Genetic and Antigenic Characterization and Retrospective Surveillance of Bovine Influenza D Viruses Identified in Hokkaido, Japan from 2018 to 2020

Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples, RNA Extraction, Diagnostic Test, and Ethics Statement
2.2. Cell Culture, Virus Isolation, and Velification
2.3. Sequence and Phylogenetic Analyses
2.4. Comparison of Antigenicity between Two BIDV Isolates Using A Neutralization Assay
2.5. Retrospective Surveillance
3. Results
3.1. Diagnosis of Cattle from Three BRD Outbreaks
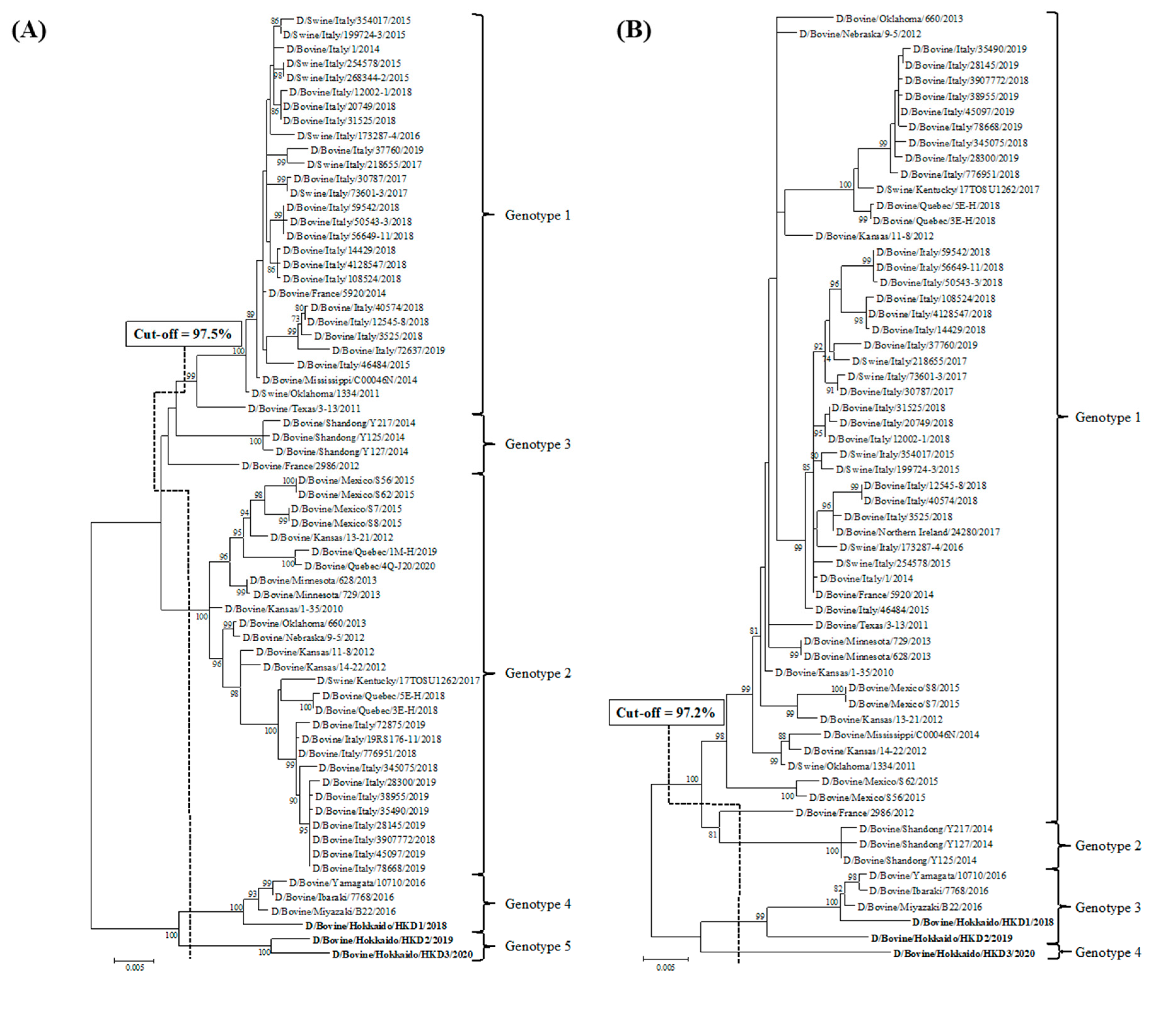
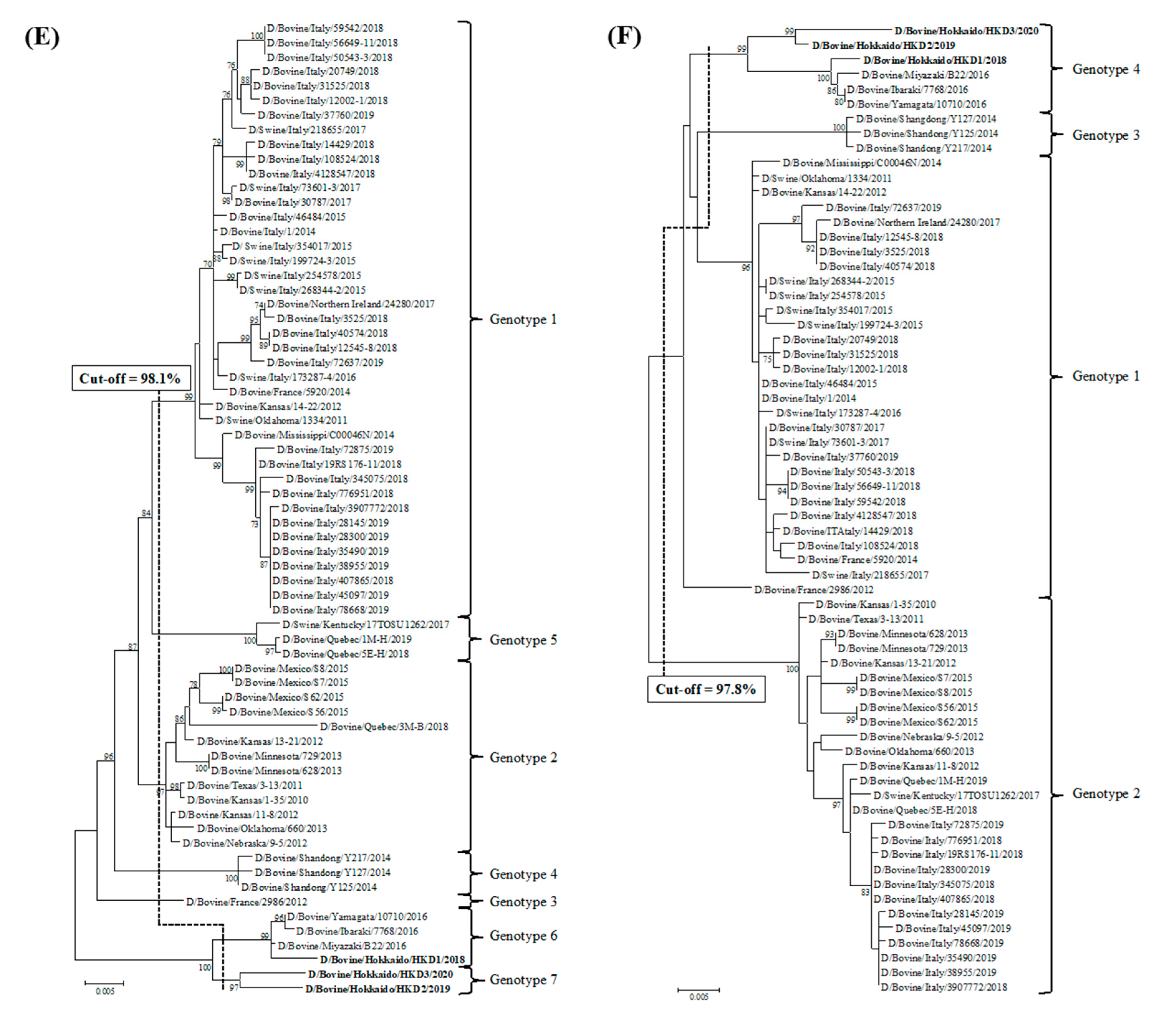
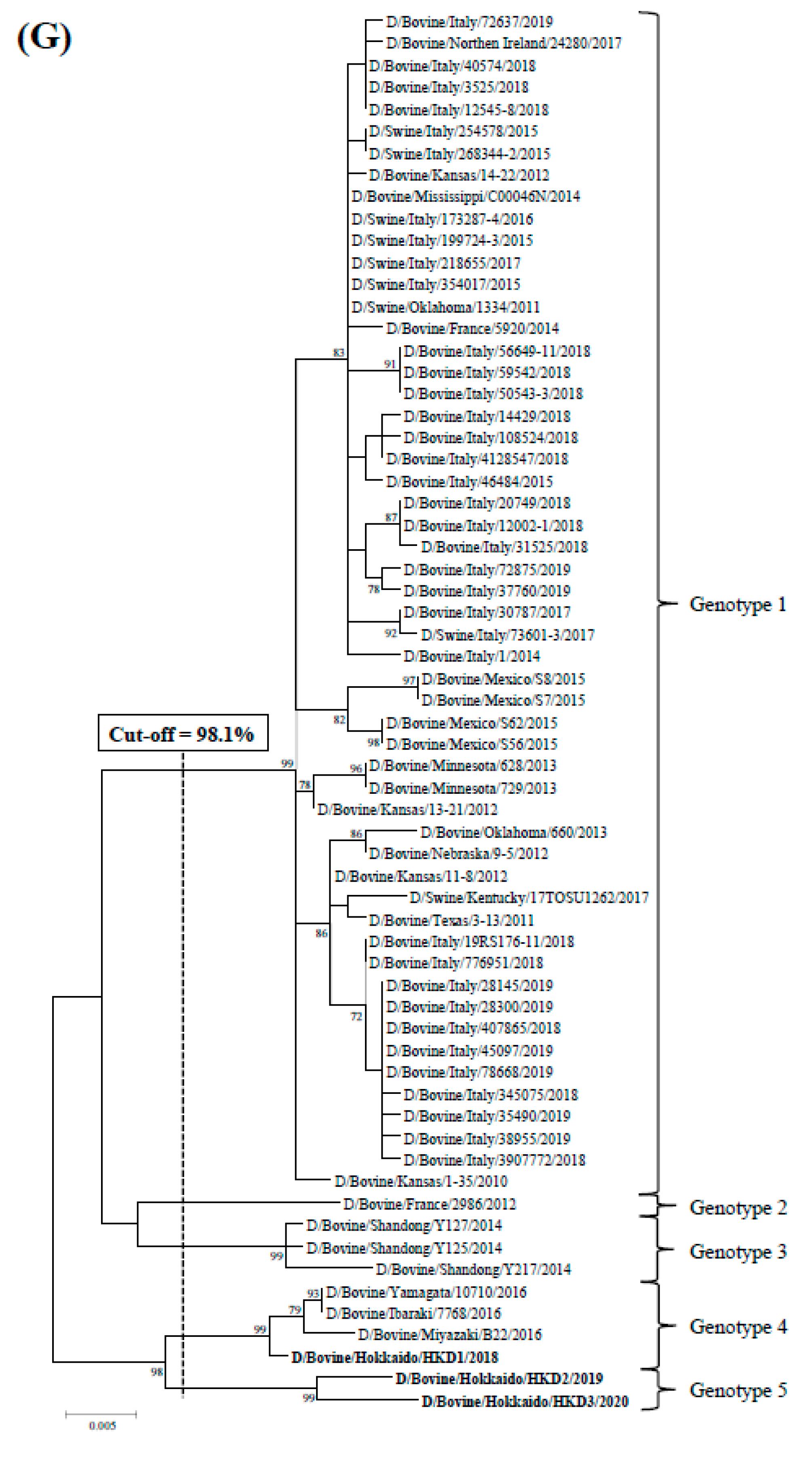
3.2. Sequence and Phylogenetic Analyses
3.3. Comparison of Antigenicity between Two BIDV Isolates Using Neutralization Assay
3.4. Retrospective Surveillance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef] [Green Version]
- Herrler, G.; Durkop, I.; Becht, H.; Klenk, H.D. The glycoprotein of influenza C virus is the haemagglutinin, esterase and fusion factor. J. Gen. Virol. 1988, 69, 839–846. [Google Scholar] [CrossRef]
- Herrler, G.; Klenk, H.D. Structure and function of the hef glycoprotein of influenza c virus. Adv. Virus Res. 1991, 40, 213–234. [Google Scholar] [CrossRef]
- Wang, M.; Veit, M. Hemagglutinin-esterase-fusion (HEF) protein of influenza C virus. Protein Cell 2016, 7, 28–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Fu, X.; Li, G.; Kerlin, F.; Veit, M. Novel influenza D virus: Epidemiology, pathology, evolution and biological characteristics. Virulence 2017, 8, 1580–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Qi, J.; Khedri, Z.; Diaz, S.; Yu, H.; Chen, X.; Varki, A.; Shi, Y.; Gao, G.F. An Open Receptor-Binding Cavity of Hemagglutinin-Esterase-Fusion Glycoprotein from Newly-Identified Influenza D Virus: Basis for Its Broad Cell Tropism. PLoS Pathog. 2016, 12, e1005411. [Google Scholar] [CrossRef] [Green Version]
- Hause, B.M.; Ducatez, M.; Collin, E.A.; Ran, Z.; Liu, R.; Sheng, Z.; Armien, A.; Kaplan, B.; Chakravarty, S.; Hoppe, A.D.; et al. Isolation of a Novel Swine Influenza Virus from Oklahoma in 2011 Which Is Distantly Related to Human Influenza C Viruses. PLoS Pathog. 2013, 9, e1003176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hause, B.M.; Collin, E.A.; Liu, R.; Huang, B.; Sheng, Z.; Lu, W.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a novel influenza virus in cattle and swine: Proposal for a new genus in the Orthomyxoviridae family. MBio 2014, 5, e00031-14. [Google Scholar] [CrossRef] [Green Version]
- Collin, E.A.; Sheng, Z.; Lang, Y.; Ma, W.; Hause, B.M.; Li, F. Cocirculation of Two Distinct Genetic and Antigenic Lineages of Proposed Influenza D Virus in Cattle. J. Virol. 2015, 89, 1036–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, L.; Olivier, A.K.; Genova, S.; Epperson, W.B.; Smith, D.R.; Schneider, L.; Barton, K.; McCuan, K.; Webby, R.J.; Wan, X.-F. Pathogenesis of Influenza D Virus in Cattle. J. Virol. 2016, 90, 5636–5642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hause, B.M.; Huntimer, L.; Falkenberg, S.; Henningson, J.; Lechtenberg, K.; Halbur, T. An inactivated influenza D virus vaccine partially protects cattle from respiratory disease caused by homologous challenge. Vet. Microbiol. 2017, 199, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.F.; Kondov, N.O.; Deng, X.; Van Eenennaam, A.; Neibergs, H.L.; Delwart, E. A Metagenomics and Case-Control Study To Identify Viruses Associated with Bovine Respiratory Disease. J. Virol. 2015, 89, 5340–5349. [Google Scholar] [CrossRef] [Green Version]
- Mitra, N.; Cernicchiaro, N.; Torres, S.; Li, F.; Hause, B.M. Metagenomic characterization of the virome associated with bovine respiratory disease in feedlot cattle identified novel viruses and suggests an etiologic role for influenza D virus. J. Gen. Virol. 2016, 97, 1771–1784. [Google Scholar] [CrossRef] [PubMed]
- Quast, M.; Sreenivasan, C.; Sexton, G.; Nedland, H.; Singrey, A.; Fawcett, L.; Miller, G.; Lauer, D.; Voss, S.; Pollock, S.; et al. Serological evidence for the presence of influenza D virus in small ruminants. Vet. Microbiol. 2015, 180, 281–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, E.; Cook, E.A.J.; Lbacha, H.A.; Oliva, J.; Awoume, F.; Aplogan, G.L.; Hymann, E.C.; Muloi, D.; Deem, S.L.; Alali, S.; et al. Serologic evidence for influenza c and d virus among ruminants and Camelids, Africa, 1991–2015. Emerg. Infect. Dis. 2017, 23, 1556–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedland, H.; Wollman, J.; Sreenivasan, C.; Quast, M.; Singrey, A.; Fawcett, L.; Christopher-Hennings, J.; Nelson, E.; Kaushik, R.S.; Wang, D.; et al. Serological evidence for the co-circulation of two lineages of influenza D viruses in equine populations of the Midwest United States. Zoonoses Public Health 2018, 65, e148–e154. [Google Scholar] [CrossRef]
- White, S.K.; Ma, W.; McDaniel, C.J.; Gray, G.C.; Lednicky, J.A. Serologic evidence of exposure to influenza D virus among persons with occupational contact with cattle. J. Clin. Virol. 2016, 81, 31–33. [Google Scholar] [CrossRef]
- Trombetta, C.M.; Marchi, S.; Manini, I.; Kistner, O.; Li, F.; Piu, P.; Manenti, A.; Biuso, F.; Sreenivasan, C.; Druce, J.; et al. Influenza D virus: Serological evidence in the Italian population from 2005 to 2017. Viruses 2019, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.M.; Wang, S.C.; Peng, C.; Yu, J.M.; Zhuang, Q.Y.; Hou, G.Y.; Liu, S.; Li, J.P.; Chen, J.M. Identification of a potential novel type of influenza virus in Bovine in China. Virus Genes 2014, 49, 493–496. [Google Scholar] [CrossRef]
- Ducatez, M.F.; Pelletier, C.; Meyer, G. Influenza d virus in cattle, France, 2011–2014. Emerg. Infect. Dis. 2015, 21, 368–371. [Google Scholar] [CrossRef]
- Ferguson, L.; Eckard, L.; Epperson, W.B.; Long, L.P.; Smith, D.; Huston, C.; Genova, S.; Webby, R.; Wan, X.F. Influenza D virus infection in Mississippi beef cattle. Virology 2015, 486, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiapponi, C.; Faccini, S.; De Mattia, A.; Baioni, L.; Barbieri, I.; Rosignoli, C.; Nigrelli, A.; Foni, E. Detection of influenza D virus among swine and cattle, Italy. Emerg. Infect. Dis. 2016, 22, 352–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horimoto, T.; Hiono, T.; Mekata, H.; Odagiri, T.; Lei, Z.; Kobayashi, T.; Norimine, J.; Inoshima, Y.; Hikono, H.; Murakami, K.; et al. Nationwide distribution of bovine influenza D virus infection in Japan. PLoS ONE 2016, 11, e0163828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Endoh, M.; Kobayashi, T.; Takenaka-Uema, A.; Chambers, J.K.; Uchida, K.; Nishihara, M.; Hause, B.; Horimoto, T. Influenza d virus infection in herd of cattle, Japan. Emerg. Infect. Dis. 2016, 22, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Flynn, O.; Gallagher, C.; Mooney, J.; Irvine, C.; Ducatez, M.; Hause, B.; McGrath, G.; Ryan, E. Influenza D Virus in Cattle, Ireland. Emerg. Infect. Dis. 2018, 24, 389–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snoeck, C.J.; Oliva, J.; Pauly, M.; Losch, S.; Wildschutz, F.; Muller, C.P.; Hübschen, J.M.; Ducatez, M.F. Influenza D virus circulation in cattle and swine, Luxembourg, 2012–2016. Emerg. Infect. Dis. 2018, 24, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekata, H.; Yamamoto, M.; Hamabe, S.; Tanaka, H.; Omatsu, T.; Mizutani, T.; Hause, B.M.; Okabayashi, T. Molecular epidemiological survey and phylogenetic analysis of bovine influenza D virus in Japan. Transbound. Emerg. Dis. 2018, 65, e355–e360. [Google Scholar] [CrossRef]
- Vilček, S.; Herring, A.J.; Herring, J.A.; Nettleton, P.F.; Lowings, J.P.; Paton, D.J. Pestiviruses isolated from pigs, cattle and sheep can be allocated into at least three genogroups using polymerase chain reaction and restriction endonuclease analysis. Arch. Virol. 1994, 136, 309–323. [Google Scholar] [CrossRef]
- Vilcek, S.; Elvander, M.; Ballagi-Pordany, A.; Belak, S. Development of nested PCR assays for detection of bovine respiratory syncytial virus in clinical samples. J. Clin. Microbiol. 1994, 32, 2225–2231. [Google Scholar] [CrossRef] [Green Version]
- Tsunemitsu, H.; Smith, D.R.; Saif, L.J. Experimental inoculation of adult dairy cows with bovine coronavirus and detection of coronavirus in feces by RT-PCR. Arch. Virol. 1999, 144, 167–175. [Google Scholar] [CrossRef]
- Kishimoto, M.; Tsuchiaka, S.; Rahpaya, S.S.; Hasebe, A.; Otsu, K.; Sugimura, S.; Kobayashi, S.; Komatsu, N.; Nagai, M.; Omatsu, T.; et al. Development of a one-run real-time PCR detection system for pathogens associated with bovine respiratory disease complex. J. Vet. Med. Sci. 2017, 79, 517–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Yaegashi, G.; Fukunari, K.; Suzuki, T. Design of a multiplex quantitative reverse transcription-PCR system to simultaneously detect 16 pathogens associated with bovine respiratory and enteric diseases. J. Appl. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Umit, O.; M Ali, T.; Robin, N. Detection and Antibiotic Susceptibility of Mycoplasma bovis and Other Respiratory Disease Pathogens from Pneumonic Lung Samples in a Calf Rearing Unit. Madridge J. Vet. Med. Res. 2019, 1, 8–12. [Google Scholar]
- Zafar, A.; Shakeel, B.; Ferhat, A.; Muhammad, A.A.; Muhammad, S.; Mohammad, M.T.; Muhammad, A.M.; Nadeem, R.; Shahid, A.; Kamran, T.; et al. Prevalence of Mycoplasma bovis in Respiratory Tract of Cattle Slaughtered in Balochistan, Pakistan. Pak. Vet. J. 2013, 34, 46–49. [Google Scholar]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis Across Computing Platforms. Mol. Biol. Evol 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group a rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef] [Green Version]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gómara, M.; Maes, P.; Patton, J.T.; et al. Full Genome-Based Classification of Rotaviruses Reveals a Common Origin between Human Wa-Like and Porcine Rotavirus Strains and Human DS-1-Like and Bovine Rotavirus Strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Alam M, M.; Kojima, K.; Mise, K.; Ishino, M.; Ayako, S. Genomic diversity and evolution of rotaviruses: An overview. In Genetic Diversity and Mollecular Epidemiology of Rotaviruses; Kobayashi, N., Ed.; Research Signpost: Kerala, India, 2003; pp. 75–89. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BRD Outbreaks | Age [Days] | RT-PCR | qRT-PCR | Virus Isolation | Bacteria Isolation | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| BVDV | BRSV | BCoV | BIDV | Mycoplasma bovis | Pasteurella multocida | Mannheimia haemolytica | ||||
| 1 | 1 | 70 | − | − | − | − | − | − | + | − |
| 2 | 70 | − | − | − | − | BPIV3 | + | + | − | |
| 3 | 73 | − | + | + | − | BCoV | − | + | − | |
| 4 | 74 | − | − | − | + | − | − | + | − | |
| 5 | 82 | − | − | + | + | BIDV (HKD1) | − | + | − | |
| 2 | 1 | 90 | − | − | − | + | BIDV (HKD2) | − | − | − |
| 2 | 100 | − | + | − | + | BIDV | − | − | − | |
| 3 | 131 | − | + | − | + | − | − | − | + | |
| 4 | 123 | − | − | − | + | BIDV | − | − | − | |
| 5 | 114 | − | − | − | + | − | − | − | − | |
| 3 | 1 | 99 | − | − | − | − | − | + | − | − |
| 2 | 42 | − | − | + | − | BCoV | + | − | − | |
| 3 | 63 | − | + | + | + | − | + | − | + | |
| 4 | 35 | − | + | + | − | − | − | − | − | |
| 5 | 55 | − | + | + | + | BIDV (HKD3) | − | − | + | |
| 6 | 61 | − | − | + | − | − | + | + | − | |
| PB1 | PB2 | P3 | HEF | NP | M | NS | |
|---|---|---|---|---|---|---|---|
| Within three BIDVs | 95.9–97.4 | 96.7–98.8 | 97.7–98.7 | 92.5–98.5 | 97.4–98.3 | 96.8–98.6 | 97.3–98.6 |
| vs. other BIDVs detected in Japan | 96.4–99.1 | 96.8–98.9 | 97.7–99.4 | 93.6–98.2 | 97.7–99.3 | 96.8–99.5 | 97.1–99.5 |
| vs. other IDVs detected in other countries | 94.6–96.3 | 94.2–95.9 | 95.0–96.4 | 93.0–95.1 | 94.1–96.3 | 94.9–97.9 | 95.2–97.1 |
| Strains | Genes | PB2 | PB1 | P3 | HEF | NP | M | NS |
|---|---|---|---|---|---|---|---|---|
| Cut-off Value (%) | 97.5 | 97.2 | 97.6 | 97.4 | 98.1 | 97.8 | 98.1 | |
| Total Number of Genotypes | 5 | 4 | 5 | 6 | 7 | 4 | 5 | |
| D/bovine/Hokkaido/HKD1/2018 | genotype 4 | genotype 3 | genotype 4 | genotype 5 | genotype 6 | genotype 4 | genotype 4 | |
| D/bovine/Hokkaido/HKD2/2019 | genotype 5 | genotype 3 | genotype 5 | genotype 6 | genotype 7 | genotype 4 | genotype 5 | |
| D/bovine/Hokkaido/HKD3/2020 | genotype 5 | genotype 4 | genotype 5 | genotype 6 | genotype 7 | genotype 4 | genotype 5 | |
| D/bovine/Ibaraki/7768/2016 | genotype 4 | genotype 3 | genotype 4 | genotype 5 | genotype 6 | genotype 4 | genotype 4 | |
| D/swine/Oklahoma/1334/2011 | genotype 1 | genotype 1 | genotype 1 | genotype 1 | genotype 1 | genotype 1 | genotype 1 | |
| D/bovine/Oklahoma/660/2013 | genotype 2 | genotype 1 | genotype 1 | genotype 3 | genotype 2 | genotype 2 | genotype 1 | |
| D/bovine/France/2986/2012 | genotype 3 | genotype 1 | genotype 3 | genotype 2 | genotype 3 | genotype 1 | genotype 2 | |
| D/bovine /Shandong/Y125/2014 | genotype 3 | genotype 2 | genotype 1 | genotype 1 | genotype 4 | genotype 3 | genotype 3 | |
| Viral Neutralizing Antibody Titers for Serum Samples from Farm A | Viral Neutralizing Antibody Titers for Serum Samples from Farm B | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample Number | HKD1 Isolate | HKD2 Isolate | Sample Number | HKD1 Isolate | HKD2 Isolate | ||||
| Pre | Post | Pre | Post | Pre | Post | Pre | Post | ||
| 1 | 64 | 512 | 128 | 128 | 1 | <2 | 4 | <2 | 64 |
| 2 | 8 | 256 | 8 | 16 | 2 | <2 | 4 | <2 | 64 |
| 3 | 8 | 128 | 16 | 32 | 3 | 2 | 2 | 32 | 128 |
| 4 | 32 | 4096 | 32 | 512 | 4 | <2 | 8 | <2 | 128 |
| 5 | 32 | 256 | 64 | 64 | 5 | 2 | 4 | 8 | 128 |
| 6 | 64 | 512 | 64 | 64 | |||||
| 7 | <2 | 32 | <2 | 4 | |||||
| 8 | 8 | 64 | 16 | 16 | |||||
| Collection Year | Viral Neutralizing Antibody Titers Against HKD1 | Total Number of Positive Samples | Ratio of Positive Samples (%) | Total Number of Samples | |||||
| <10 (Negative) | 10 | 20 | 40 | 80 | ≥160 | ||||
| 2009 | 53 | 2 | 6 | 6 | 9 | 20 | 43 | 45 | 96 |
| 2010 | 37 | 1 | 4 | 13 | 14 | 27 | 59 | 61 | 96 |
| 2011 | 31 | 3 | 6 | 11 | 13 | 32 | 65 | 68 | 96 |
| 2012 | 40 | 0 | 6 | 9 | 13 | 28 | 56 | 58 | 96 |
| 2013 | 39 | 3 | 1 | 9 | 11 | 33 | 57 | 59 | 96 |
| 2014 | 37 | 4 | 6 | 11 | 7 | 31 | 59 | 61 | 96 |
| 2015 | 51 | 3 | 1 | 9 | 7 | 25 | 45 | 47 | 96 |
| 2016 | 52 | 0 | 3 | 9 | 7 | 25 | 44 | 46 | 96 |
| 2017 | 50 | 3 | 1 | 3 | 11 | 38 | 46 | 48 | 96 |
| 2018 | 39 | 1 | 1 | 1 | 11 | 43 | 57 | 59 | 96 |
| Total | 429 | 20 | 35 | 81 | 103 | 292 | 531 | 55 | 960 |
| Collection Year | Viral Neutralizing Antibody Titers Against HKD2 | Total Number of Positive Samples | Ratio of Positive Samples | Total Number of Samples | |||||
| <10 (Negative) | 10 | 20 | 40 | 80 | ≥160 | ||||
| 2009 | 53 | 6 | 9 | 9 | 10 | 9 | 43 | 45 | 96 |
| 2010 | 33 | 0 | 1 | 16 | 17 | 29 | 63 | 66 | 96 |
| 2011 | 28 | 2 | 9 | 8 | 18 | 31 | 68 | 71 | 96 |
| 2012 | 39 | 0 | 3 | 10 | 20 | 24 | 57 | 59 | 96 |
| 2013 | 38 | 4 | 1 | 10 | 8 | 35 | 58 | 60 | 96 |
| 2014 | 36 | 3 | 6 | 7 | 11 | 33 | 60 | 63 | 96 |
| 2015 | 52 | 1 | 6 | 10 | 8 | 19 | 44 | 46 | 96 |
| 2016 | 50 | 1 | 5 | 5 | 9 | 26 | 46 | 48 | 96 |
| 2017 | 47 | 5 | 3 | 2 | 8 | 31 | 49 | 51 | 96 |
| 2018 | 37 | 2 | 4 | 5 | 6 | 42 | 59 | 61 | 96 |
| Total | 413 | 24 | 47 | 82 | 115 | 279 | 547 | 57 | 960 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayakawa, J.; Masuko, T.; Takehana, T.; Suzuki, T. Genetic and Antigenic Characterization and Retrospective Surveillance of Bovine Influenza D Viruses Identified in Hokkaido, Japan from 2018 to 2020. Viruses 2020, 12, 877. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080877
Hayakawa J, Masuko T, Takehana T, Suzuki T. Genetic and Antigenic Characterization and Retrospective Surveillance of Bovine Influenza D Viruses Identified in Hokkaido, Japan from 2018 to 2020. Viruses. 2020; 12(8):877. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080877
Chicago/Turabian StyleHayakawa, Jun, Tomomi Masuko, Tae Takehana, and Tohru Suzuki. 2020. "Genetic and Antigenic Characterization and Retrospective Surveillance of Bovine Influenza D Viruses Identified in Hokkaido, Japan from 2018 to 2020" Viruses 12, no. 8: 877. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080877