Molecular Characterization of Porcine Epidemic Diarrhea Virus and Its New Genetic Classification Based on the Nucleocapsid Gene

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, PEDV Detection by PCR, and Complete Sequencing
2.2. Genetic Analysis of Recombination
2.3. Bayesian Phylogenetic Analysis
2.4. p-Distance Analysis
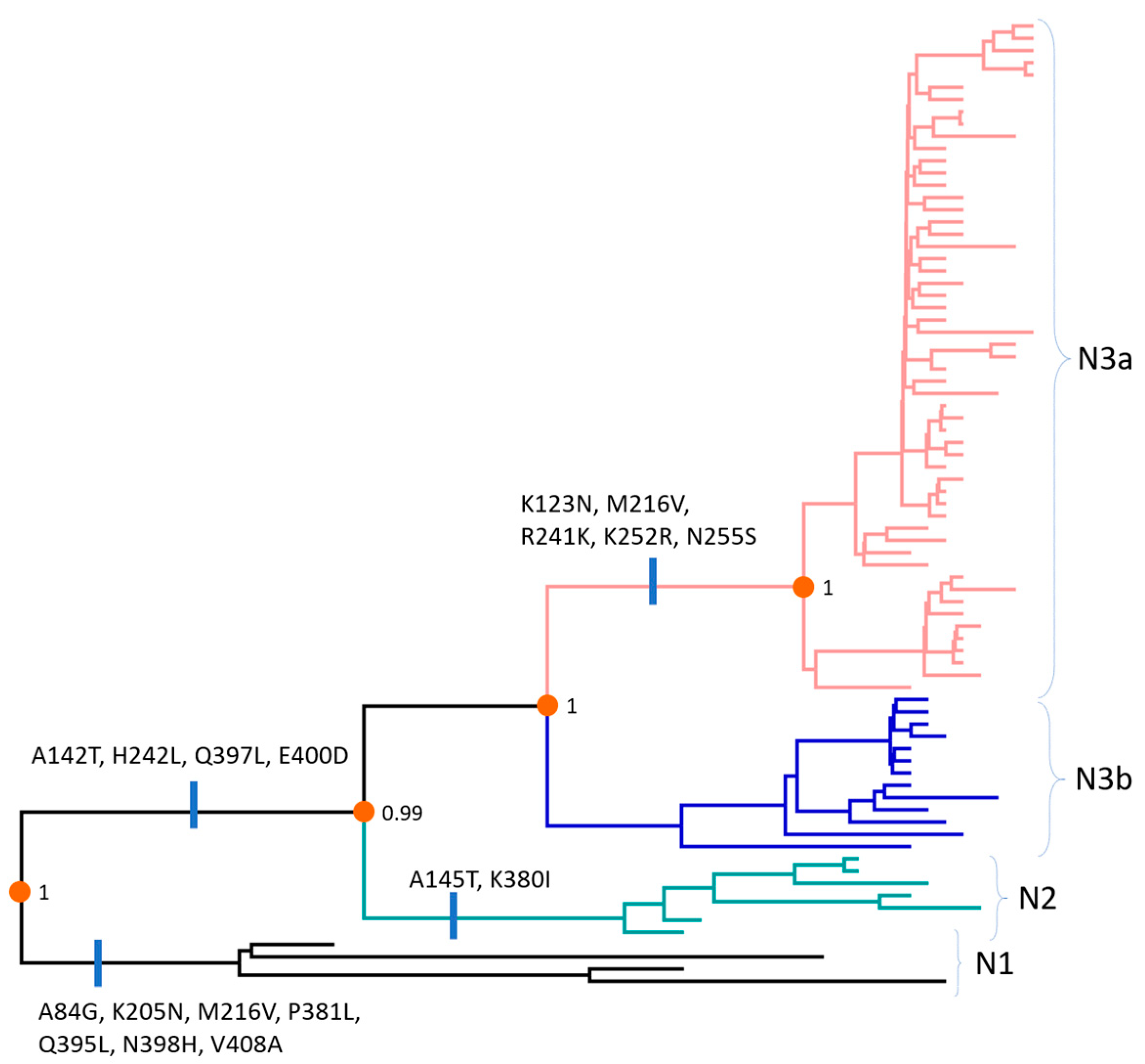
2.5. Inferring Ancestral Amino Acid Changes
2.6. Amino Acids and Antigenic Index Analysis of N Gene
2.7. Antigenic Index Analysis of B-Cell Epitopes in Korean PEDV Strains
3. Results
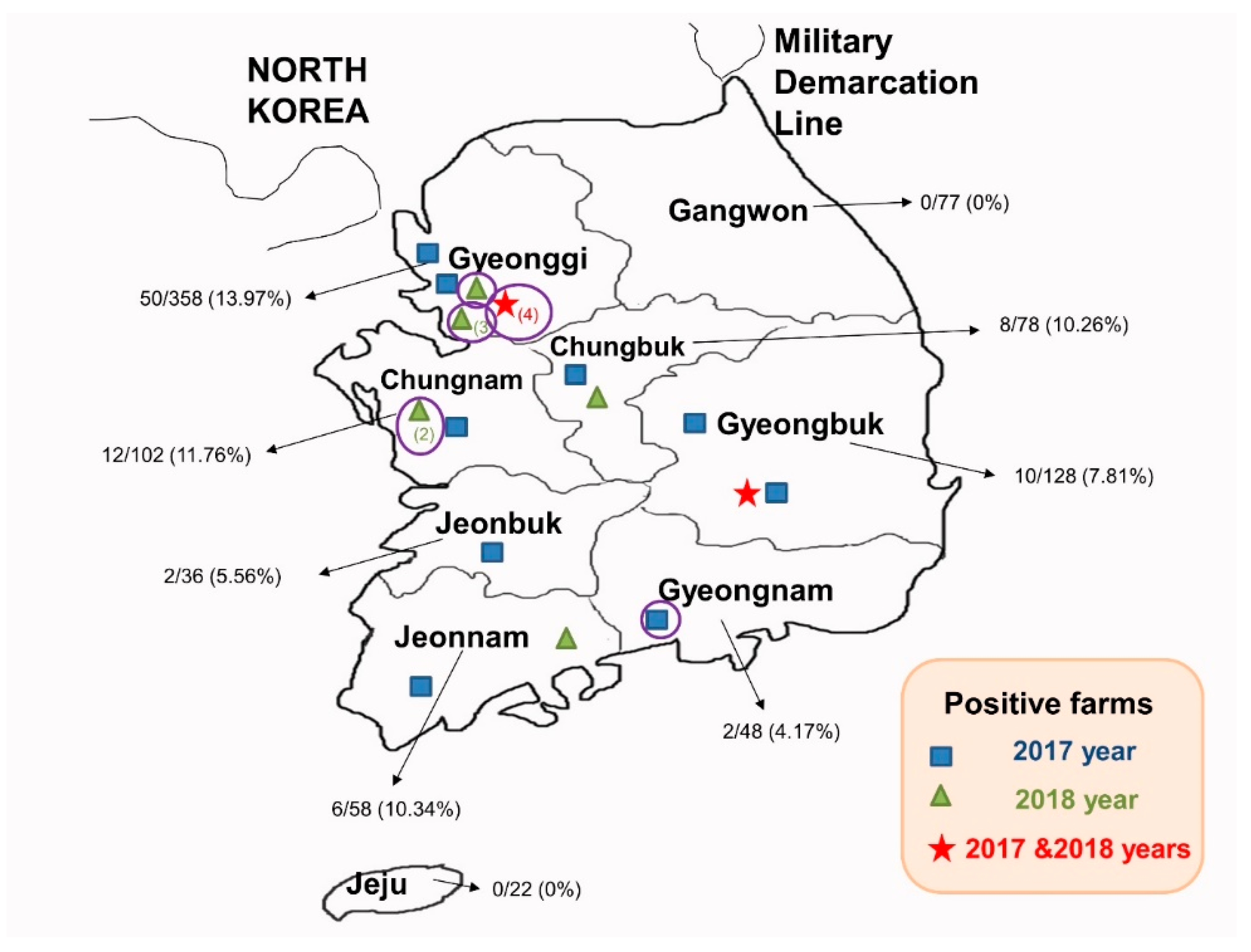
3.1. The Detection of PEDV in Korea from 2017 to 2018
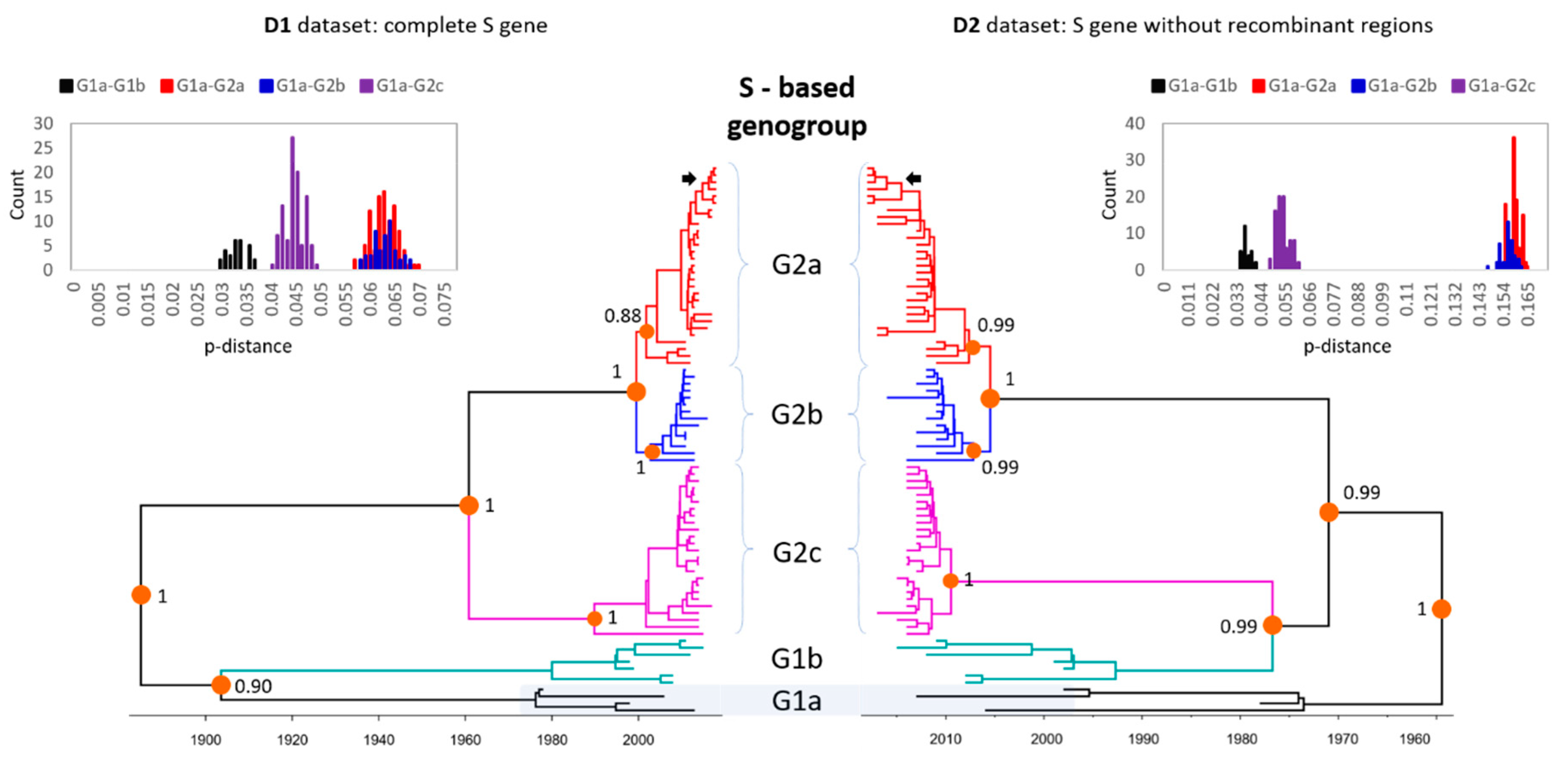
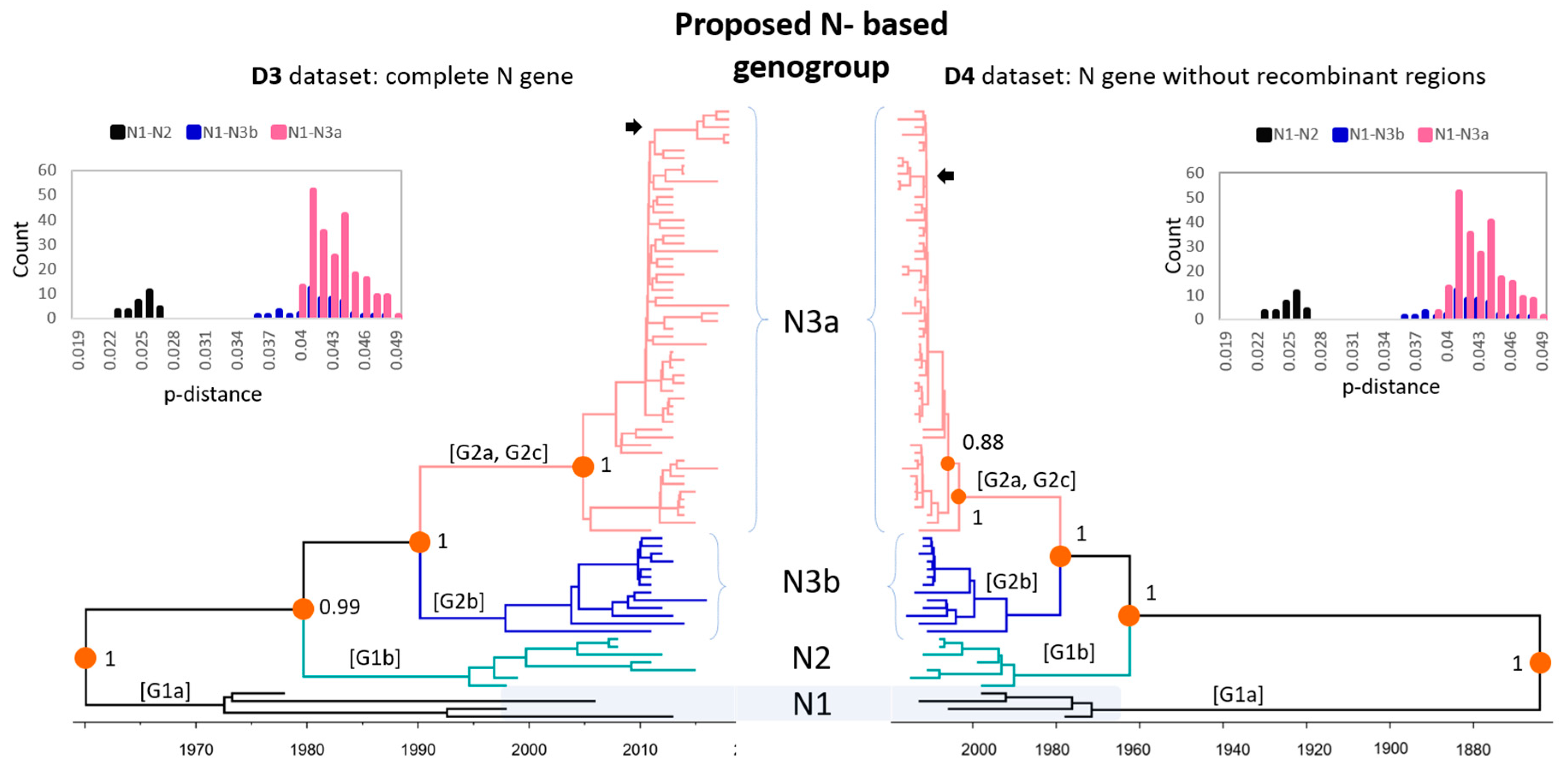
3.2. Phylogenetic Analysis of Global PEDV Strains
3.3. Evolutionary Rates of PEDV Genes
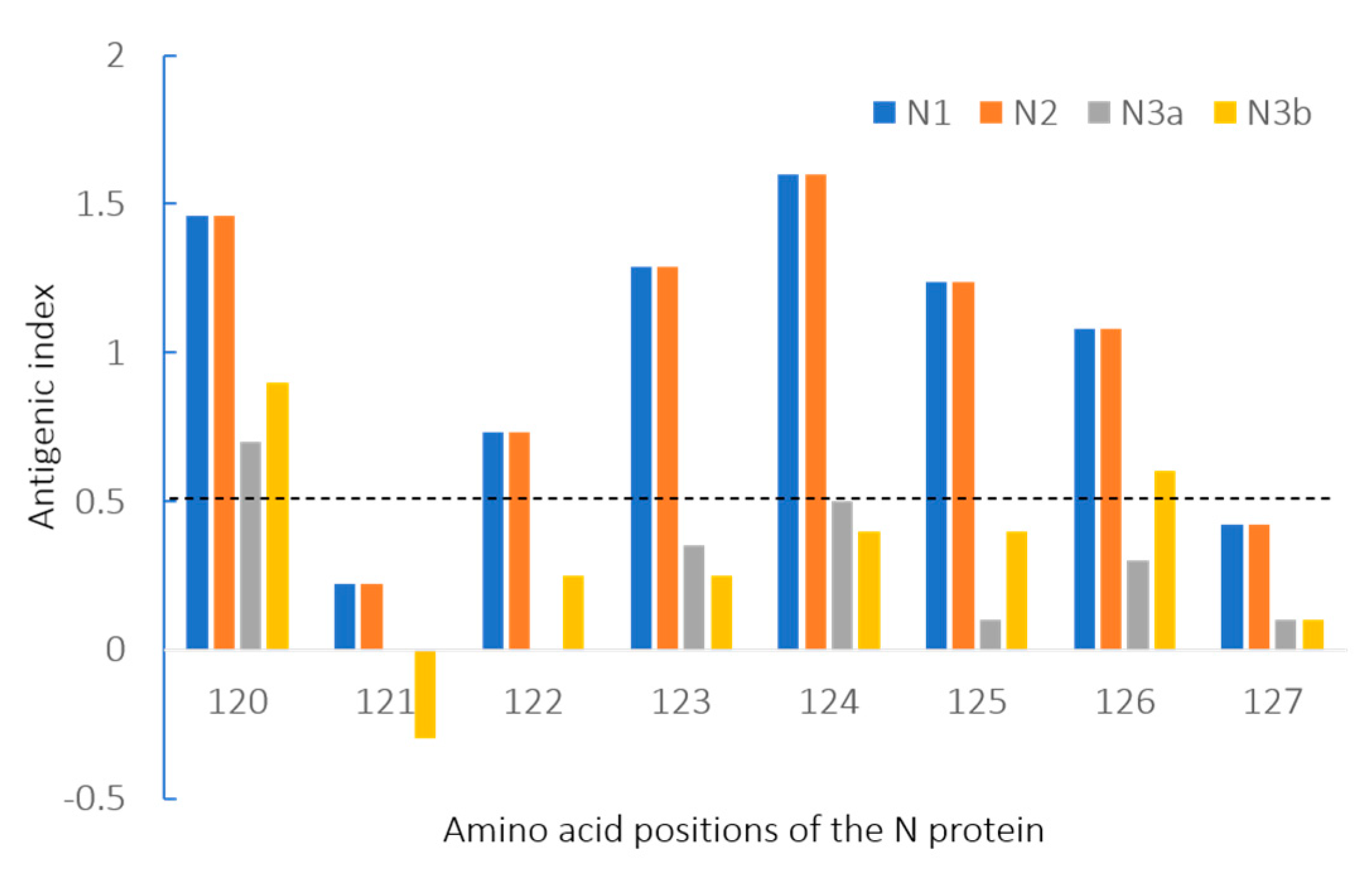
3.4. Amino Acids and Antigenic Index Analysis of N Gene Sequences
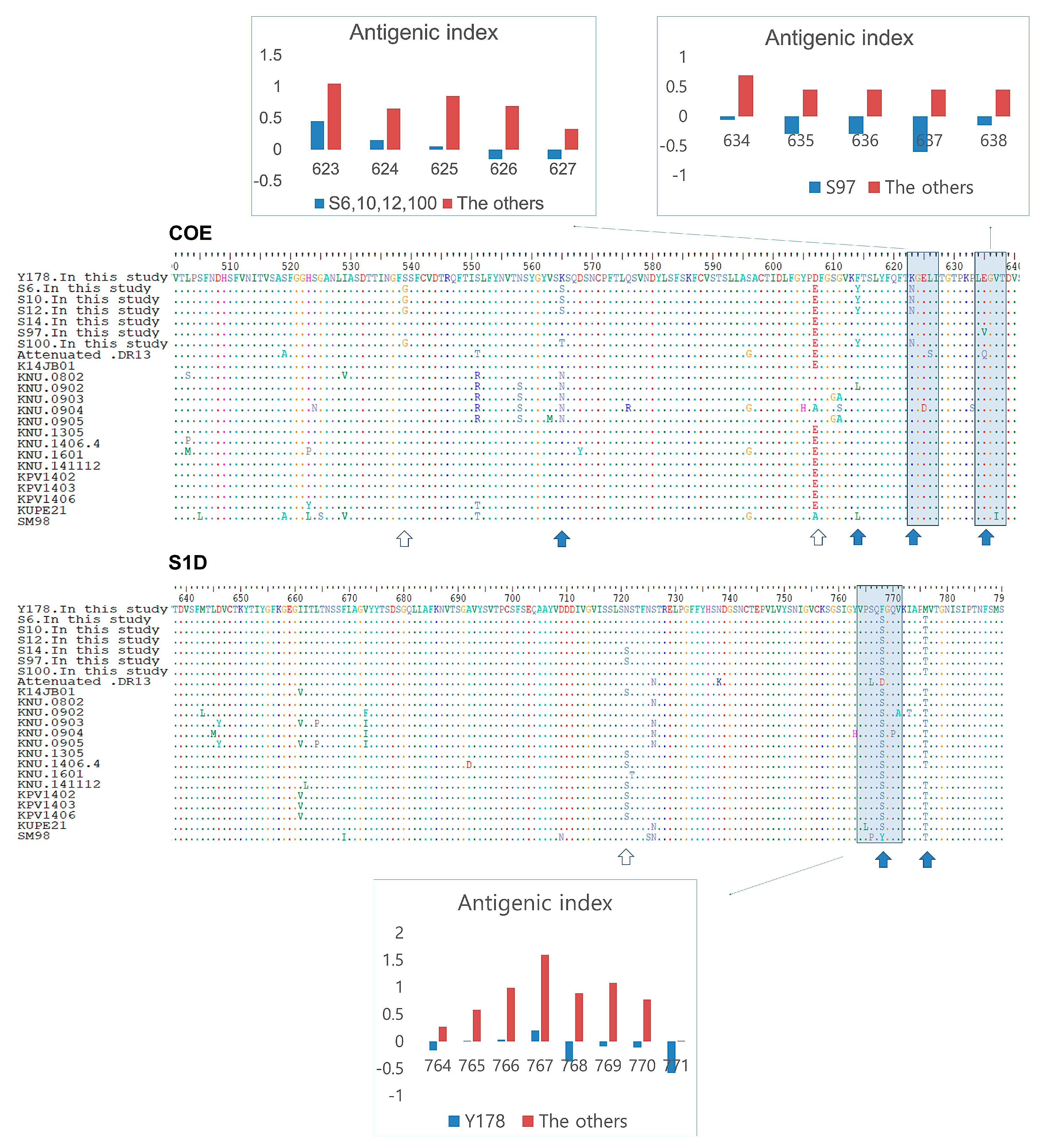
3.5. Antigenic Index Analysis of S Protein B-Cell Epitopes in Korean PEDV Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Debouck, P.; Pensaert, M. Experimental infection of pigs with a new porcine enteric coronavirus, CV 777. Am. J. Vet. Res. 1980, 41, 219–223. [Google Scholar]
- Pensaert, M.B.; De Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Kweon, C.; Kwon, B.; Jung, T.S.; Kee, Y.; Hur, D.; Hwang, E.; Rhee, J.; An, S. Isolation of porcine epidemic diarrhea virus (PEDV) in Korea. Korean J. Vet. Res. 1993, 33, 249–254. [Google Scholar]
- Chung, H.-C.; Nguyen, V.-G.; Moon, H.-J.; Lee, J.-H.; Park, S.-J.; Lee, G.-E.; Kim, H.-K.; Noh, Y.-S.; Lee, C.-H.; Goede, D.; et al. Isolation of Porcine Epidemic Diarrhea Virus during Outbreaks in South Korea, 2013–2014. Emerg. Infect. Dis. 2015, 21, 2238–2240. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, H.; Liu, Y.; Pan, Y.; Deng, F.; Song, Y.; Tang, X.; He, Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012, 18, 1350. [Google Scholar] [CrossRef]
- Cima, G. PED virus reinfecting U.S. herds. Virus estimated to have killed 7 million-plus pigs. J. Am. Vet. Med. Assoc. 2014, 245, 166. [Google Scholar] [PubMed]
- Vlasova, A.N.; Marthaler, D.; Wang, Q.; Culhane, M.R.; Rossow, K.D.; Rovira, A.; Collins, J.; Saif, L.J. Distinct Characteristics and Complex Evolution of PEDV Strains, North America, May 2013–February 2014. Emerg. Infect. Dis. 2014, 20, 1620–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Fang, L.; Ye, X.; Chen, J.; Xu, S.; Zhu, X.; Miao, Y.; Wang, D.; Xiao, S. Evolutionary and genotypic analyses of global porcine epidemic diarrhea virus strains. Transbound. Emerg. Dis. 2019, 66, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and re-emerging coronaviruses in pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef]
- Song, D.; Park, B. Porcine epidemic diarrhoea virus: A comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes 2012, 44, 167–175. [Google Scholar] [CrossRef]
- Jung, K.; Saif, L.J. Porcine epidemic diarrhea virus infection: Etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet. J. 2015, 204, 134–143. [Google Scholar] [CrossRef]
- Lai, M.M.; Cavanagh, D. The molecular biology of coronaviruses. In Advances in Virus Research; Elsevier: Berkeley, CA, USA, 1997; Volume 48, pp. 1–100. [Google Scholar]
- Saif, L.J. Coronavirus immunogens. Vet. Microbiol. 1993, 37, 285–297. [Google Scholar] [CrossRef]
- Utiger, A.; Tobler, K.; Bridgen, A.; Ackermann, M. Identification of the membrane protein of porcine epidemic diarrhea virus. Virus Genes 1995, 10, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Van Kuppeveld, F.J.; He, Q.; Rottier, P.J.; Bosch, B.-J. Cellular entry of the porcine epidemic diarrhea virus. Virus Res. 2016, 226, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Shi, H.; Chen, J.; Zhang, X.; Ji, Z.; Yuan, J.; Shi, D.; Cao, L.; Zhu, X.; Dong, H.; et al. Neutralization of genotype 2 porcine epidemic diarrhea virus strains by a novel monoclonal antibody. Virology 2017, 507, 257–262. [Google Scholar] [CrossRef]
- Chung, H.-C.; Nguyen, V.G.; Le Huynh, T.M.; Moon, H.-J.; Kang, B.-K.; Kim, S.-J.; Kim, H.-K.; Park, S.-J.; Park, K.-T.; Park, Y.-H.; et al. Molecular characterization of a Korean porcine epidemic diarrhea virus strain NB1. Can. J. Vet. Res. 2019, 83, 97–103. [Google Scholar]
- Van Diep, N.; Sueyoshi, M.; Norimine, J.; Hirai, T.; Myint, O.; Teh, A.P.P.; Izzati, U.Z.; Fuke, N.; Yamaguchi, R. Molecular characterization of US-like and Asian non-S INDEL strains of porcine epidemic diarrhea virus (PEDV) that circulated in Japan during 2013–2016 and PEDVs collected from recurrent outbreaks. BMC Vet. Res. 2018, 14, 96. [Google Scholar] [CrossRef]
- Su, Y.; Liu, Y.; Chen, Y.; Zhao, B.; Ji, P.; Xing, G.; Jiang, D.; Liu, C.; Song, Y.; Wang, G.; et al. Detection and phylogenetic analysis of porcine epidemic diarrhea virus in central China based on the ORF3 gene and the S1 gene. Virol. J. 2016, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Fang, L.; Jing, H.; Zeng, S.; Wang, D.; Liu, L.; Zhang, H.; Luo, R.; Chen, H.; Xiao, S. Porcine Epidemic Diarrhea Virus Nucleocapsid Protein Antagonizes Beta Interferon Production by Sequestering the Interaction between IRF3 and TBK1. J. Virol. 2014, 88, 8936–8945. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Lv, M.; Chen, J.-F.; Shi, H.-Y.; Zhang, S.; Zhang, X.; Feng, L. Molecular Characterizations of Subcellular Localization Signals in the Nucleocapsid Protein of Porcine Epidemic Diarrhea Virus. Viruses 2014, 6, 1253–1273. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhang, H.; Zhang, Q.; Huang, Y.; Dong, J.; Liang, Y.; Liu, H.-J.; Tong, D. Porcine epidemic diarrhea virus N protein prolongs S-phase cell cycle, induces endoplasmic reticulum stress, and up-regulates interleukin-8 expression. Vet. Microbiol. 2013, 164, 212–221. [Google Scholar] [CrossRef]
- Lee, J.H.; Chung, H.-C.; Nguyen, V.-G.; Moon, H.J.; Kim, H.K.; Park, S.J.; Lee, C.H.; Lee, G.E.; Park, B.K. Detection and Phylogenetic Analysis of Porcine Deltacoronavirus in Korean Swine Farms, 2015. Transbound. Emerg. Dis. 2016, 63, 248–252. [Google Scholar] [CrossRef]
- Song, D.S.; Kang, B.K.; Oh, J.S.; Ha, G.W.; Yang, J.S.; Moon, H.J.; Jang, Y.-S.; Park, B.K. Multiplex Reverse Transcription-PCR for Rapid Differential Detection of Porcine Epidemic Diarrhea Virus, Transmissible Gastroenteritis Virus, and Porcine Group a Rotavirus. J. Vet. Diagn. Investig. 2006, 18, 278–281. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Kou, Q.; Ge, X.; Zhou, L.; Guo, X.; Yang, H. Phylogenetic analysis of porcine epidemic diarrhea virus field strains prevailing recently in China. Arch. Virol. 2013, 158, 711–715. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Bouckaert, R.R.; Drummond, A.J. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A. Bayesian random local clocks, or one rate to rule them all. BMC Biol. 2010, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, N.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Tamura, K.; Jakobsen, I.B.; Nei, M. MEGA2: Molecular evolutionary genetics analysis software. Bioinformatics 2001, 17, 1244–1245. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, K.-L.; Wang, L.-Y.; Lin, C.-L.; Liu, H.-F. New Phylogenetic Groups of Torque Teno Virus Identified in Eastern Taiwan Indigenes. PLoS ONE 2016, 11, e0149901. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Tamuri, A.U. Treesub: Annotating Ancestral Substitution on a Tree. 2013. Available online: https://github.com/tamuri/treesub (accessed on 19 July 2020).
- Jameson, B.; Wolf, H. The antigenic index: A novel algorithm for predicting antigenic determinants. Bioinformatics 1988, 4, 181–186. [Google Scholar] [CrossRef]
- Fan, J.-H.; Zuo, Y.-Z.; Li, J.-H.; Pei, L.-H. Heterogeneity in membrane protein genes of porcine epidemic diarrhea viruses isolated in China. Virus Genes 2012, 45, 113–117. [Google Scholar] [CrossRef]
- Sun, D.B.; Feng, L.; Shi, H.Y.; Chen, J.F.; Liu, S.W.; Chen, H.Y.; Wang, Y.F. Spike protein region (aa 636789) of porcine epidemic diarrhea virus is essential for induction of neutralizing antibodies. Acta Virol. 2007, 51, 149–156. [Google Scholar]
- Cruz, D.J.M.; Kim, C.-J.; Shin, H. The GPRLQPY motif located at the carboxy-terminal of the spike protein induces antibodies that neutralize Porcine epidemic diarrhea virus. Virus Res. 2008, 132, 192–196. [Google Scholar] [CrossRef]
- Wang, K.; Xie, C.; Zhang, J.; Zhang, W.; Yang, D.; Yu, L.; Jiang, Y.; Yang, S.; Gao, F.; Yang, Z.; et al. The Identification and Characterization of Two Novel Epitopes on the Nucleocapsid Protein of the Porcine Epidemic Diarrhea Virus. Sci. Rep. 2016, 6, 39010. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Chen, F.; Yuan, Y.; Zeng, X.; Wei, Z.; Zhu, L.; Sun, B.; Xie, Q.; Cao, Y.; Xue, C.; et al. Sequence and phylogenetic analysis of nucleocapsid genes of porcine epidemic diarrhea virus (PEDV) strains in China. Arch. Virol. 2013, 158, 1267–1273. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Liu, X.; Lang, H.; Wang, Z.; Shi, D.; Shi, H.; Zhang, X.; Feng, L. Genetic variation of nucleocapsid genes of porcine epidemic diarrhea virus field strains in China. Arch. Virol. 2013, 158, 1397–1401. [Google Scholar] [CrossRef]
- Hou, X.-L.; Yu, L.-Y.; Liu, J. Development and evaluation of enzyme-linked immunosorbent assay based on recombinant nucleocapsid protein for detection of porcine epidemic diarrhea (PEDV) antibodies. Vet. Microbiol. 2007, 123, 86–92. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Peng, J.-Y.; Cheng, Y.-H.; Chang, Y.-C.; Wu, Y.-T.; Tsai, P.-S.; Chiou, H.-Y.; Jeng, C.-R.; Chang, H.-W. Development and comparison of enzyme-linked immunosorbent assays based on recombinant trimeric full-length and truncated spike proteins for detecting antibodies against porcine epidemic diarrhea virus. BMC Vet. Res. 2019, 15, 1–13. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Number | Sample Collection * | Farm Information | Other Pathogens *** (TGEV, RotaV, PDCoV, PRRSV, PPV, JEV) | GenBank | ||||
|---|---|---|---|---|---|---|---|---|
| Stage | Collected Date | Province | Farm Name | No. of Sows | Vaccinated ** | |||
| Y178 | Suckling | 21-Feb-2017 | Gyeongnam | HA | 1500 | PRRSV (MLV), PPV (VLP) | Negative | MH891588 |
| S6 | Suckling | 09-Jan-2018 | Chungnam | DB | 200 | PRRSV (MLV), PPV (VLP) | Negative | MH891589 |
| S10 | Suckling | 06-Feb-2018 | Gyeonggido | WD | 200 | PPV (VLP), PRRSV (MLV), PEDV (killed) | Negative | MH891590 |
| S12 | Suckling | 06-Feb-2018 | Gyeonggido | WT | 200 | PPV (VLP), PRRSV (MLV), PEDV (killed) | Negative | MH891584 |
| S14 | Suckling | 07-Feb-2018 | Gyeonggido | SW | 400 | PPV (VLP), PRRSV (MLV), JEV (MLV) | Negative | MH891585 |
| S97 | Suckling | 17-Apr-2018 | Gyeonggido | NG | 400 | PPV (VLP), PRRSV (MLV), PEDV (Killed) | Negative | MH891586 |
| S100 | Suckling | 19-Apr-2018 | Gyeonggido | YG | 700 | PPV (VLP), PRRSV (MLV), JEV (MLV), PEDV (Killed) | Negative | MH891587 |
| 2017 Year/Stage * | Suckling | Weaned | Growing | Finishing | Gilt | Sow | Total |
|---|---|---|---|---|---|---|---|
| Number of samples | 325 | 162 | 42 | 37 | 43 | 63 | 672 |
| Positive samples | 33 | 15 | 2 | 3 | 2 | 3 | 58 |
| % | 10.15 | 9.26 | 4.76 | 8.11 | 4.65 | 4.76 | 8.63 |
| 2018 year/stage * | Suckling | Weaned | Grower | Finisher | Gilt | Sow | Total |
| Number of samples | 76 | 58 | 34 | 20 | 18 | 29 | 235 |
| Positive samples | 16 | 8 | 2 | 2 | 2 | 2 | 32 |
| % | 21.05 | 13.79 | 5.88 | 10.00 | 11.11 | 6.90 | 13.62 |
| Total/stage * | Suckling | Weaned | Grower | Finisher | Gilt | Sow | Total |
| Number of samples | 401 | 220 | 76 | 57 | 61 | 92 | 907 |
| Positive samples | 49 | 23 | 4 | 5 | 4 | 5 | 90 |
| % | 12.22 | 10.45 | 5.26 | 8.77 | 6.56 | 5.43 | 9.92 |
| Dataset | Data Best Fit Molecular Clock * | Data Best Fit Tree Prior | Geometric Mean Rate (×10−4) ** | 95% HPD Interval (×10−4) *** |
|---|---|---|---|---|
| D1: Complete S gene | RLC | Constant | 5.23 | 3.81–6.53 |
| D2: S gene without recombinant regions | RLC | Exponential | 7.98 | 6.18–10.02 |
| D3: Complete N gene | RLC | Constant | 6.58 | 4.34–9.03 |
| D4: N gene without recombinant regions | RLC | BSP | 3.24 | 2.12–4.52 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-J.; Nguyen, V.-G.; Huynh, T.-M.-L.; Park, Y.-H.; Park, B.-K.; Chung, H.-C. Molecular Characterization of Porcine Epidemic Diarrhea Virus and Its New Genetic Classification Based on the Nucleocapsid Gene. Viruses 2020, 12, 790. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080790
Kim S-J, Nguyen V-G, Huynh T-M-L, Park Y-H, Park B-K, Chung H-C. Molecular Characterization of Porcine Epidemic Diarrhea Virus and Its New Genetic Classification Based on the Nucleocapsid Gene. Viruses. 2020; 12(8):790. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080790
Chicago/Turabian StyleKim, Sung-Jae, Van-Giap Nguyen, Thi-My-Le Huynh, Yong-Ho Park, Bong-Kyun Park, and Hee-Chun Chung. 2020. "Molecular Characterization of Porcine Epidemic Diarrhea Virus and Its New Genetic Classification Based on the Nucleocapsid Gene" Viruses 12, no. 8: 790. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080790