Phosphorylation of the Arginine-Rich C-Terminal Domains of the Hepatitis B Virus (HBV) Core Protein as a Fine Regulator of the Interaction between HBc and Nucleic Acid

 , and
, and
Abstract
:
1. Introduction
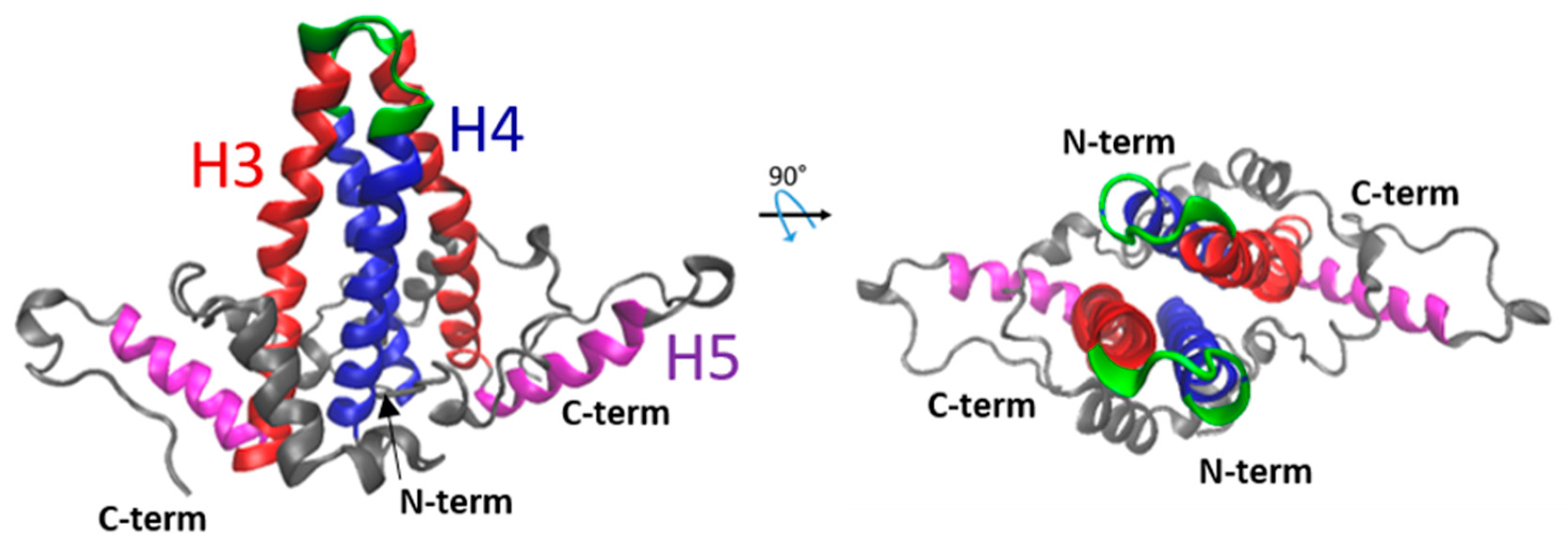
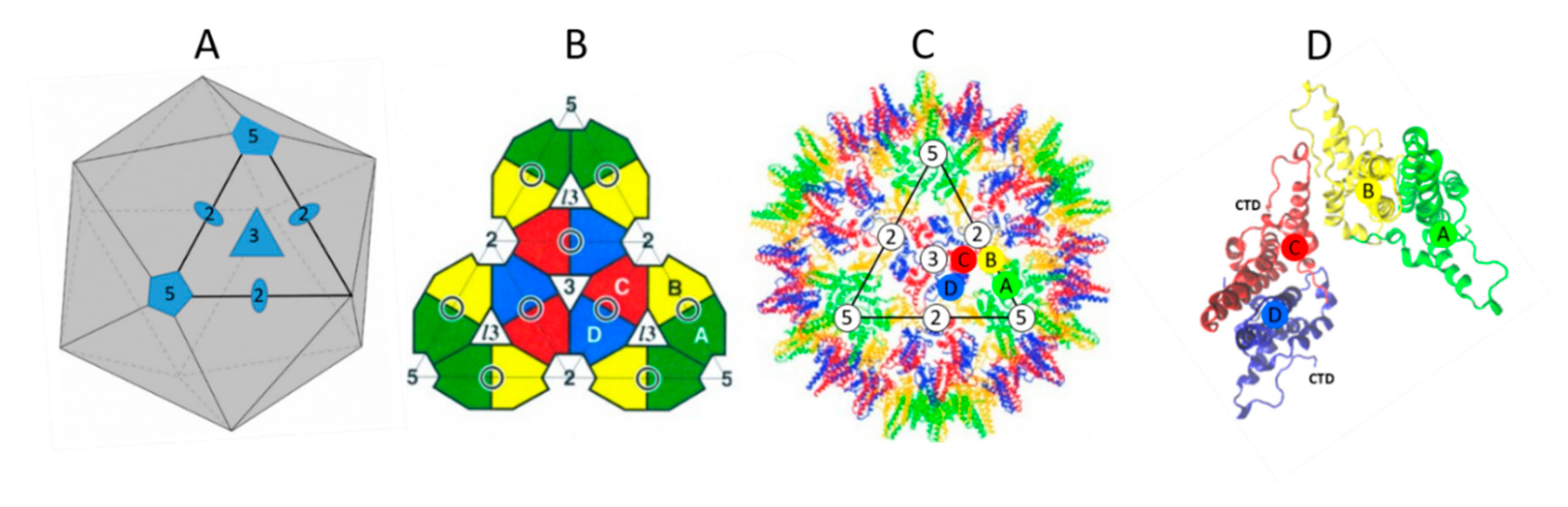
2. The NTD Is Essential for Capsid Formation and Envelope Protein Recruitment
3. The CTD Has Multiple Roles
3.1. Phosphorylation of the CTD (P-CTD)
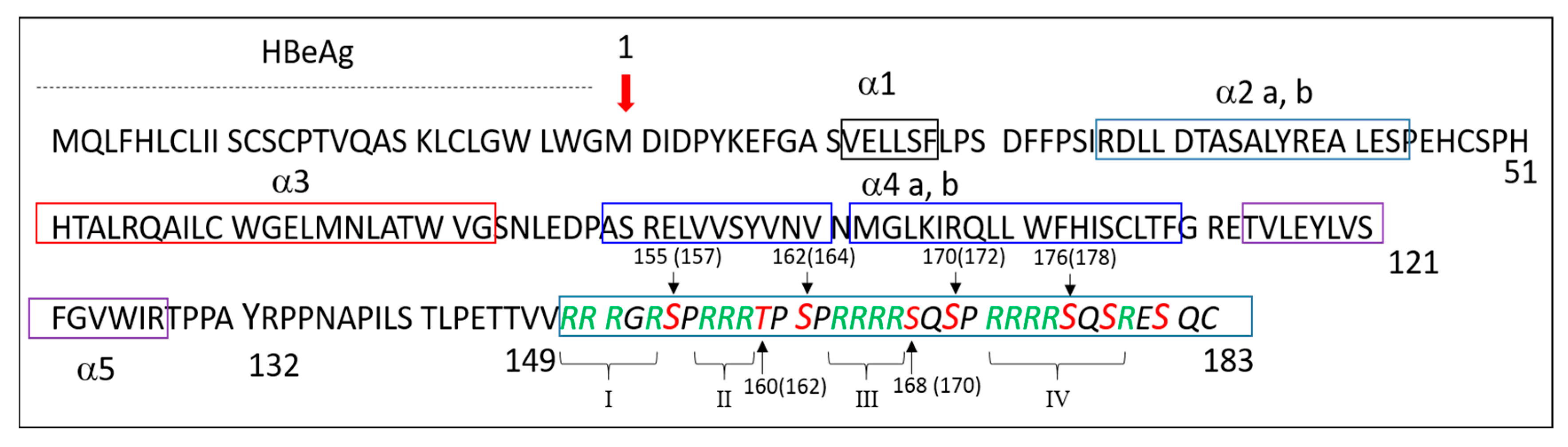
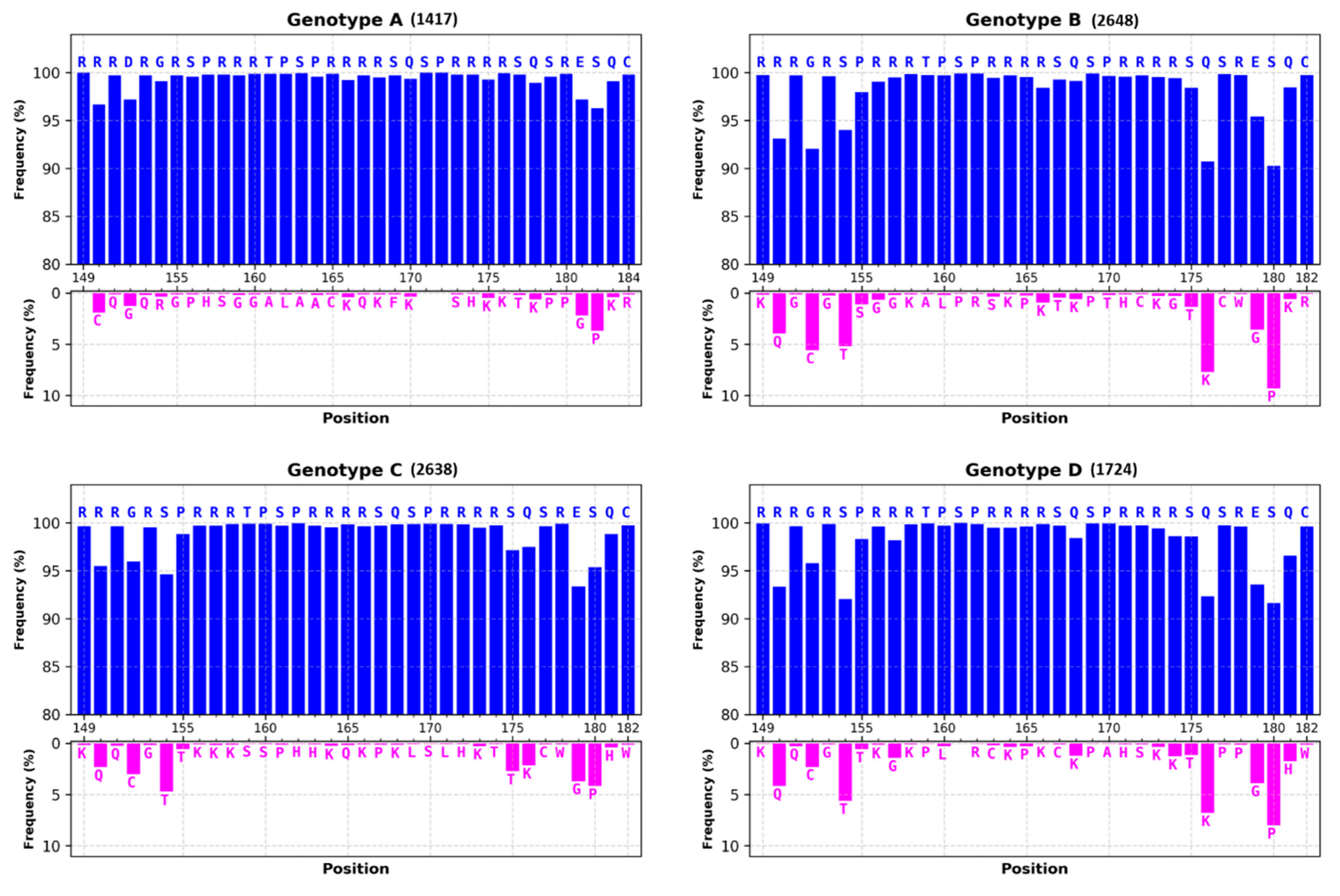
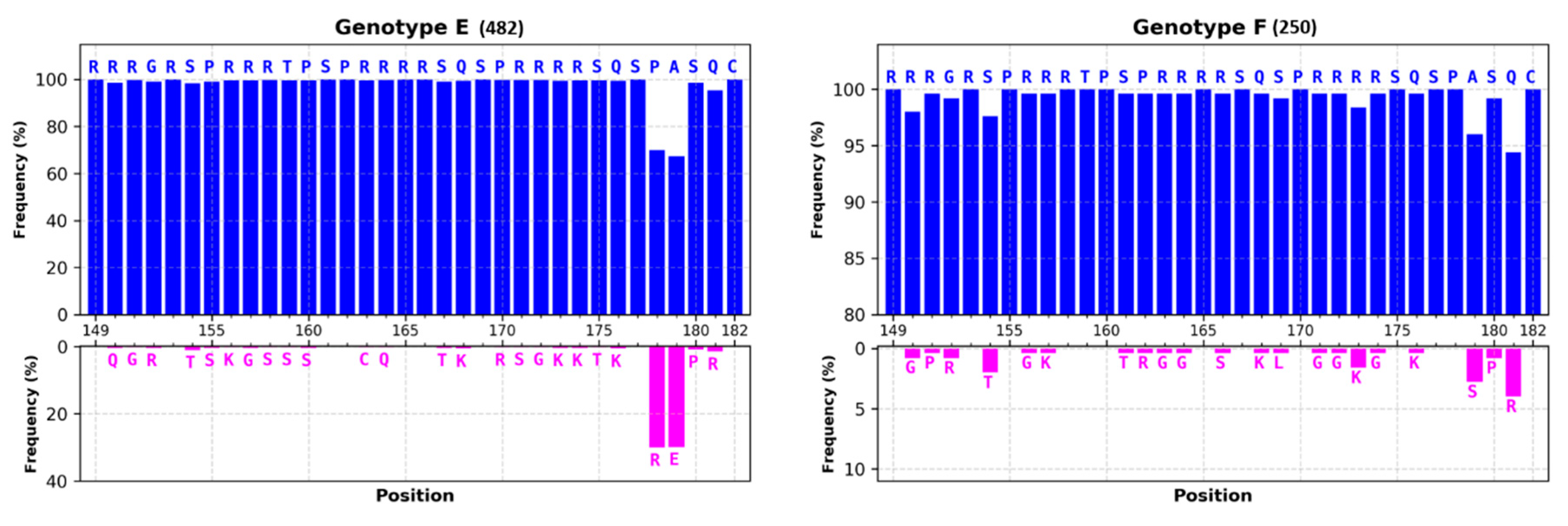
3.1.1. Most of HBc Phospho-Acceptors Are in the CTD
3.1.2. Phosphorylation Can Be Mimicked by Acidic Residues (mP-HBc)
3.1.3. Kinase Identity
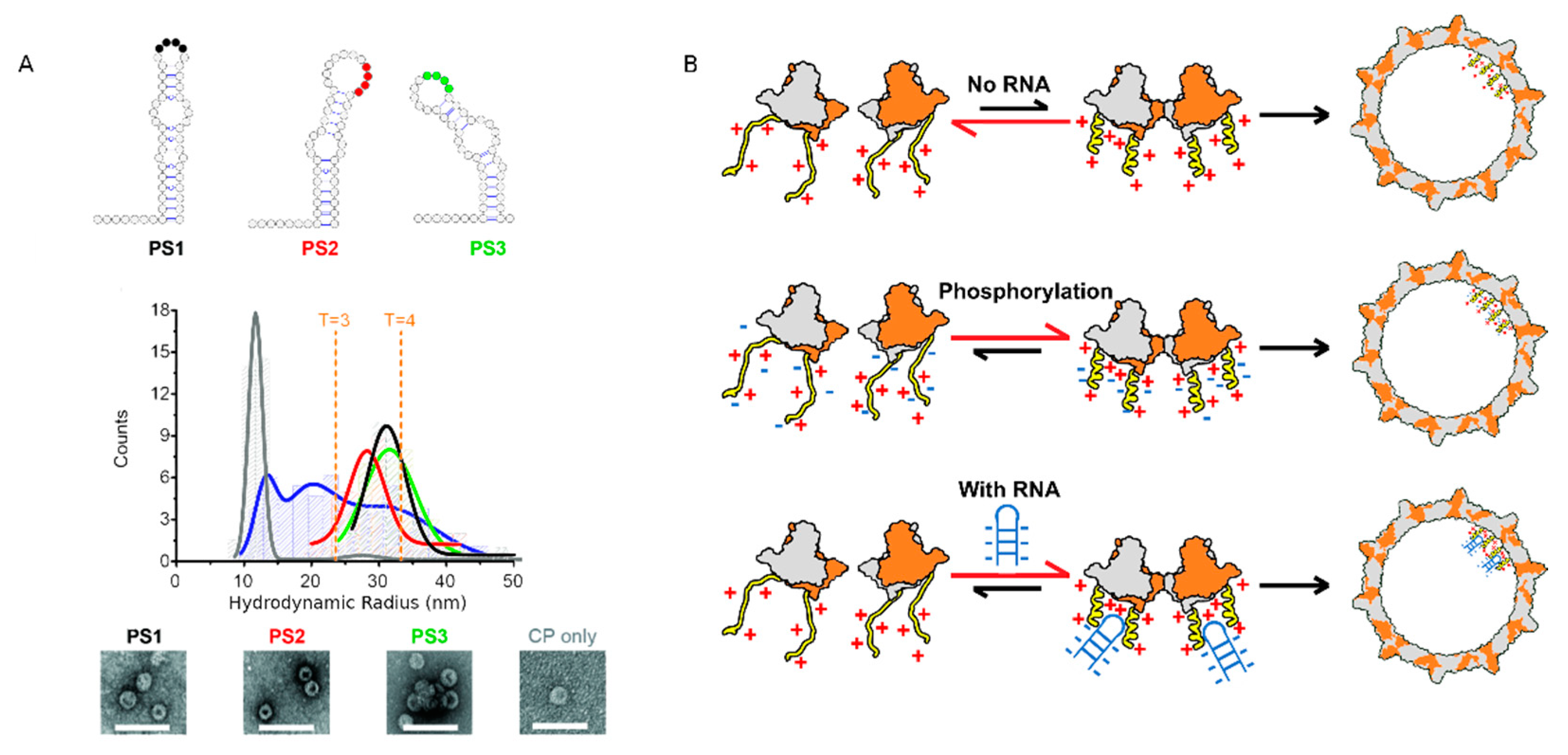
3.2. Phosphorylation Is Not Essential for Capsid Assembly
4. The CTD Interacts with Nucleic Acids (NAs)
4.1. Role of Arginine Rich Domains (ARDs) in HBV Replication
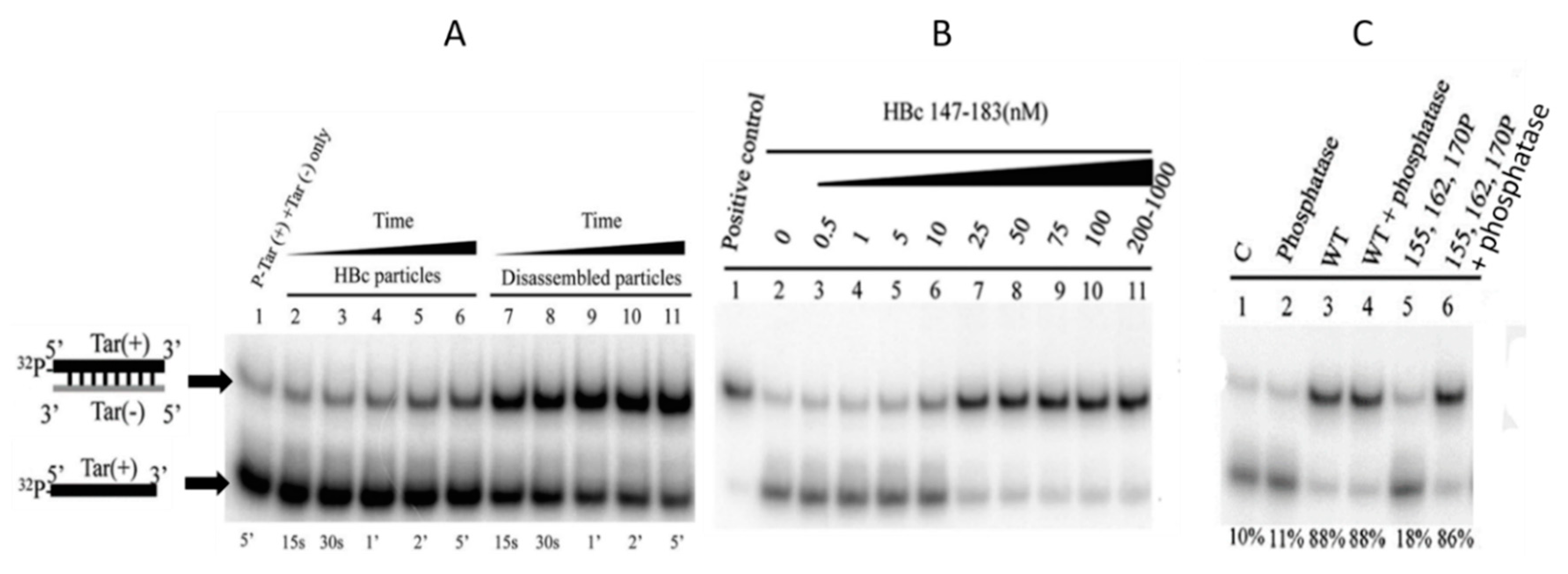
4.2. ARDs Endowed with NA Chaperone Activity
4.3. Interaction of HBc with dsDNA
4.4. Specific Interaction between the CTD and NAs
5. RNA Packaging Is Tuned by Phosphorylation and by the Nature of NAs
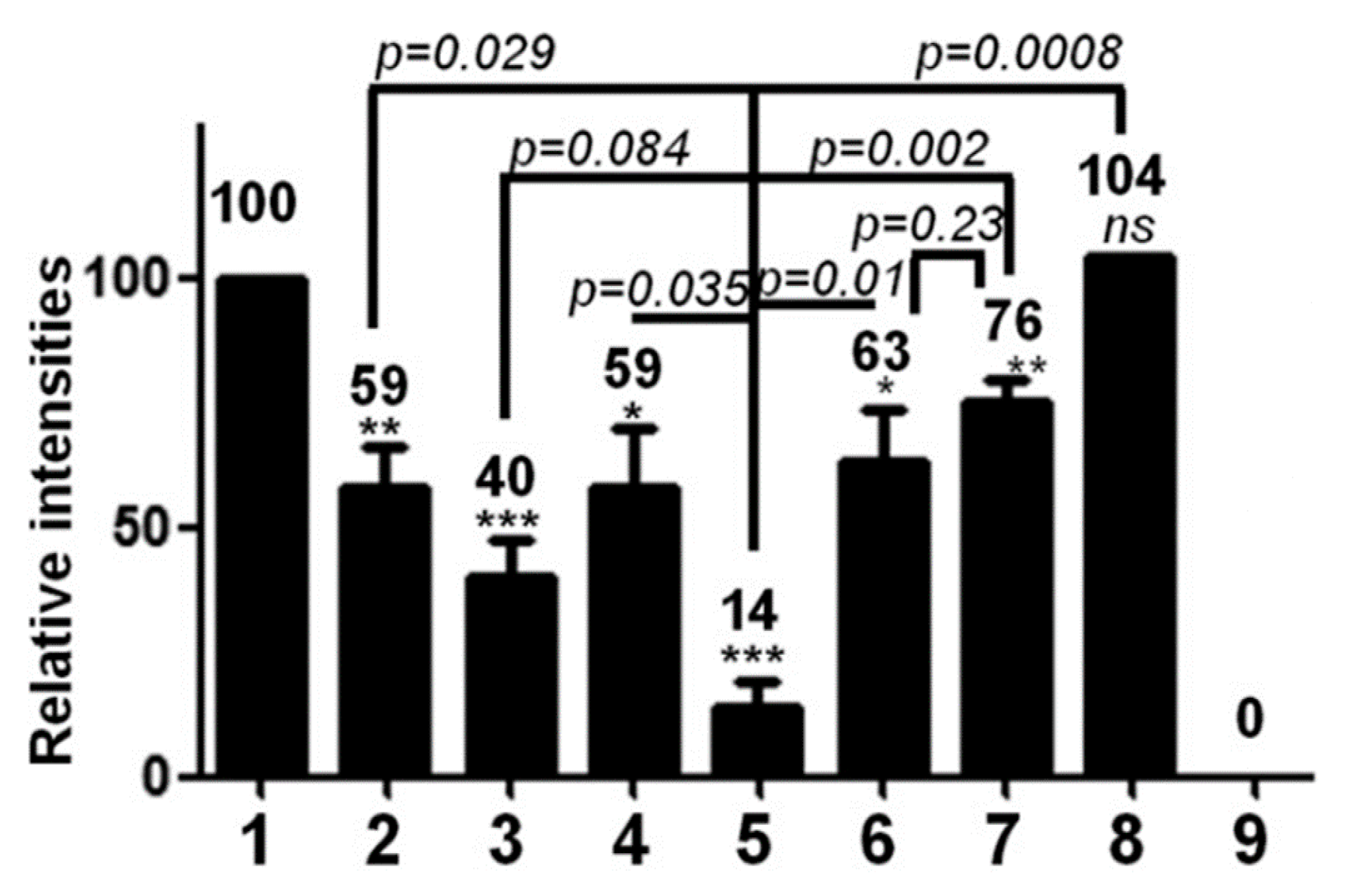
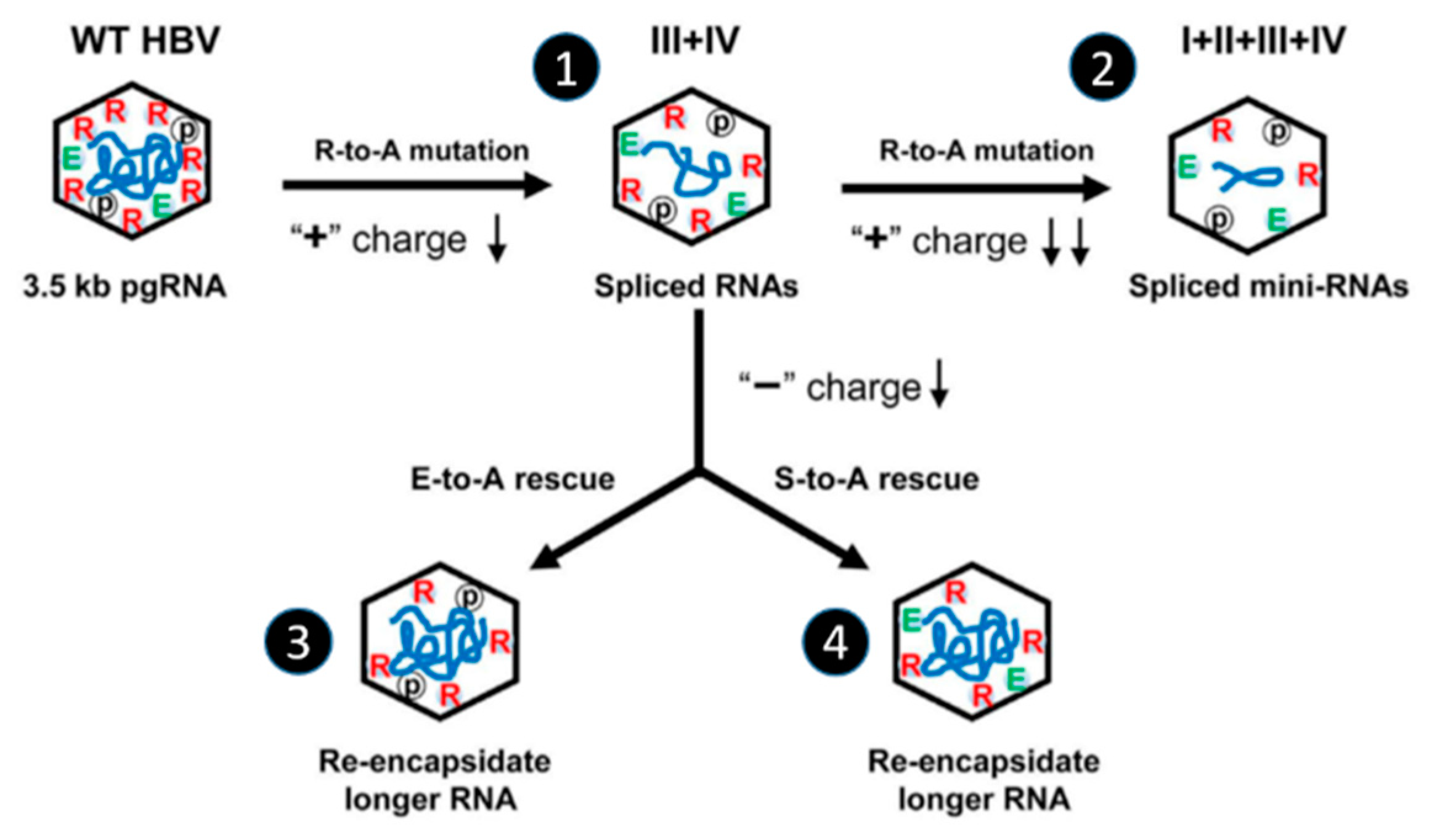
5.1. Effect of Phospho-Acceptor Modifications on RNA Packaging
5.2. Phosphorylation Causes a Decrease in HBc-NA Interaction In Vitro
5.3. Phosphorylation and RT Activity
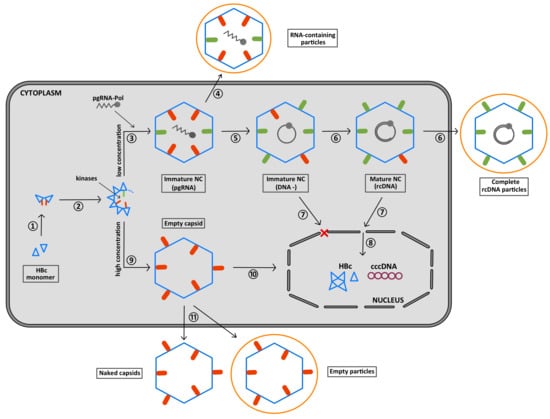
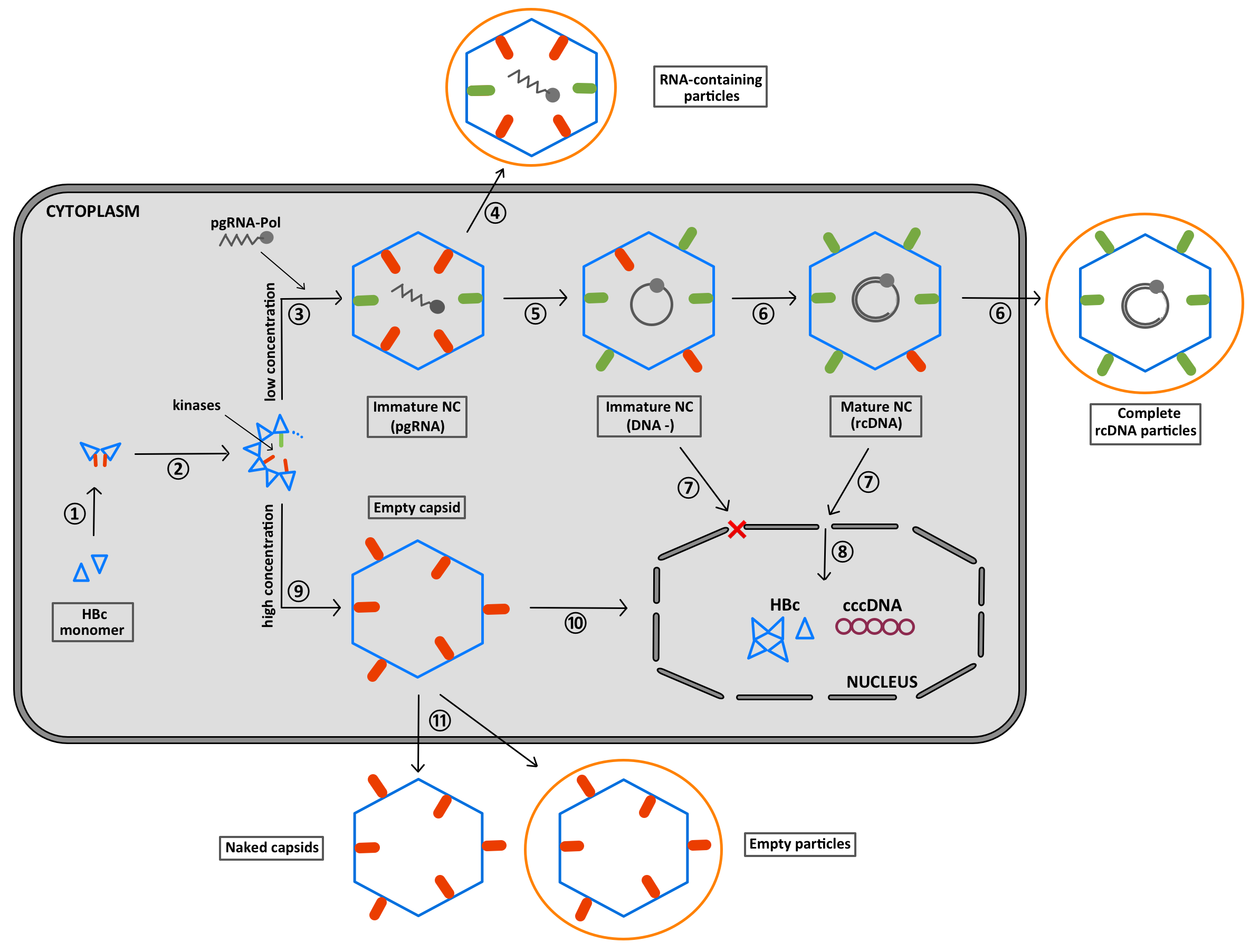
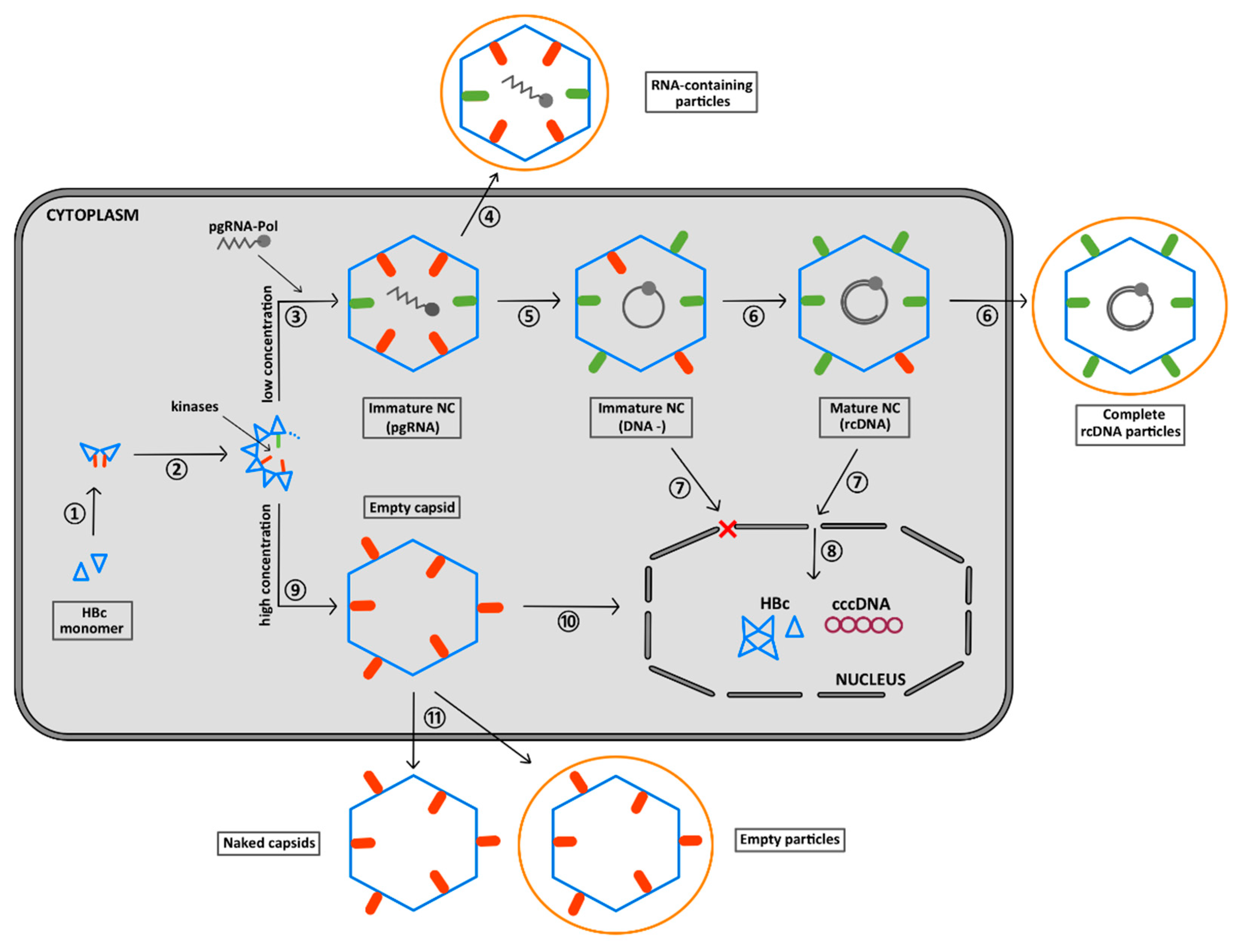
6. The CTD Localization during the Virus Replication Cycle
7. Summary of the Role of the CTD Phosphorylation during HBV Morphogenesis
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Neuveut, C.; Wei, Y.; Buendia, M.A. Mechanisms of HBV-related hepatocarcinogenesis. J. Hepatol. 2010, 52, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.J.; Block, T.M.; McMahon, B.J.; Ghany, M.G.; Urban, S.; Guo, J.T.; Locarnini, S.; Zoulim, F.; Chang, K.M.; Lok, A.S. Present and future therapies of hepatitis B: From discovery to cure. Hepatology 2015, 62, 1893–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Rat, V.; Pinson, X.; Seigneuret, F.; Durand, S.; Herrscher, C.; Lemoine, R.; Burlaud-Gaillard, J.; Raynal, P.Y.; Hourioux, C.; Roingeard, P.; et al. Hepatitis B Virus Core Protein Domains Essential for Viral Capsid Assembly in a Cellular Context. J. Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [Green Version]
- Blumberg, B.S. Australia antigen and the biology of hepatitis B. Science 1977, 197, 17–25. [Google Scholar] [CrossRef] [Green Version]
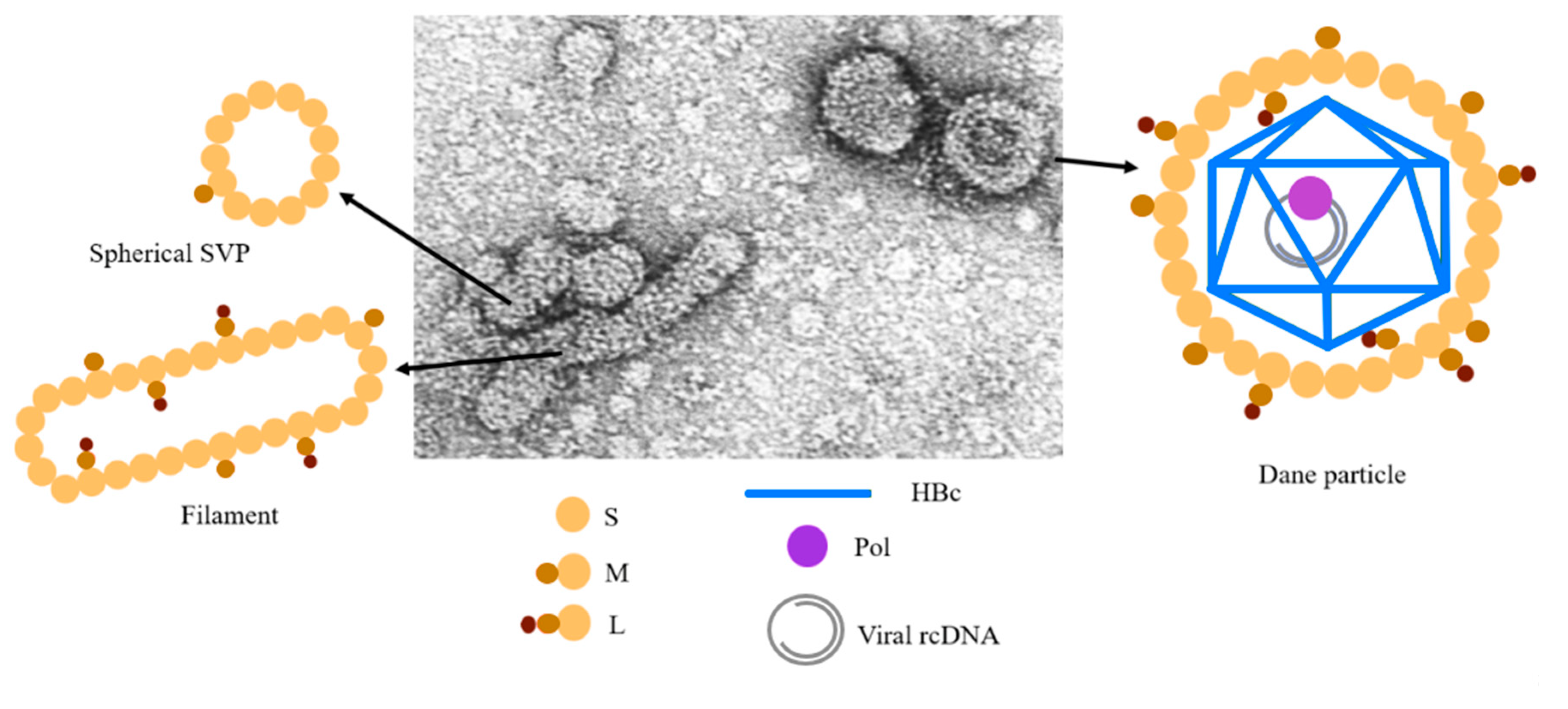
- Bruss, V. Hepatitis B virus morphogenesis. World J. Gastroenterol. 2007, 13, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, K. Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, R.J.; Beales, L.; Blond, D.; Simon, M.N.; Lin, B.Y.; Chisari, F.V.; Stuart, D.I.; Rowlands, D.J. Hepatitis B small surface antigen particles are octahedral. Proc. Natl. Acad. Sci. USA 2005, 102, 14783–14788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patient, R.; Hourioux, C.; Roingeard, P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell. Microbiol. 2009, 11, 1561–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cova, L. Advances and challenges in the development of therapeutic DNA vaccines against hepatitis B virus infection. Curr. Gene Ther. 2014, 14, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Nguyen, D.; Mentzer, L.; Adams, C.; Lee, H.; Ashley, R.; Hafenstein, S.; Hu, J. Secretion of genome-free hepatitis B virus--single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog. 2011, 7, e1002255. [Google Scholar] [CrossRef]
- Peiffer, K.H.; Akhras, S.; Himmelsbach, K.; Hassemer, M.; Finkernagel, M.; Carra, G.; Nuebling, M.; Chudy, M.; Niekamp, H.; Glebe, D.; et al. Intracellular accumulation of subviral HBsAg particles and diminished Nrf2 activation in HBV genotype G expressing cells lead to an increased ROI level. J. Hepatol. 2015, 62, 791–798. [Google Scholar] [CrossRef]
- Chen, M.T.; Billaud, J.N.; Sällberg, M.; Guidotti, L.G.; Chisari, F.V.; Jones, J.; Hughes, J.; Milich, D.R. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. USA 2004, 101, 14913–14918. [Google Scholar] [CrossRef] [Green Version]
- Zlotnick, A.; Mukhopadhyay, S. Virus assembly, allostery and antivirals. Trends Microbiol. 2011, 19, 14–23. [Google Scholar] [CrossRef]
- Schinazi, R.F.; Ehteshami, M.; Bassit, L.; Asselah, T. Towards HBV curative therapies. Liver Int. Off. J. Int. Assoc. Study Liver 2018, 38 (Suppl. S1), 102–114. [Google Scholar] [CrossRef] [Green Version]
- Asselah, T.; Loureiro, D.; Boyer, N.; Mansouri, A. Targets and future direct-acting antiviral approaches to achieve hepatitis B virus cure. Lancet Gastroenterol. Hepatol. 2019, 4, 883–892. [Google Scholar] [CrossRef]
- Liu, K.; Hu, J. Secretion of empty or complete hepatitis B virions: Envelopment of empty capsids versus mature nucleocapsids. Future Virol. 2019, 14, 95–105. [Google Scholar] [CrossRef]
- Gallucci, L.; Kann, M. Nuclear Import of Hepatitis B Virus Capsids and Genome. Viruses 2017, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, B.; Zlotnick, A. The Structural Biology of Hepatitis B Virus: Form and Function. Annu. Rev. Virol. 2016, 3, 429–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynne, S.A.; Crowther, R.A.; Leslie, A.G. The crystal structure of the human hepatitis B virus capsid. Mol. Cell 1999, 3, 771–780. [Google Scholar] [CrossRef]
- Rost, M.; Mann, S.; Lambert, C.; Doring, T.; Thome, N.; Prange, R. Gamma-adaptin, a novel ubiquitin-interacting adaptor, and Nedd4 ubiquitin ligase control hepatitis B virus maturation. J. Biol. Chem. 2006, 281, 29297–29308. [Google Scholar] [CrossRef] [Green Version]
- Fujii, K.; Hurley, J.H.; Freed, E.O. Beyond Tsg101: The role of Alix in ‘ESCRTing’ HIV-1. Nat. Rev. 2007, 5, 912–916. [Google Scholar] [CrossRef]
- Bottcher, B.; Wynne, S.A.; Crowther, R.A. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 1997, 386, 88–91. [Google Scholar] [CrossRef]
- Conway, J.F.; Cheng, N.; Zlotnick, A.; Wingfield, P.T.; Stahl, S.J.; Steven, A.C. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 1997, 386, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Konig, S.; Beterams, G.; Nassal, M. Mapping of homologous interaction sites in the hepatitis B virus core protein. J. Virol. 1998, 72, 4997–5005. [Google Scholar] [CrossRef] [Green Version]
- Koschel, M.; Thomssen, R.; Bruss, V. Extensive mutagenesis of the hepatitis B virus core gene and mapping of mutations that allow capsid formation. J. Virol. 1999, 73, 2153–2160. [Google Scholar] [CrossRef] [Green Version]
- Crowther, R.A.; Kiselev, N.A.; Bottcher, B.; Berriman, J.A.; Borisova, G.P.; Ose, V.; Pumpens, P. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 1994, 77, 943–950. [Google Scholar] [CrossRef]
- Zlotnick, A.; Cheng, N.; Conway, J.F.; Booy, F.P.; Steven, A.C.; Stahl, S.J.; Wingfield, P.T. Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry 1996, 35, 7412–7421. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, B.; Nassal, M. Structure of Mutant Hepatitis B Core Protein Capsids with Premature Secretion Phenotype. J. Mol. Biol. 2018, 430, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Dhason, M.S.; Zlotnick, A. Structural organization of pregenomic RNA and the carboxy-terminal domain of the capsid protein of hepatitis B virus. PLoS Pathog. 2012, 8, e1002919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koschel, M.; Oed, D.; Gerelsaikhan, T.; Thomssen, R.; Bruss, V. Hepatitis B virus core gene mutations which block nucleocapsid envelopment. J. Virol. 2000, 74, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ponsel, D.; Bruss, V. Mapping of amino acid side chains on the surface of hepatitis B virus capsids required for envelopment and virion formation. J. Virol. 2003, 77, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Seitz, S.; Urban, S.; Antoni, C.; Böttcher, B. Cryo-electron microscopy of hepatitis B virions reveals variability in envelope capsid interactions. EMBO J. 2007, 26, 4160–4167. [Google Scholar] [CrossRef]
- Pastor, F.; Herrscher, C.; Patient, R.; Moreau, A.; Burlaud-Gaillard, J.; Seigneuret, F.; de Rocquigny, H.; Roingeard, P.; Hourioux, C. Direct interaction between hepatitis B virus core and envelope proteins analyzed in a cellular context. Sci. Rep. 2019, in press. [Google Scholar] [CrossRef] [Green Version]
- Ludgate, L.; Liu, K.; Luckenbaugh, L.; Streck, N.; Eng, S.; Voitenleitner, C.; Delaney, W.E.t.; Hu, J. Cell-Free Hepatitis B Virus Capsid Assembly Dependent on the Core Protein C-Terminal Domain and Regulated by Phosphorylation. J. Virol. 2016, 90, 5830–5844. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; White, S.J.; Thompson, R.F.; Bingham, R.; Weiss, E.U.; Maskell, D.P.; Zlotnick, A.; Dykeman, E.; Tuma, R.; Twarock, R.; et al. HBV RNA pre-genome encodes specific motifs that mediate interactions with the viral core protein that promote nucleocapsid assembly. Nat. Microbiol. 2017, 2, 17098. [Google Scholar] [CrossRef] [Green Version]
- Zlotnick, A.; Porterfield, J.Z.; Wang, J.C. To build a virus on a nucleic acid substrate. Biophys. J. 2013, 104, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Heger-Stevic, J.; Zimmermann, P.; Lecoq, L.; Bottcher, B.; Nassal, M. Hepatitis B virus core protein phosphorylation: Identification of the SRPK1 target sites and impact of their occupancy on RNA binding and capsid structure. PLoS Pathog. 2018, 14, e1007488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazina, E.V.; Fielding, J.E.; Lin, B.; Anderson, D.A. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J. Virol. 2000, 74, 4721–4728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beames, B.; Lanford, R.E. Carboxy-terminal truncations of the HBV core protein affect capsid formation and the apparent size of encapsidated HBV RNA. Virology 1993, 194, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 1992, 66, 4107–4116. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Ou, J.H. Phosphorylation and nuclear localization of the hepatitis B virus core protein: Significance of serine in the three repeated SPRRR motifs. J. Virol. 1995, 69, 1025–1029. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.T.; Li, J.; Liao, W.; Ou, J. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 1999, 259, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Le Pogam, S.; Chua, P.K.; Newman, M.; Shih, C. Exposure of RNA templates and encapsidation of spliced viral RNA are influenced by the arginine-rich domain of human hepatitis B virus core antigen (HBcAg 165–173). J. Virol. 2005, 79, 1871–1887. [Google Scholar] [CrossRef] [Green Version]
- Porterfield, J.Z.; Dhason, M.S.; Loeb, D.D.; Nassal, M.; Stray, S.J.; Zlotnick, A. Full-length hepatitis B virus core protein packages viral and heterologous RNA with similarly high levels of cooperativity. J. Virol. 2010, 84, 7174–7184. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Hwang, S.G.; Chwae, Y.J.; Park, S.; Shin, H.J.; Kim, K. Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J. Virol. 2014, 88, 8754–8767. [Google Scholar] [CrossRef] [Green Version]
- Diab, A.; Foca, A.; Fusil, F.; Lahlali, T.; Jalaguier, P.; Amirache, F.; N’Guyen, L.; Isorce, N.; Cosset, F.L.; Zoulim, F.; et al. Polo-like-kinase 1 is a proviral host factor for hepatitis B virus replication. Hepatology 2017, 66, 1750–1765. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xi, J.; Gao, L.; Hu, J. Role of Hepatitis B virus capsid phosphorylation in nucleocapsid disassembly and covalently closed circular DNA formation. PLoS Pathog. 2020, 16, e1008459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, P.Y.; Yang, C.J.; Chu, T.H.; Chang, C.H.; Chiang, C.; Tang, F.M.; Lee, C.Y.; Shih, C. HBV maintains electrostatic homeostasis by modulating negative charges from phosphoserine and encapsidated nucleic acids. Sci. Rep. 2016, 6, 38959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciechowski, M.; Grycuk, T.; Antosiewicz, J.M.; Lesyng, B. Prediction of secondary ionization of the phosphate group in phosphotyrosine peptides. Biophys. J. 2003, 84, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Daub, H.; Blencke, S.; Habenberger, P.; Kurtenbach, A.; Dennenmoser, J.; Wissing, J.; Ullrich, A.; Cotten, M. Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J. Virol. 2002, 76, 8124–8137. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, J.C.; Zlotnick, A. A kinase chaperones hepatitis B virus capsid assembly and captures capsid dynamics in vitro. PLoS Pathog. 2011, 7, e1002388. [Google Scholar] [CrossRef]
- Wittkop, L.; Schwarz, A.; Cassany, A.; Grun-Bernhard, S.; Delaleau, M.; Rabe, B.; Cazenave, C.; Gerlich, W.; Glebe, D.; Kann, M. Inhibition of protein kinase C phosphorylation of hepatitis B virus capsids inhibits virion formation and causes intracellular capsid accumulation. Cell Microbiol. 2010, 12, 962–975. [Google Scholar] [CrossRef]
- Ludgate, L.; Ning, X.; Nguyen, D.H.; Adams, C.; Mentzer, L.; Hu, J. Cyclin-dependent kinase 2 phosphorylates s/t-p sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J. Virol. 2012, 86, 12237–12250. [Google Scholar] [CrossRef] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.T.; Wong, S.W.; Fung, Y.K.; Ou, J.H. Cell cycle regulation of nuclear localization of hepatitis B virus core protein. Proc. Natl. Acad. Sci. USA 1993, 90, 6459–6463. [Google Scholar] [CrossRef] [Green Version]
- Klumpp, K.; Lam, A.M.; Lukacs, C.; Vogel, R.; Ren, S.; Espiritu, C.; Baydo, R.; Atkins, K.; Abendroth, J.; Liao, G.; et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc. Natl. Acad. Sci. USA 2015, 112, 15196–15201. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Hu, Z.; Cheng, J.; Wu, S.; Luo, Y.; Chang, J.; Hu, J.; Guo, J.T. Hepatitis B Virus Core Protein Dephosphorylation Occurs during Pregenomic RNA Encapsidation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katen, S.; Zlotnick, A. The thermodynamics of virus capsid assembly. Methods Enzym. 2009, 455, 395–417. [Google Scholar] [CrossRef] [Green Version]
- Selzer, L.; Kant, R.; Wang, J.C.; Bothner, B.; Zlotnick, A. Hepatitis B Virus Core Protein Phosphorylation Sites Affect Capsid Stability and Transient Exposure of the C-terminal Domain. J. Biol. Chem. 2015, 290, 28584–28593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallina, A.; Bonelli, F.; Zentilin, L.; Rindi, G.; Muttini, M.; Milanesi, G. A recombinant hepatitis B core antigen polypeptide with the protamine-like domain deleted self-assembles into capsid particles but fails to bind nucleic acids. J. Virol. 1989, 63, 4645–4652. [Google Scholar] [CrossRef] [Green Version]
- Petit, M.A.; Pillot, J. HBc and HBe antigenicity and DNA-binding activity of major core protein P22 in hepatitis B virus core particles isolated from the cytoplasm of human liver cells. J. Virol. 1985, 53, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnbaum, F.; Nassal, M. Hepatitis B virus nucleocapsid assembly: Primary structure requirements in the core protein. J. Virol. 1990, 64, 3319–3330. [Google Scholar] [CrossRef] [Green Version]
- Hudson, W.H.; Ortlund, E.A. The structure, function and evolution of proteins that bind DNA and RNA. Nat. Rev. Mol. Cell Biol. 2014, 15, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M.; Leifer, I.; Wingert, I.; Dallmeier, K.; Prinz, S.; Vorreiter, J. A Structural Model for Duck Hepatitis B Virus Core Protein Derived by Extensive Mutagenesis. J. Virol. 2007, 81, 13218–13229. [Google Scholar] [CrossRef] [Green Version]
- Hatton, T.; Zhou, S.; Standring, D.N. RNA- and DNA-binding activities in hepatitis B virus capsid protein: A model for their roles in viral replication. J. Virol. 1992, 66, 5232–5241. [Google Scholar] [CrossRef] [Green Version]
- Kock, J.; Nassal, M.; Deres, K.; Blum, H.E.; von Weizsacker, F. Hepatitis B virus nucleocapsids formed by carboxy-terminally mutated core proteins contain spliced viral genomes but lack full-size DNA. J. Virol. 2004, 78, 13812–13818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewellyn, E.B.; Loeb, D.D. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J. Virol. 2011, 85, 1298–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, R.; Barta, A.; Semrad, K. Strategies for RNA folding and assembly. Nat. Rev. Mol. Cell Biol. 2004, 5, 908–919. [Google Scholar] [CrossRef]
- Rajkowitsch, L.; Chen, D.; Stampfl, S.; Semrad, K.; Waldsich, C.; Mayer, O.; Jantsch, M.F.; Konrat, R.; Blasi, U.; Schroeder, R. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007, 4, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herschlag, D. RNA chaperones and the RNA folding problem. J. Biol. Chem. 1995, 270, 20871–20874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godet, J.; Mely, Y. Biophysical studies of the nucleic acid chaperone properties of the HIV-1 nucleocapsid protein. RNA Biol. 2010, 7, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofari, G.; Darlix, J.L. The ubiquitous nature of RNA chaperone proteins. Prog. Nucleic Acid Res. Mol. Biol. 2002, 72, 223–268. [Google Scholar]
- Tompa, P.; Csermely, P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004, 18, 1169–1175. [Google Scholar] [CrossRef]
- Chu, T.H.; Liou, A.T.; Su, P.Y.; Wu, H.N.; Shih, C. Nucleic acid chaperone activity associated with the arginine-rich domain of human hepatitis B virus core protein. J. Virol. 2014, 88, 2530–2543. [Google Scholar] [CrossRef] [Green Version]
- Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 2011, 6, 4–11. [Google Scholar] [CrossRef]
- Darlix, J.L.; de Rocquigny, H.; Mauffret, O.; Mely, Y. Retrospective on the all-in-one retroviral nucleocapsid protein. Virus Res. 2014, 193, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rein, A.; Datta, S.A.; Jones, C.P.; Musier-Forsyth, K. Diverse interactions of retroviral Gag proteins with RNAs. Trends Biochem. Sci. 2011, 36, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Mitra, M.; Naufer, M.N.; McCauley, M.J.; Gorelick, R.J.; Rouzina, I.; Musier-Forsyth, K.; Williams, M.C. Differential contribution of basic residues to HIV-1 nucleocapsid protein’s nucleic acid chaperone function and retroviral replication. Nucleic Acids Res. 2014, 42, 2525–2537. [Google Scholar] [CrossRef] [PubMed]
- Machida, A.; Ohnuma, H.; Tsuda, F.; Yoshikawa, A.; Hoshi, Y.; Tanaka, T.; Kishimoto, S.; Akahane, Y.; Miyakawa, Y.; Mayumi, M. Phosphorylation in the carboxyl-terminal domain of the capsid protein of hepatitis B virus: Evaluation with a monoclonal antibody. J. Virol. 1991, 65, 6024–6030. [Google Scholar] [CrossRef] [Green Version]
- Dhason, M.S.; Wang, J.C.; Hagan, M.F.; Zlotnick, A. Differential assembly of Hepatitis B Virus core protein on single- and double-stranded nucleic acid suggest the dsDNA-filled core is spring-loaded. Virology 2012, 430, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef]
- Chong, C.K.; Cheng, C.Y.S.; Tsoi, S.Y.J.; Huang, F.Y.; Liu, F.; Seto, W.K.; Lai, C.L.; Yuen, M.F.; Wong, D.K. Role of hepatitis B core protein in HBV transcription and recruitment of histone acetyltransferases to cccDNA minichromosome. Antivir. Res. 2017, 144, 1–7. [Google Scholar] [CrossRef]
- Lott, L.; Beames, B.; Notvall, L.; Lanford, R.E. Interaction between hepatitis B virus core protein and reverse transcriptase. J. Virol. 2000, 74, 11479–11489. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, P.; Johnson, J.; Smith, E.; Chen, Y.; Mueller, J.D. Brightness analysis. Methods Enzym. 2013, 518, 71–98. [Google Scholar] [CrossRef]
- Melegari, M.; Wolf, S.K.; Schneider, R.J. Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J. Virol. 2005, 79, 9810–9820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.T.; Ou, J.H. Phosphorylation of hepatitis B virus precore and core proteins. J. Virol. 1991, 65, 2327–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, J.; Zweidler, A.; Summers, J. Characterization of the major duck hepatitis B virus core particle protein. J. Virol. 1989, 63, 1371–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabe, B.; Vlachou, A.; Pante, N.; Helenius, A.; Kann, M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 2003, 100, 9849–9854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, D.H.; Berg, E.A.; O’Connor, P.B.; Costello, C.E.; Hu, J. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc. Natl. Acad. Sci. USA 2005, 102, 9020–9025. [Google Scholar] [CrossRef] [Green Version]
- Ning, X.; Basagoudanavar, S.H.; Liu, K.; Luckenbaugh, L.; Wei, D.; Wang, C.; Wei, B.; Zhao, Y.; Yan, T.; Delaney, W.; et al. Capsid Phosphorylation State and Hepadnavirus Virion Secretion. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Basagoudanavar, S.H.; Perlman, D.H.; Hu, J. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J. Virol. 2007, 81, 1641–1649. [Google Scholar] [CrossRef] [Green Version]
- Schlicht, H.J.; Bartenschlager, R.; Schaller, H. The duck hepatitis B virus core protein contains a highly phosphorylated C terminus that is essential for replication but not for RNA packaging. J. Virol. 1989, 63, 2995–3000. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Satoh, S.; Ohori, H. DNA-binding activity of hepatitis B e antigen polypeptide lacking the protaminelike sequence of nucleocapsid protein of human hepatitis B virus. J. Virol. 1988, 62, 3517–3521. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Yang, S.Q.; Standring, D.N. Characterization of hepatitis B virus capsid particle assembly in Xenopus oocytes. J. Virol. 1992, 66, 3086–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Standring, D.N.; Ou, J.H.; Masiarz, F.R.; Rutter, W.J. A signal peptide encoded within the precore region of hepatitis B virus directs the secretion of a heterogeneous population of e antigens in Xenopus oocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 8405–8409. [Google Scholar] [CrossRef] [Green Version]
- Seifer, M.; Standring, D.N. A protease-sensitive hinge linking the two domains of the hepatitis B virus core protein is exposed on the viral capsid surface. J. Virol. 1994, 68, 5548–5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotnick, A.; Cheng, N.; Stahl, S.J.; Conway, J.F.; Steven, A.C.; Wingfield, P.T. Localization of the C terminus of the assembly domain of hepatitis B virus capsid protein: Implications for morphogenesis and organization of encapsidated RNA. Proc. Natl. Acad. Sci. USA 1997, 94, 9556–9561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, M.; Diez, M.; Eisfeld, J.; Nassal, M. In vitro assembly of mosaic hepatitis B virus capsid-like particles (CLPs): Rescue into CLPs of assembly-deficient core protein fusions and FRET-suited CLPs. FEBS Lett. 2005, 579, 5211–5216. [Google Scholar] [CrossRef] [Green Version]
- Kann, M.; Sodeik, B.; Vlachou, A.; Gerlich, W.H.; Helenius, A. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J. Cell Biol. 1999, 145, 45–55. [Google Scholar] [CrossRef]
- Li, H.C.; Huang, E.Y.; Su, P.Y.; Wu, S.Y.; Yang, C.C.; Lin, Y.S.; Chang, W.C.; Shih, C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 2010, 6, e1001162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondot, M.L.; Bruss, V.; Kann, M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 2016, 64, S49–S59. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jin, L.; Jih, J.; Shih, C.; Zhou, Z.H. 3.5A cryoEM structure of hepatitis B virus core assembled from full-length core protein. PLoS ONE 2013, 8, e69729. [Google Scholar] [CrossRef] [Green Version]
- Ning, X.; Luckenbaugh, L.; Liu, K.; Bruss, V.; Sureau, C.; Hu, J. Common and Distinct Capsid and Surface Protein Requirements for Secretion of Complete and Genome-Free Hepatitis B Virions. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Watts, N.R.; Conway, J.F.; Cheng, N.; Stahl, S.J.; Belnap, D.M.; Steven, A.C.; Wingfield, P.T. The morphogenic linker peptide of HBV capsid protein forms a mobile array on the interior surface. EMBO J. 2002, 21, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Ceres, P.; Zlotnick, A. Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 2002, 41, 11525–11531. [Google Scholar] [CrossRef]
- Albin, C.; Robinson, W.S. Protein kinase activity in hepatitis B virus. J. Virol. 1980, 34, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, K.S.; Hu, C.P.; Chang, C. Hepatitis B core antigen forms oligomers and complexes with the p gene product in hepatitis B virus core particles. J. Gastroenterol. Hepatol. 1993, 8, S114–S118. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Lin, L. RNA-protein interactions in hepadnavirus reverse transcription. Front. Biosci. 2009, 14, 1606–1618. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Nickens, D.G.; Lentz, T.B.; Loeb, D.D.; Zlotnick, A. Encapsidated hepatitis B virus reverse transcriptase is poised on an ordered RNA lattice. Proc. Natl. Acad. Sci. USA 2014, 111, 11329–11334. [Google Scholar] [CrossRef] [Green Version]
- Rabe, B.; Delaleau, M.; Bischof, A.; Foss, M.; Sominskaya, I.; Pumpens, P.; Cazenave, C.; Castroviejo, M.; Kann, M. Nuclear entry of hepatitis B virus capsids involves disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog. 2009, 5, e1000563. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, J.C.; Pierson, E.E.; Keifer, D.Z.; Delaleau, M.; Gallucci, L.; Cazenave, C.; Kann, M.; Jarrold, M.F.; Zlotnick, A. Importin beta Can Bind Hepatitis B Virus Core Protein and Empty Core-Like Particles and Induce Structural Changes. PLoS Pathog. 2016, 12, e1005802. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Rocquigny, H.; Rat, V.; Pastor, F.; Darlix, J.L.; Hourioux, C.; Roingeard, P. Phosphorylation of the Arginine-Rich C-Terminal Domains of the Hepatitis B Virus (HBV) Core Protein as a Fine Regulator of the Interaction between HBc and Nucleic Acid. Viruses 2020, 12, 738. https://0-doi-org.brum.beds.ac.uk/10.3390/v12070738
de Rocquigny H, Rat V, Pastor F, Darlix JL, Hourioux C, Roingeard P. Phosphorylation of the Arginine-Rich C-Terminal Domains of the Hepatitis B Virus (HBV) Core Protein as a Fine Regulator of the Interaction between HBc and Nucleic Acid. Viruses. 2020; 12(7):738. https://0-doi-org.brum.beds.ac.uk/10.3390/v12070738
Chicago/Turabian Stylede Rocquigny, Hugues, Virgile Rat, Florentin Pastor, Jean Luc Darlix, Christophe Hourioux, and Philippe Roingeard. 2020. "Phosphorylation of the Arginine-Rich C-Terminal Domains of the Hepatitis B Virus (HBV) Core Protein as a Fine Regulator of the Interaction between HBc and Nucleic Acid" Viruses 12, no. 7: 738. https://0-doi-org.brum.beds.ac.uk/10.3390/v12070738