictly correlate with efficient viral RNA replication and inhibition of type I interferon signaling. J. Virol. 2013, 87, 4545–4557. [Google Scholar] [CrossRef] [Green Version]
- Tay, M.Y.; Fraser, J.E.; Chan, W.K.; Moreland, N.J.; Rathore, A.P.; Wang, C.; Vasudevan, S.G.; Jans, D.A. Nuclear localization of dengue virus (DENV) 1-4 non-structural protein 5; protection against all 4 DENV serotypes by the inhibitor Ivermectin. Antivir. Res. 2013, 99, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Audetat, K.A.; Galbraith, M.D.; Odell, A.T.; Lee, T.; Pandey, A.; Espinosa, J.M.; Dowell, R.D.; Taatjes, D.J. A kinase-independent role for cyclin-dependent kinase 19 in p53 response. Mol. Cell Biol. 2017, 37, e00626-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.J.; Hwang, H.S.; Park, J.H.; Bang, S.H.; Kang, W.J.; Yun, M.; Lee, J.D. Evaluation of the role of hexokinase type II in cellular proliferation and apoptosis using human hepatocellular carcinoma cell lines. J. Nucl. Med. 2009, 50, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, S.; Weiss, J.N.; Ribalet, B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE 2011, 6, e17674. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Lopez, N.; Singh, R. Autophagy and lipid droplets in the liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef]
- Reid, D.W.; Campos, R.K.; Child, J.R.; Zheng, T.; Chan, K.W.K.; Bradrick, S.S.; Vasudevan, S.G.; Garcia-Blanco, M.A.; Nicchitta, C.V. Dengue virus selectively annexes endoplasmic reticulum-associated translation machinery as a strategy for co-opting host cell protein synthesis. J. Virol. 2018, 92, e01766-17. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, C.; Lebreton, A.; Young Ng, C.; Lim, J.Y.; Liu, W.; Vasudevan, S.; Labow, M.; Gu, F.; Gaither, L.A. Cholesterol biosynthesis modulation regulates dengue viral replication. Virology 2009, 389, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, A.S.; Martins, I.C.; Santos, N.C. Methods for lipid droplet biophysical characterization in flaviviridae infections. Front. Microbiol. 2018, 9, 1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Randall, G. Lipid droplet metabolism during dengue virus infection. Trends Microbiol. 2018, 26, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assuncao-Miranda, I.; Barbosa-Lima, G.; Da Poian, A.T.; Bozza, P.T.; Gamarnik, A.V. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef] [PubMed]
- Datan, E.; Roy, S.G.; Germain, G.; Zali, N.; McLean, J.E.; Golshan, G.; Harbajan, S.; Lockshin, R.A.; Zakeri, Z. Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 2016, 7, e2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Acebes, M.A.; Blazquez, A.B.; Saiz, J.C. Reconciling west nile virus with the autophagic pathway. Autophagy 2015, 11, 861–864. [Google Scholar] [CrossRef] [Green Version]
- Mateo, R.; Nagamine, C.M.; Spagnolo, J.; Mendez, E.; Rahe, M.; Gale, M., Jr.; Yuan, J.; Kirkegaard, K. Inhibition of cellular autophagy deranges dengue virion maturation. J. Virol. 2013, 87, 1312–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Bhattacharyya, S.; Nain, M.; Kaur, M.; Sood, V.; Gupta, V.; Khasa, R.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 2014, 10, 1637–1651. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Jezek, J.; Chang, K.T.; Joshi, A.M.; Strich, R. Mitochondrial translocation of cyclin C stimulates intrinsic apoptosis through Bax recruitment. EMBO Rep. 2019, 20, e47425. [Google Scholar] [CrossRef] [PubMed]
- Jezek, J.; Smethurst, D.G.J.; Stieg, D.C.; Kiss, Z.A.C.; Hanley, S.E.; Ganesan, V.; Chang, K.T.; Cooper, K.F.; Strich, R. Cyclin C: The story of a non-cycling cyclin. Biology 2019, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieg, D.C.; Willis, S.D.; Ganesan, V.; Ong, K.L.; Scuorzo, J.; Song, M.; Grose, J.; Strich, R.; Cooper, K.F. A complex molecular switch directs stress-induced cyclin C nuclear release through SCF(Grr1)-mediated degradation of Med13. Mol. Biol. Cell 2018, 29, 363–375. [Google Scholar] [CrossRef]
- El-Bacha, T.; Midlej, V.; Pereira da Silva, A.P.; Silva da Costa, L.; Benchimol, M.; Galina, A.; Da Poian, A.T. Mitochondrial and bioenergetic dysfunction in human hepatic cells infected with dengue 2 virus. Biochim. Biophys. Acta 2007, 1772, 1158–1166. [Google Scholar] [CrossRef] [Green Version]
- Waggoner, J.J.; Katzelnick, L.C.; Burger-Calderon, R.; Gallini, J.; Moore, R.H.; Kuan, G.; Balmaseda, A.; Pinsky, B.A.; Harris, E. Antibody-dependent enhancement of severe disease is mediated by serum viral load in pediatric dengue virus infections. J. Infect. Dis. 2020, 221, 1846–1854. [Google Scholar] [CrossRef]
- Nguyen, M.N.; Duong, T.H.; Trung, V.T.; Nguyen, T.H.; Tran, C.N.; Long, V.T.; Dui le, T.; Nguyen, H.L.; Farrar, J.J.; Holmes, E.C.; et al. Host and viral features of human dengue cases shape the population of infected and infectious Aedes aegypti mosquitoes. Proc. Natl. Acad. Sci. USA 2013, 110, 9072–9077. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse | Source |
|---|---|---|---|
| CDK8 | GGGATCTCTATGTCGGCATGTAG | AAATGACGTTTGGATGCTTAAGC | [28] |
| CDK19 | GCCACGGCTAGGGCCT | GCGAGAACTGGAGTGCTGATAA | [28] |
| CyclinC | ATGGCAGGGAACTTTTGGCAG | ACCGTAGCAGTGGCAATAACT | Birkenheuer, unpublished |
| DENV | ACAAGTCGAACAACCTGGTCCAT | GCCGCACCATTGGTCTTCTC | [41] |
| HK2 | CAAAGTGACAGTGGGTGTGG3 | GCCAGGTCCTTCACTGTCTC3 | [42] |
| LC3 | AAGGCTTTCAGAGAGACCCTG | CCGTTTACCCTGCGTTTGTG | [43] |
| ENO1 | GTCTCTTCAGGCGTGCAAGC | GATGAGACACCATGACGCCC | [33] |
| PFKL | GGCATTTATGTGGGTGCCAAAGTC | CAGTTGGCCTGCTTGATGTTCTCA | [44] |
| PKM2 | CCACTTGCAATTATTTGAGGAA | GTGAGCAGACCTGCCAGACT | [45] |
| GAPDH | GCCATCAATGACCCCTTCAT | CGCTCCTGGAAGATGGTG | [35] |
| SDHA | GACAACTGGAGGTGGCATT | CCGTCATGTAGTGGATGGCA | [40] |
| Gene | Designation | Sequence |
|---|---|---|
| CDK8 | TRCN0000000489 | CCGGATGTCCAGTAGCCAAGTTCCACTCGAGTGGAACTTGGCTACTGGACATTTTTT |
| CDK19 | TRCN0000195069 | CCGGAGGACTGATAGCTCTTCTTTACTCGAGTAAAGAAGAGCTATCAGTCCTTTTTT |
| Cyclin C | TRCN0000020189 | CCGGGCATCCAAAGTAGAGGAATTTCTCGAGAAATTCCTCTACTTTGGATGCTTTTT |
| Nontarget | SHC002 | CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT |
| Antibody | Source | Catalog Number |
|---|---|---|
| Rabbit anti ß Actin | Cell Signaling | 4967 |
| Goat anti CDK8 | Santa Cruz Biotechnology | sc 1521 |
| Rabbit anti Cyclin C | Novus Biologicals | NB1202950 |
| Mouse anti Cytochrome C oxidase subunit IV | Molecular Probes | A21347 |
| Mouse anti Phospho Histone H3 (Ser10) | Cell Signaling | 9706 |
| Rabbit anti Histone H3 | Cell Signaling | 9715 |
| Rabbit anti Hexokinase 2 | Proteintech | AP#22029 |
| Rabbit anti LC3B | Novus Biologicals | NBP246892SS |
| Antibody | Source | Catalog Number |
|---|---|---|
| Mouse anti Flavivirus group antigen protein E, Clone 4G2 | Novus Biologicals | NBP252079 |
| Mouse anti DENV2 NS5 protein | GeneTex | GTX629447 |
| Rabbit anti DENV capsid protein | GeneTex | GTX103343 |
| Rabbit anti DENV prM protein | GeneTex | GTX128093 |
| Mouse anti DENV NS3 protein | GeneTex | GTX124252 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butler, M.; Chotiwan, N.; Brewster, C.D.; DiLisio, J.E.; Ackart, D.F.; Podell, B.K.; Basaraba, R.J.; Perera, R.; Quackenbush, S.L.; Rovnak, J. Cyclin-Dependent Kinases 8 and 19 Regulate Host Cell Metabolism during Dengue Virus Serotype 2 Infection. Viruses 2020, 12, 654. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060654
Butler M, Chotiwan N, Brewster CD, DiLisio JE, Ackart DF, Podell BK, Basaraba RJ, Perera R, Quackenbush SL, Rovnak J. Cyclin-Dependent Kinases 8 and 19 Regulate Host Cell Metabolism during Dengue Virus Serotype 2 Infection. Viruses. 2020; 12(6):654. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060654
Chicago/Turabian StyleButler, Molly, Nunya Chotiwan, Connie D. Brewster, James E. DiLisio, David F. Ackart, Brendan K. Podell, Randall J. Basaraba, Rushika Perera, Sandra L. Quackenbush, and Joel Rovnak. 2020. "Cyclin-Dependent Kinases 8 and 19 Regulate Host Cell Metabolism during Dengue Virus Serotype 2 Infection" Viruses 12, no. 6: 654. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060654