Low-Level Ionizing Radiation Induces Selective Killing of HIV-1-Infected Cells with Reversal of Cytokine Induction Using mTOR Inhibitors

 , ,
, , 
Abstract
:1. Introduction
2. Methods
2.1. Reagents and Cell Culture Procedures
2.2. Enrichment of EVs and Virions Using Nanotrap Particles (NTs)
2.3. Human Cohort Information
2.4. Cell Viability Assay
2.5. X-ray Irradiation
2.6. Isolation of RNA, Generation of cDNA, and Real-Time Quantitative PCR (RT-qPCR)
2.7. SDS Page and Western Blot Analysis
2.8. PBMC Infection with Dual-Tropic 89.6 HIV-1 and Induction of Latency
2.9. Statistical analysis
3. Results
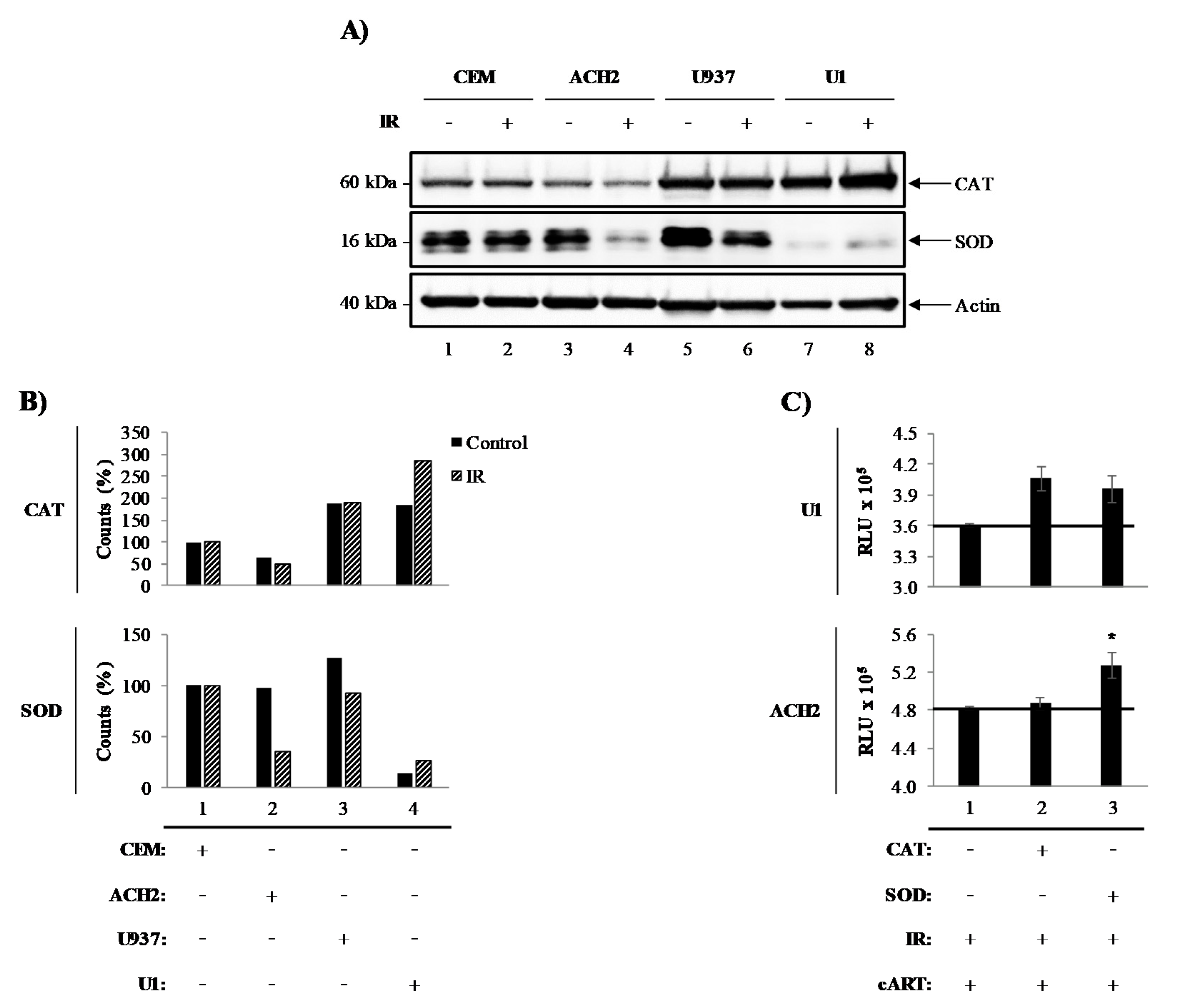
3.1. Catalase and Superoxide Dismutase Mediate Survival of Irradiated HIV-1-Infected Cells
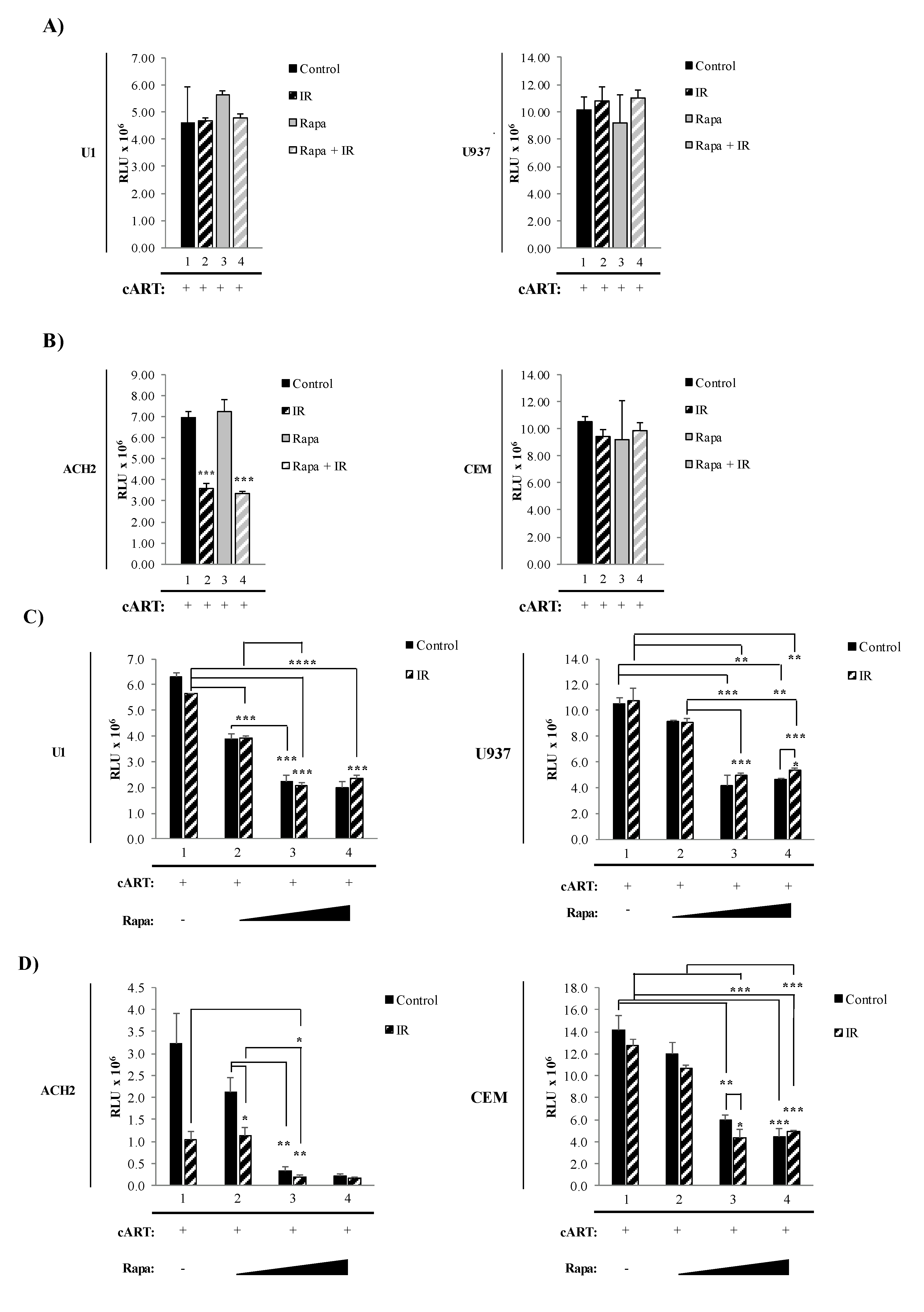
3.2. Combined Treatment with IR and Mtor Inhibitor Selectively Induces Death of HIV-1-Infected T-Cells
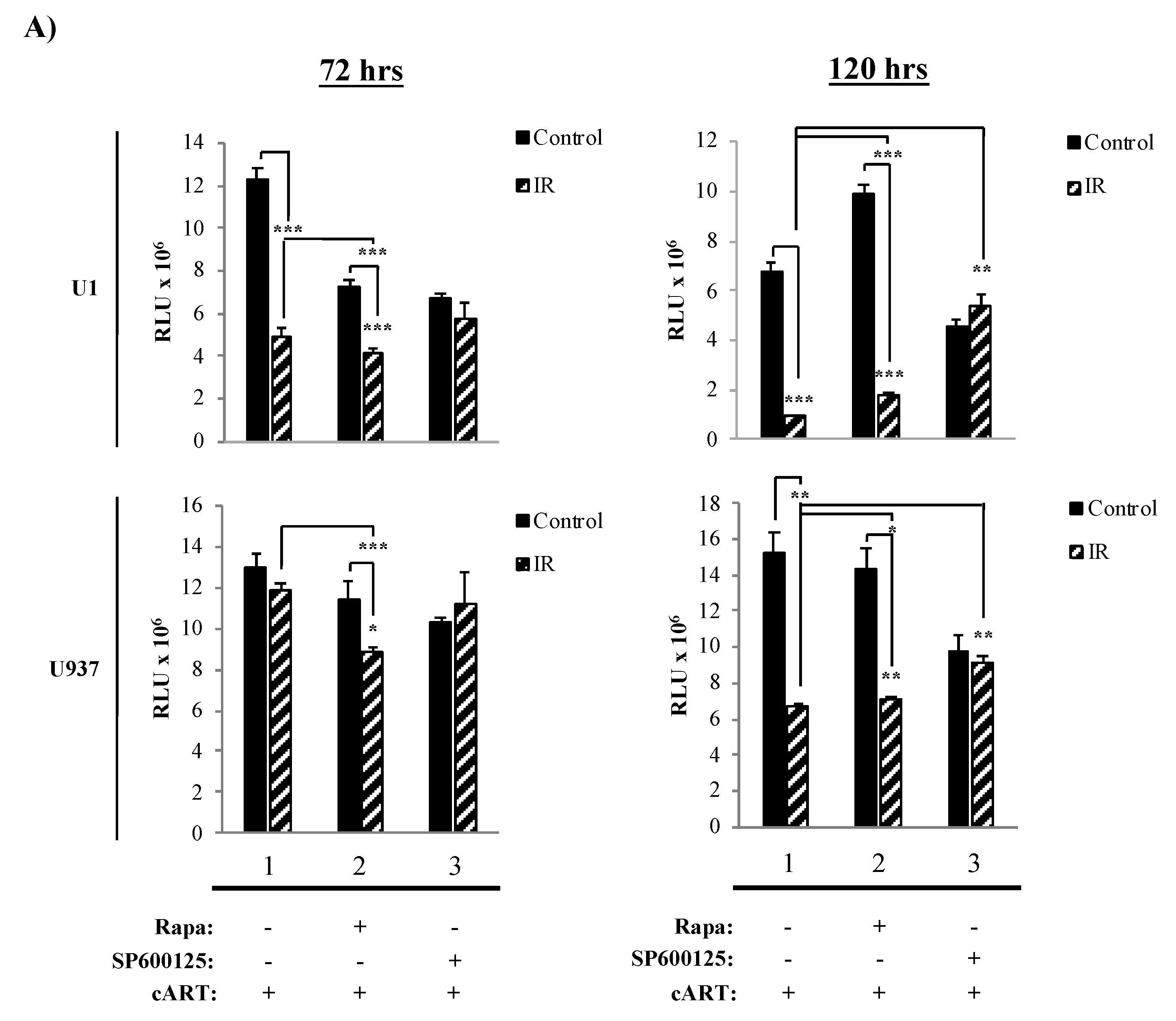
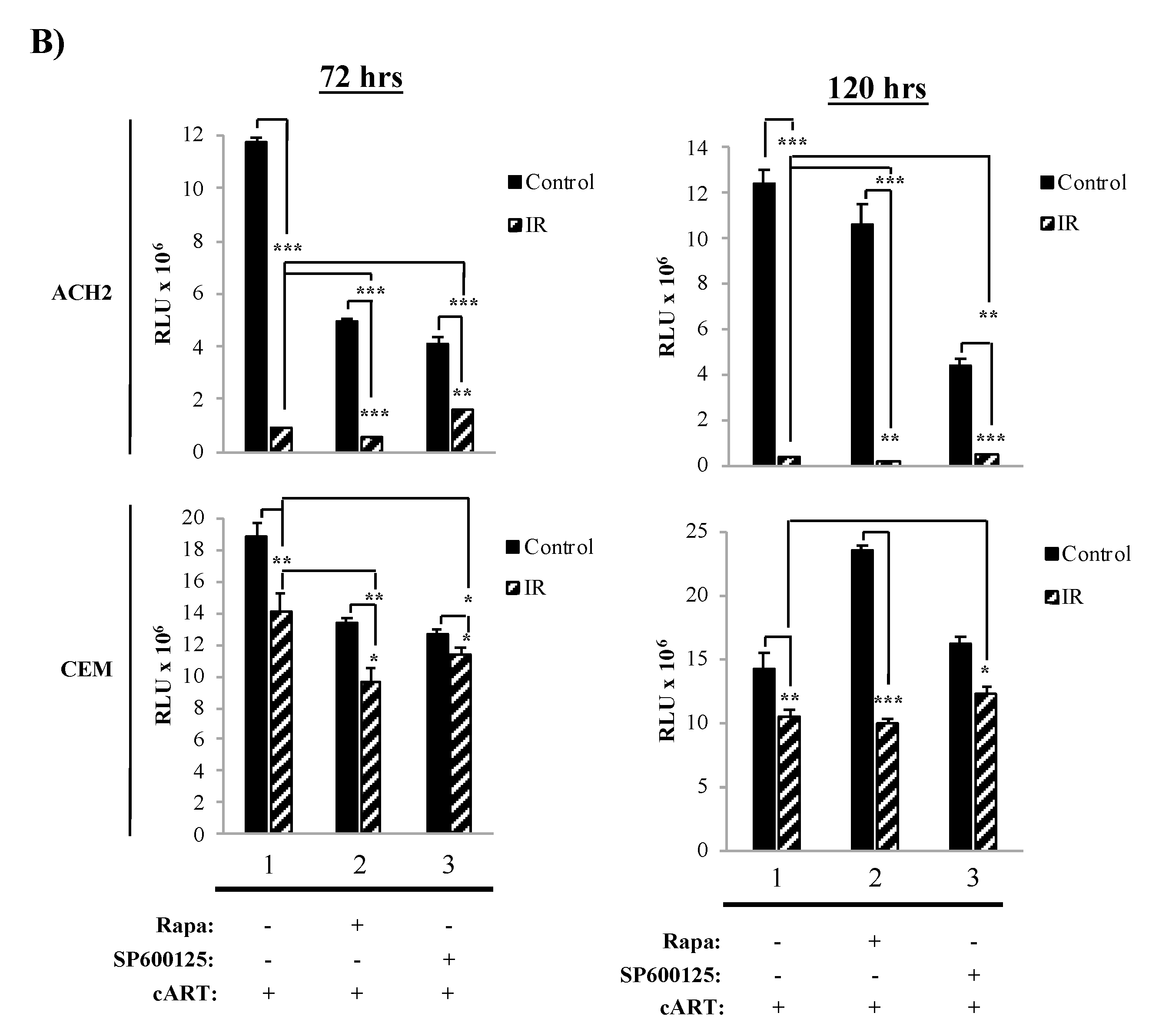
3.3. The Combination of IR, Serum Starvation, and Mtor Inhibition Induces Cell Death in HIV-1-Infected Immune Cells
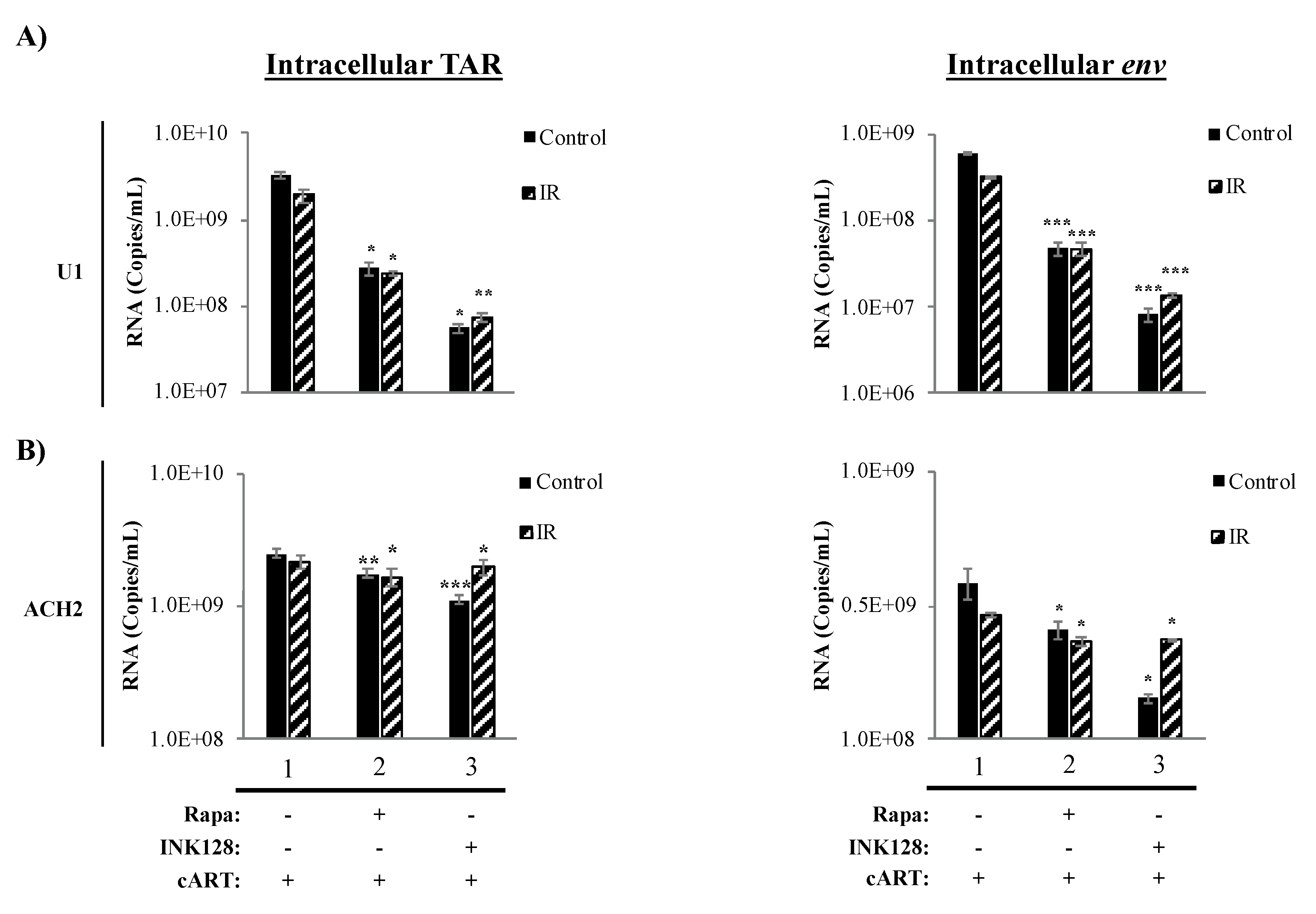
3.4. Inhibition of mTOR Causes a Reduction in Intracellular TAR and Env RNA in HIV-1-Infected Myeloid Cells
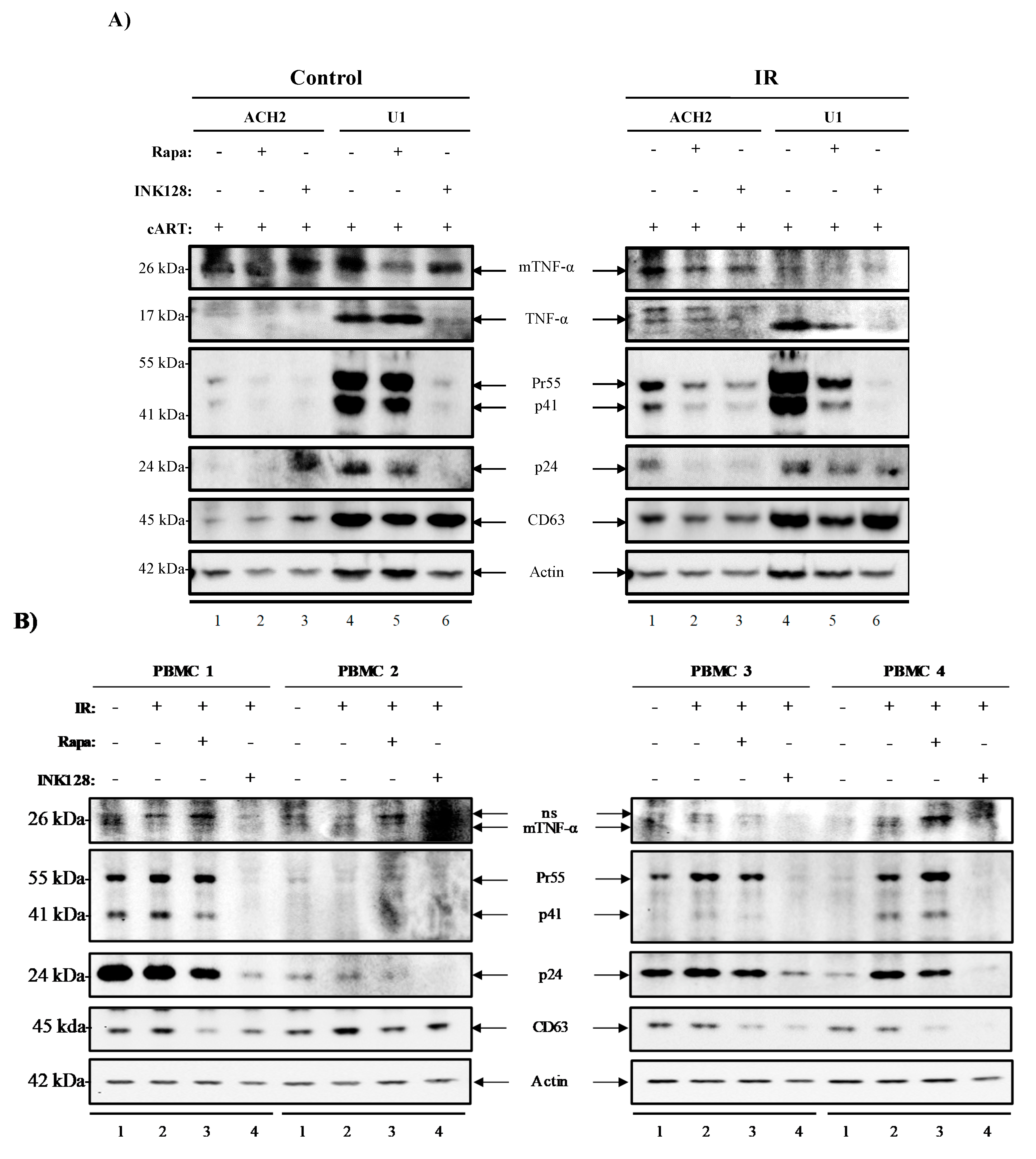
3.5. Inhibition of mTOR Suppresses the Release of Evs Carrying TNF-α and Viral Proteins in HIV-1-Infected Myeloid Cells
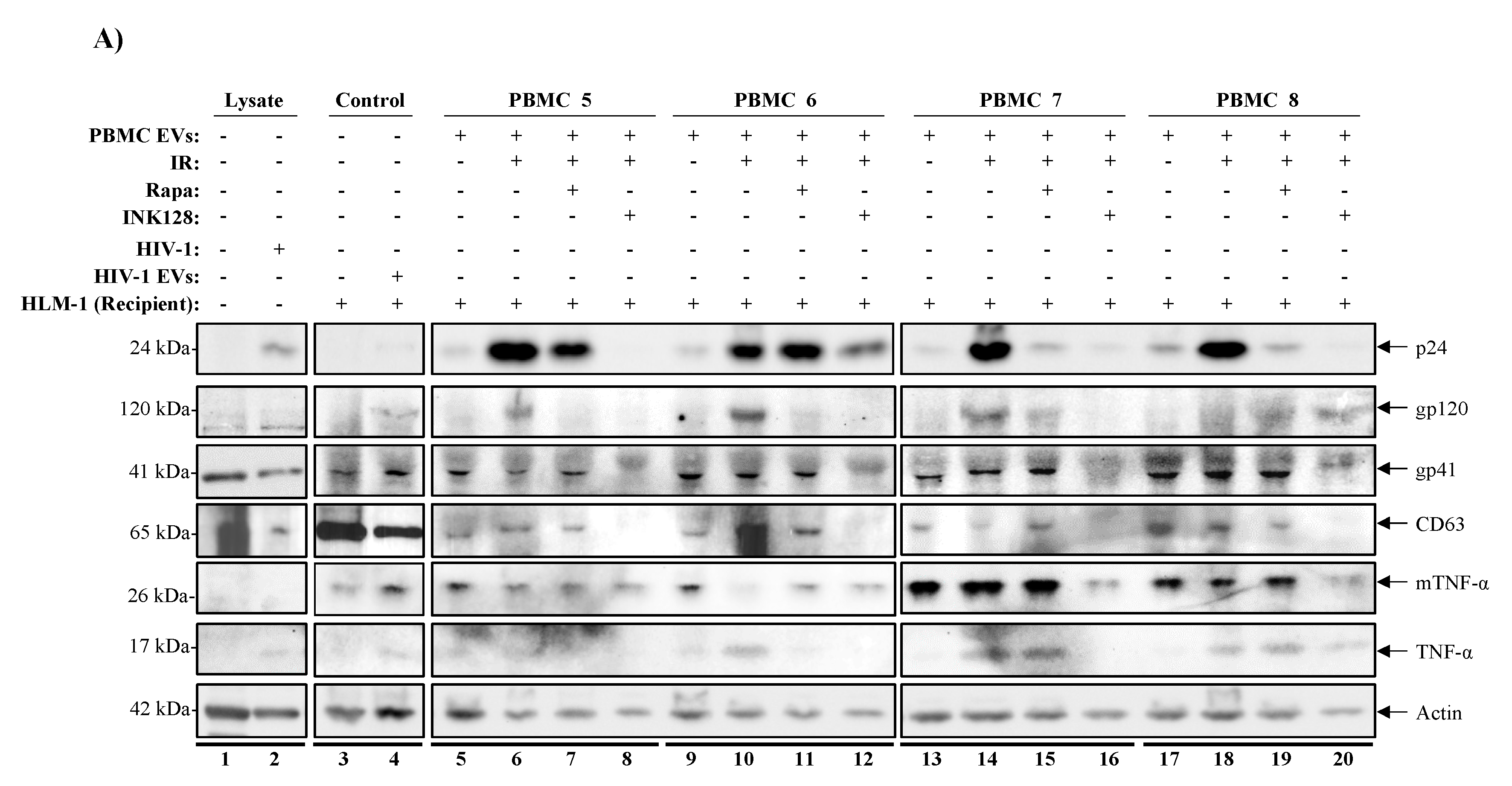
3.6. Inhibition of mTOR Suppresses TNF-α and Viral Protein Expression in Evs from Latently HIV-1-Infected Primary Cells
3.7. mTORi Drugs Mitigate TNF-α Mediated HIV-1 Promoter Activation
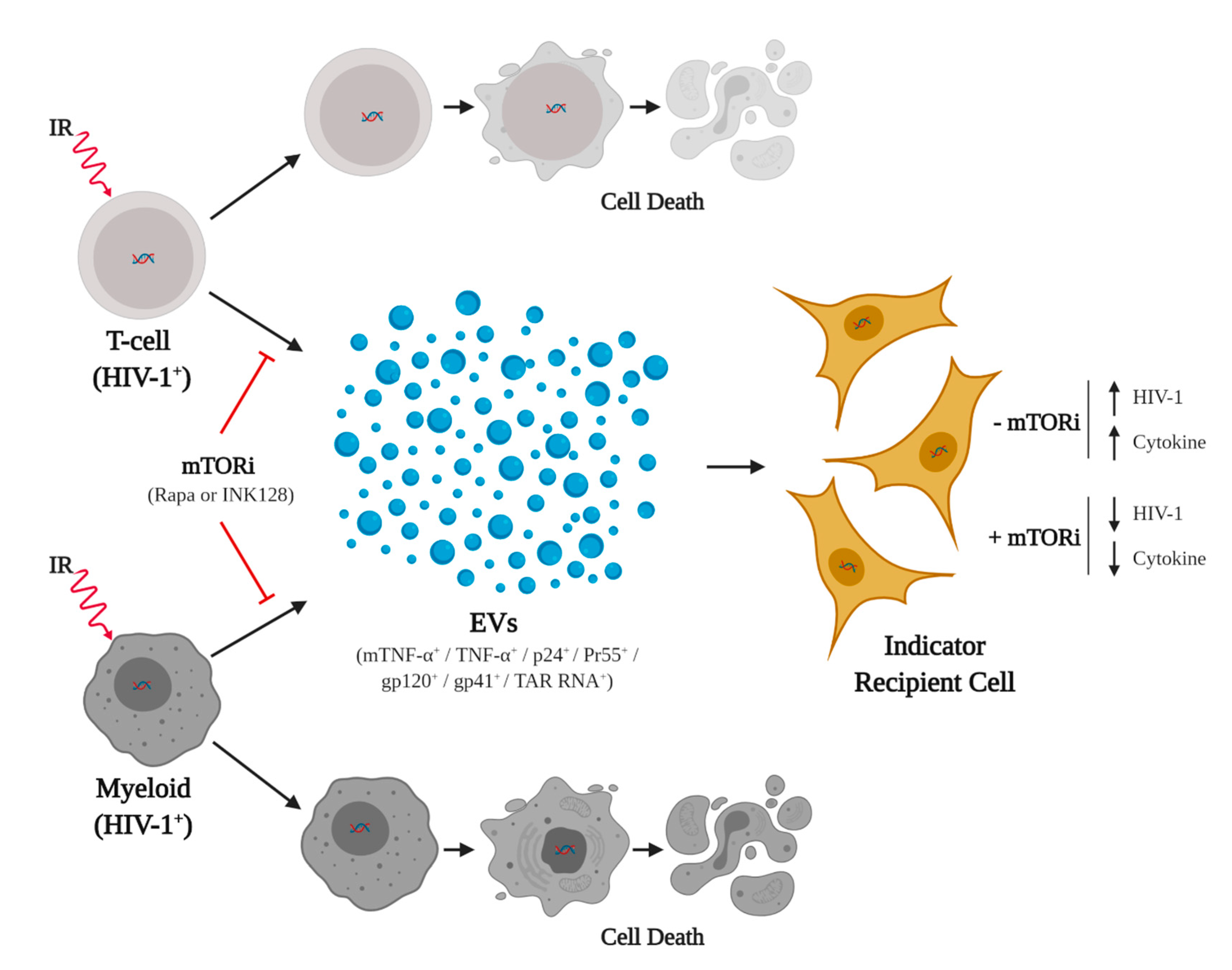
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Global AIDS Update 2019; UNAIDS: Geneva, Switzerland, 2019; p. 316.
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-based measures of viral persistence are Associated with immune activation and programmed cell death protein 1 (PD-1)–expressing CD4+ T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. PNAS 2016, 113, 8783–8788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iordanskiy, S.; Van Duyne, R.; Sampey, G.C.; Woodson, C.M.; Fry, K.; Saifuddin, M.; Guo, J.; Wu, Y.; Romerio, F.; Kashanchi, F.; et al. Therapeutic doses of irradiation activate viral transcription and induce apoptosis in HIV-1 infected cells. Virology 2015, 485, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, J.A.; Spencer, B.; Swinton, M.; Qvale, E.M.; Marquine, M.J.; Alexeeva, A.; Gough, S.; Soontornniyomkij, B.; Valera, E.; Masliah, E.; et al. Alterations in brain TREM2 and Amyloid-β levels are associated with neurocognitive impairment in HIV-infected persons on antiretroviral therapy. J. Neurochem. 2018. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.M.; Borodowsky, I.; Fernandez, B.; Gonzalez, L.; Kumar, M. Human immunodeficiency virus type 1 RNA levels in different regions of human brain: Quantification using real-time reverse transcriptase-polymerase chain reaction. J. Neuro Virol. 2007, 13, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.B.; Kozakewich, E.; Clouser, R.; Kershaw, C.; Hale, A.J. Occam’s razor need not apply: Advanced HIV infection presenting with five simultaneous opportunistic infections and central nervous system lymphoma. ID Cases 2018, 13, e00437. [Google Scholar] [CrossRef]
- Price, R.W. Neurological complications of HIV infection. Lancet 1996, 348, 445–452. [Google Scholar] [CrossRef]
- Robertson, K.; Oladeji, B.; Jiang, H.; Kumwenda, J.; Supparatpinyo, K.; Campbell, T.; Hakim, J.; Tripathy, S.; Hosseinipour, M.; Marra, C.M.; et al. HIV-1 and TB Co-infection in multinational resource limited settings: Increased neurological dysfunction. Clin. Infect. Dis. 2018. [Google Scholar] [CrossRef]
- Rajasuriar, R.; Khoury, G.; Kamarulzaman, A.; French, M.A.; Cameron, P.U.; Lewin, S.R. Persistent immune activation in chronic HIV infection: Do any interventions work? AIDS 2013, 27, 1199–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chege, D.; Sheth, P.M.; Kain, T.; Kim, C.J.; Kovacs, C.; Loutfy, M.; Halpenny, R.; Kandel, G.; Chun, T.W.; Ostrowski, M.; et al. Sigmoid Th17 populations, the HIV latent reservoir, and microbial translocation in men on long-term antiretroviral therapy. AIDS 2011, 25, 741–749. [Google Scholar] [CrossRef]
- Hunt, P.W.; Cao, H.L.; Muzoora, C.; Ssewanyana, I.; Bennett, J.; Emenyonu, N.; Kembabazi, A.; Neilands, T.B.; Bangsberg, D.R.; Deeks, S.G.; et al. Impact of CD8+ T-cell activation on CD4+ T-cell recovery and mortality in HIV-infected Ugandans initiating antiretroviral therapy. AIDS 2011, 25, 2123–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandler, N.G.; Wand, H.; Roque, A.; Law, M.; Nason, M.C.; Nixon, D.E.; Pedersen, C.; Ruxrungtham, K.; Lewin, S.R.; Emery, S.; et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J. Infect. Dis. 2011, 203, 780–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyrer, C.; Pozniak, A. HIV Drug resistance-An emerging threat to epidemic control. N. Engl. J. Med. 2017, 377, 1605–1607. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Liu, K.; Liu, H.; Hu, Y.; Zhang, Z.; Qin, J.; Xu, Q.; Peng, K.; Jin, X.; Wang, J.H.; et al. Trend of HIV-1 drug resistance in China: A systematic review and meta-analysis of data accumulated over 17 years (2001–2017). E Clin. Med. 2020, 18, 100238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ances, B.M.; Ellis, R.J. Dementia and neurocognitive disorders due to HIV-1 infection. Semin. Neurol. 2007, 27, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Gelman, B.B. Neuropathology of HAND with suppressive antiretroviral therapy: Encephalitis and neurodegeneration reconsidered. Curr. HIV/AIDS Rep. 2015, 12, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Celis, V.; Valiente-Echeverría, F.; Soto-Rifo, R.; Toro-Ascuy, D. New challenges of HIV-1 infection: How HIV-1 attacks and resides in the central nervous system. Cells 2019, 8, 1245. [Google Scholar] [CrossRef] [Green Version]
- Baldeweg, T.; Catalan, J.; Pugh, K.; Gruzelier, J.; Lovett, E.; Scurlock, H.; Burgess, A.; Riccio, M.; Hawkins, D. Neurophysiological changes associated with psychiatric symptoms in HIV-infected individuals without AIDS. Biol. Psychiatry 1997, 41, 474–487. [Google Scholar] [CrossRef]
- Akpamagbo, Y.A.; DeMarino, C.; Pleet, M.L.; Schwab, A.; Rodriguez, M.; Barclay, R.A.; Sampey, G.; Iordanskiy, S.; El-Hage, N.; Kashanchi, F.; et al. HIV-1 transcription inhibitors increase the synthesis of viral non-coding RNA that contribute to latency. Curr. Pharm. Des. 2017, 23, 4133–4144. [Google Scholar] [CrossRef]
- Carpio, L.; Klase, Z.; Coley, W.; Guendel, I.; Choi, S.; Van Duyne, R.; Narayanan, A.; Kehn-Hall, K.; Meijer, L.; Kashanchi, F.; et al. MicroRNA machinery is an integral component of drug-induced transcription inhibition in HIV-1 infection. J. RNAi Gene Silenc. 2010, 6, 386–400. [Google Scholar]
- De Marino, C.; Pleet, M.L.; Cowen, M.; Barclay, R.A.; Akpamagbo, Y.; Erickson, J.; Ndembe, N.; Charurat, M.; Jumare, J.; Bwala, S.; et al. Antiretroviral drugs alter the content of extracellular vesicles from HIV-1-infected cells. Sci. Rep. 2018, 8, 1–20. [Google Scholar]
- Klase, Z.; Kale, P.; Winograd, R.; Gupta, M.V.; Heydarian, M.; Berro, R.; McCaffrey, T.; Kashanchi, F. HIV-1 TAR element is processed by dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol. Biol. 2007, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected cells stimulate production of pro-inflammatory cytokines through trans-activating response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.O.; Scott, T.A.; DeMarino, C.; Pleet, M.L.; Vo, T.T.; Saifuddin, M.; Kovalskyy, D.; Erickson, J.; Cowen, M.; Barclay, R.A.; et al. Effect of transcription inhibition and generation of suppressive viral non-coding RNAs. Retrovirology 2019, 16, 13. [Google Scholar] [CrossRef]
- Margolis, D.M.; Garcia, J.V.; Hazuda, D.J.; Haynes, B.F. Latency reversal and viral clearance to cure HIV-1. Science 2016, 353. [Google Scholar] [CrossRef] [Green Version]
- Mbonye, U.; Karn, J. The Molecular basis for human immunodeficiency virus latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Søgaard, O.S. Reversal of latency as part of a cure for HIV-1. Trends Microbiol. 2016, 24, 90–97. [Google Scholar] [CrossRef]
- Richman, D.D.; Margolis, D.M.; Delaney, M.; Greene, W.C.; Hazuda, D.; Pomerantz, R.J. The challenge of finding a cure for HIV infection. Science 2009, 323, 1304–1307. [Google Scholar] [CrossRef] [Green Version]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; Svensson, J.P.; Silva, I.T.D.; Cohn, L.B.; Gramatica, A.; Deeks, S.; Greene, W.C.; Pillai, S.K.; et al. Distinct Chromatin Functional States Correlate with HIV Latency Reactivation in Infected Primary CD4+ T cells. Available online: https://elifesciences.org/articles/34655 (accessed on 15 August 2018).
- Pouget, J.P.; Georgakilas, A.G.; Ravanat, J.L. Targeted and off-target (bystander and abscopal) effects of radiation therapy: Redox mechanisms and risk/benefit analysis. Antioxid Redox Signal 2018, 29, 1447–1487. [Google Scholar] [CrossRef]
- Wozny, A.S.; Vares, G.; Alphonse, G.; Lauret, A.; Monini, C.; Magné, N.; Cuerq, C.; Fujimori, A.; Monboisse, J.C.; Beuve, M.; et al. ROS production and distribution: A new paradigm to explain the differential effects of X-ray and carbon ion irradiation on cancer stem cell migration and invasion. Cancers 2019, 11, 468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Zhou, T.; He, F.; Rong, Y.; Lee, S.H.; Wu, S.; Zuo, L. Reactive oxygen species formation and bystander effects in gradient irradiation on human breast cancer cells. Oncotarget 2016, 7, 41622–41636. [Google Scholar] [CrossRef] [Green Version]
- Ao, Z.; Zhu, R.; Tan, X.; Liu, L.; Chen, L.; Liu, S.; Yao, X. Activation of HIV-1 expression in latently infected CD4+ T cells by the small molecule PKC412. Virol. J. 2016, 13, 177. [Google Scholar] [CrossRef] [Green Version]
- Barton, K.; Hiener, B.; Winckelmann, A.; Rasmussen, T.A.; Shao, W.; Byth, K.; Lanfear, R.; Solomon, A.; McMahon, J.; Harrington, S.; et al. Broad activation of latent HIV-1 in vivo. Nat. Commun. 2016, 7, 12731. [Google Scholar] [CrossRef]
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Driessche, B.V.; Corazza, F.; Gatot, J.S.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-aza-2’-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8, 117–138. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+JQ1 and ingenol-B+JQ1 to potently reactivate viral gene expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef]
- Gutiérrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Pérez-Elías, M.J.; Martín, M.E.; Barbas, C.; Ruipérez, J.; Muñoz, E.; Muñoz-Fernández, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef]
- Quivy, V.; Lint, C.V. Diversity of acetylation targets and roles in transcriptional regulation: The human immunodeficiency virus type 1 promoter as a model system. Biochem. Pharmacol. 2002, 64, 925–934. [Google Scholar] [CrossRef]
- Schwartz, C.; Bouchat, S.; Marban, C.; Gautier, V.; Van Lint, C.; Rohr, O.; Le Douce, V. On the way to find a cure: Purging latent HIV-1 reservoirs. Biochem. Pharmacol. 2017, 146, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; Bosque, A. Targeting cellular and tissue HIV reservoirs with toll-like receptor agonists. Front. Immunol. 2019, 10, 2450. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, I.; Hashemi, F.B. Strategies to eradicate HIV from infected patients: Elimination of latent provirus reservoirs. Cell. Mol. Life Sci. 2019, 76, 3583–3600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavigner, M.; Watkins, B.; Lawson, B.; Lee, S.T.; Chahroudi, A.; Kean, L.; Silvestri, G. Persistence of virus reservoirs in ART-treated SHIV-infected Rhesus Macaques after autologous hematopoietic stem cell Ttransplant. PLoS Pathog. 2014, 10, e1004406. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.O.; DeMarino, C.; Pleet, M.L.; Cowen, M.; Branscome, H.; Al Sharif, S.; Jones, J.; Dutartre, H.; Lepene, B.; Liotta, L.A.; et al. HTLV-1 extracellular vesicles promote cell-to-cell contact. Front. Microbiol. 2019, 10, 2147. [Google Scholar] [CrossRef] [PubMed]
- Alongi, F.; Giaj-Levra, N.; Sciascia, S.; Fozza, A.; Fersino, S.; Fiorentino, A.; Mazzola, R.; Ricchetti, F.; Buglione, M.; Buonfrate, D.; et al. Radiotherapy in patients with HIV: Current issues and review of the literature. Lancet Oncol. 2017, 18, e379–e393. [Google Scholar] [CrossRef]
- Iordanskiy, S.; Kashanchi, F. Potential of radiation-induced cellular stress for reactivation of latent HIV-1 and killing of infected cells. AIDS Res. Hum. Retrovir. 2016, 32, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Carter, C.A.; Ehrlich, L.S. Cell biology of HIV-1 infection of macrophages. Annu. Rev. Microbiol. 2008, 62, 425–443. [Google Scholar] [CrossRef]
- Jones, G.; Power, C. Regulation of neural cell survival by HIV-1 infection. Neurobiol. Dis. 2006, 21, 1–17. [Google Scholar] [CrossRef]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Abbas, W.; Tariq, M.; Iqbal, M.; Kumar, A.; Herbein, G. Eradication of HIV-1 from the macrophage reservoir: An uncertain goal? Viruses 2015, 7, 1578–1598. [Google Scholar] [CrossRef] [Green Version]
- Avalos, C.R.; Abreu, C.M.; Queen, S.E.; Li, M.; Price, S.; Shirk, E.N.; Engle, E.L.; Forsyth, E.; Bullock, B.T.; Mac Gabhann, F.; et al. Brain macrophages in simian immunodeficiency virus-infected, antiretroviral-suppressed macaques: A functional latent reservoir. MBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Araínga, M.; Edagwa, B.; Mosley, R.L.; Poluektova, L.Y.; Gorantla, S.; Gendelman, H.E. A mature macrophage is a principal HIV-1 cellular reservoir in humanized mice after treatment with long acting antiretroviral therapy. Retrovirology 2017, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Garcia, J.V.; et al. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, X.; Lai, F.; Lin, J.; Zhou, C.; Xu, X.; Tan, X.; Liu, S.; Li, L. A novel bromodomain inhibitor, CPI-203, serves as an HIV-1 latency-reversing agent by activating positive transcription elongation factor b. Biochem. Pharmacol. 2019, 164, 237–251. [Google Scholar] [CrossRef]
- Prins, J.M.; Jurriaans, S.; van Praag, R.M.; Blaak, H.; van Rij, R.; Schellekens, P.T.; ten Berge, I.J.; Yong, S.L.; Fox, C.H.; Roos, M.T.; et al. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS 1999, 13, 2405–2410. [Google Scholar] [CrossRef]
- Xing, S.; Siliciano, R.F. Targeting HIV latency: Pharmacologic strategies toward eradication. Drug Discov. Today 2013, 18, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.R.; Pollack, R.A.; Capoferri, A.; Ambinder, R.F.; Durand, C.M.; Siliciano, R.F. Rapamycin-mediated mTOR inhibition uncouples HIV-1 latency reversal from cytokine-associated toxicity. J. Clin. Invest. 2017, 127, 651–656. [Google Scholar] [CrossRef] [Green Version]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA. 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef] [Green Version]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.; Upton, H.; Bao, K.; Schulze-Gahmen, U.; Samelson, A.J.; He, N.; Nowak, A.; Lu, H.; Krogan, N.J.; Zhou, Q.; et al. HIV-1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold. Proc. Natl. Acad. Sci. USA 2013, 110, E123–E131. [Google Scholar] [CrossRef] [Green Version]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Endo-Munoz, L.; Warby, T.; Harrich, D.; McMillan, N.A.J. Phosphorylation of HIV Tat by PKR increases interaction with TAR RNA and enhances transcription. Virol. J. 2005, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Bieniasz, P.D.; Grdina, T.A.; Bogerd, H.P.; Cullen, B.R. Recruitment of cyclin T1/P-TEFb to an HIV type 1 long terminal repeat promoter proximal RNA target is both necessary and sufficient for full activation of transcription. Proc. Natl. Acad. Sci. USA 1999, 96, 7791–7796. [Google Scholar] [CrossRef] [Green Version]
- Ammosova, T.; Berro, R.; Jerebtsova, M.; Jackson, A.; Charles, S.; Klase, Z.; Southerland, W.; Gordeuk, V.R.; Kashanchi, F.; Nekhai, S.; et al. Phosphorylation of HIV-1 Tat by CDK2 in HIV-1 transcription. Retrovirology 2006, 3, 78. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.P. The HIV-1 tat protein: Mechanism of action and target for HIV-1 cure strategies. Curr. Pharm. Des. 2017, 23, 4098–4102. [Google Scholar] [CrossRef] [Green Version]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR complex controls HIV latency. Cell. Host. Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Li, Z.; Xue, Y.; Schulze-Gahmen, U.; Johnson, J.R.; Krogan, N.J.; Alber, T.; Zhou, Q. AFF1 is a ubiquitous P-TEFb partner to enable Tat extraction of P-TEFb from 7SK snRNP and formation of SECs for HIV transactivation. Proc. Natl. Acad. Sci. USA 2014, 111, E15–E24. [Google Scholar] [CrossRef] [Green Version]
- Asamitsu, K.; Fujinaga, K.; Okamoto, T. HIV Tat/P-TEFb interaction: A potential target for novel Anti-HIV therapies. Molecules 2018, 23, 933. [Google Scholar] [CrossRef] [Green Version]
- Budhiraja, S.; Famiglietti, M.; Bosque, A.; Planelles, V.; Rice, A.P. Cyclin T1 and CDK9 T-loop phosphorylation are downregulated during establishment of HIV-1 latency in primary resting memory CD4+ T cells. J. Virol. 2013, 87, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, A.; Sampey, G.; Van Duyne, R.; Guendel, I.; Kehn-Hall, K.; Roman, J.; Currer, R.; Galons, H.; Oumata, N.; Joseph, B.; et al. Use of ATP analogs to inhibit HIV-1 transcription. Virology 2012, 432, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Van Duyne, R.; Guendel, I.; Jaworski, E.; Sampey, G.; Klase, Z.; Chen, H.; Zeng, C.; Kovalskyy, D.; el Kouni, M.H.; Lepene, B.; et al. Effect of mimetic CDK9 inhibitors on HIV-1 activated transcription. J. Mol. Biol. 2013, 425, 812–829. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, S.; Batisse, J.; Libre, C.; Bernacchi, S.; Marquet, R.; Paillart, J.C. HIV-1 replication and the cellular eukaryotic translation apparatus. Viruses 2015, 7, 199–218. [Google Scholar] [CrossRef] [Green Version]
- Batool, A.; Majeed, S.T.; Aashaq, S.; Majeed, R.; Bhat, N.N.; Andrabi, K.I. Eukaryotic initiation factor 4E is a novel effector of mTORC1 signaling pathway in cross talk with Mnk1. Mol. Cell. Biochem. 2020, 465, 13–26. [Google Scholar] [CrossRef]
- Tsukumo, Y.; Alain, T.; Fonseca, B.D.; Nadon, R.; Sonenberg, N. Translation control during prolonged mTORC1 inhibition mediated by 4E-BP3. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Klann, K.; Tascher, G.; Münch, C. Functional translatome proteomics reveal converging and dose-dependent regulation by mTORC1 and eIF2α. Mol. Cell 2019. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in Eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Feng, X.; Molinolo, A.A.; Martin, D.; Vitale-Cross, L.; Nohata, N.; Ando, M.; Wahba, A.; Amornphimoltham, P.; Wu, X.; et al. 4E-BP1 is a tumor suppressor protein reactivated by mTOR inhibition in head and neck cancer. Cancer Res. 2019, 79, 1438–1450. [Google Scholar] [CrossRef] [Green Version]
- De Breyne, S.; Ohlmann, T. Focus on translation initiation of the HIV-1 mRNAs. Int. J. Mol. Sci. 2018, 20, 101. [Google Scholar] [CrossRef] [Green Version]
- Ohlmann, T.; Mengardi, C.; López-Lastra, M. Translation initiation of the HIV-1 mRNA. Transl. Austin 2014, 2. [Google Scholar] [CrossRef]
- Monette, A.; Valiente-Echeverría, F.; Rivero, M.; Cohen, É.A.; Lopez-Lastra, M.; Mouland, A.J. Dual mechanisms of translation initiation of the full-length HIV-1 mRNA contribute to gag synthesis. PLoS ONE 2013, 8, e68108. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000 Res. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human immunodeficiency virus type 1 nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [Green Version]
- Fields, J.; Dumaop, W.; Eleuteri, S.; Elueteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; et al. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: Implications for HIV-associated neurocognitive disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [Green Version]
- Fields, J.A.; Metcalf, J.; Overk, C.; Adame, A.; Spencer, B.; Wrasidlo, W.; Florio, J.; Rockenstein, E.; He, J.J.; Masliah, E.; et al. The anticancer drug sunitinib promotes autophagyand protects from neurotoxicity in an HIV-1 Tat model of neurodegeneration. J. Neurovirol. 2017, 23, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.K.; Kaminski, R.; Mullen, B.; Gordon, J.; Burdo, T.H.; Cheung, J.Y.; Feldman, A.M.; Madesh, M.; Khalili, K. HIV-1 Nef-induced cardiotoxicity through dysregulation of autophagy. Sci. Rep. 2017, 7, 8572. [Google Scholar] [CrossRef]
- Lapierre, J.; Rodriguez, M.; Ojha, C.R.; El-Hage, N. Critical role of beclin1 in HIV tat and morphine-induced inflammation and calcium release in glial Cells from autophagy deficient mouse. J. Neuroimmun. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Leonard, J.A.; Filzen, T.; Carter, C.C.; Schaefer, M.; Collins, K.L. HIV-1 nef disrupts intracellular trafficking of major histocompatibility complex class I, CD4, CD8, and CD28 by distinct pathways that share common elements. J. Virol. 2011, 85, 6867–6881. [Google Scholar] [CrossRef] [Green Version]
- Leymarie, O.; Lepont, L.; Berlioz-Torrent, C. Canonical and non-canonical autophagy in HIV-1 replication cycle. Viruses 2017, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog. Neurobiol. 2013, 106–107, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Sardo, L.; Iordanskiy, S.; Klase, Z.; Kashanchi, F. HIV-1 Nef blocks autophagy in human astrocytes. Cell Cycle 2015, 14, 3781–3782. [Google Scholar] [CrossRef]
- Saribas, A.S.; Khalili, K.; Sariyer, I.K. Dysregulation of autophagy by HIV-1 Nef in human astrocytes. Cell Cycle 2015, 14, 2899–2904. [Google Scholar] [CrossRef] [Green Version]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; de Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.W.; Lee, S.W.; Myung, H. Induced degradation of Tat by nucleocapsid (NC) via the proteasome pathway and its effect on HIV transcription. Viruses 2013, 5, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Nardacci, R.; Ciccosanti, F.; Marsella, C.; Ippolito, G.; Piacentini, M.; Fimia, G.M. Role of autophagy in HIV infection and pathogenesis. J. Intern. Med. 2017, 281, 422–432. [Google Scholar] [CrossRef]
- Pleet, M.L.; Branscome, H.; DeMarino, C.; Pinto, D.O.; Zadeh, M.A.; Rodriguez, M.; Sariyer, I.K.; El-Hage, N.; Kashanchi, F. Autophagy, EVs, and infections: A perfect question for a perfect time. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.G.; Vassiliou, L.V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yang, J.; Yang, L. Insights for oxidative stress and mTOR signaling in myocardial ischemia/reperfusion injury under diabetes. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Gu, L.; Smerin, D.; Mao, S.; Xiong, X. The Interrelation between Reactive Oxygen Species and Autophagy in Neurological Disorders. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Kobayashi, T.; Nishioka, A.; Kariya, S.; Hamasato, S.; Seguchi, H.; Yoshida, S. Radiation-induced reactive oxygen species formation prior to oxidative DNA damage in human peripheral T cells. Int. J. Mol. Med. 2003, 11, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Kandadi, M.R.; Yu, X.; Frankel, A.E.; Ren, J. Cardiac-specific catalase overexpression rescues anthrax lethal toxin-induced cardiac contractile dysfunction: Role of oxidative stress and autophagy. BMC Med. 2012, 10, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Deng, J.; Ye, Z.; Wang, J.; Gou, H.; Liu, W.; Zhao, M.; Liao, M.; Yi, L.; Chen, J. Absence of autophagy promotes apoptosis by modulating the ROS-dependent RLR signaling pathway in classical swine fever virus-infected cells. Autophagy 2016, 12, 1738–1758. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Sharma, M.; Pandey, R.; Saluja, D. ROS is the major player in regulating altered autophagy and lifespan in sin-3 mutants of C. elegans. Autophagy 2018, 14, 1239–1255. [Google Scholar] [CrossRef] [Green Version]
- Deori, N.M.; Kale, A.; Maurya, P.K.; Nagotu, S. Peroxisomes: Role in cellular ageing and age related disorders. Biogerontology 2018, 19, 303–324. [Google Scholar] [CrossRef]
- Hester, A.K.; Semwal, M.K.; Xiao, Y.; Cepeda, S.; Venables, T.; Griffith, A.V. Catalase expression mediates redox regulation of autophagy and promiscuous gene expression in thymic stromal cells. J. Immunol. 2017, 198, 202–211. [Google Scholar]
- Walton, P.A.; Brees, C.; Lismont, C.; Apanasets, O.; Fransen, M. The peroxisomal import receptor PEX5 functions as a stress sensor, retaining catalase in the cytosol in times of oxidative stress. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 1833–1843. [Google Scholar] [CrossRef]
- Ahsan, N.A.; Sampey, G.C.; Lepene, B.; Akpamagbo, Y.; Barclay, R.A.; Iordanskiy, S.; Hakami, R.M.; Kashanchi, F. Presence of viral RNA and proteins in exosomes from cellular clones resistant to rift valley fever virus infection. Front. Microbiol. 2016, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.R.; Pleet, M.L.; Enose-Akahata, Y.; Erickson, J.; Monaco, M.C.; Akpamagbo, Y.; Velluci, A.; Tanaka, Y.; Azodi, S.; Lepene, B.; et al. Viral antigens detectable in CSF exosomes from patients with retrovirus associated neurologic disease: Functional role of exosomes. Clin. Transl. Med. 2018, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Pleet, M.L.; Mathiesen, A.; DeMarino, C.; Akpamagbo, Y.A.; Barclay, R.A.; Schwab, A.; Iordanskiy, S.; Sampey, G.C.; Lepene, B.; Nekhai, S.; et al. Ebola VP40 in exosomes can cause immune cell dysfunction. Front. Microbiol. 2016, 7, 1765. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Feng, Z.; Yue, H.; Bazdar, D.; Mbonye, U.; Zender, C.; Harding, C.V.; Bruggeman, L.; Karn, J.; Sieg, S.F.; et al. Exosomes derived from HIV-1-infected cells promote growth and progression of cancer via HIV TAR RNA. Nat. Commun. 2018, 9, 4585. [Google Scholar] [CrossRef] [Green Version]
- Ojha, C.R.; Lapierre, J.; Rodriguez, M.; Dever, S.M.; Zadeh, M.A.; DeMarino, C.; Pleet, M.L.; Kashanchi, F.; El-Hage, N. Interplay between autophagy, exosomes and HIV-1 associated neurological disorders: New insights for diagnosis and therapeutic applications. Viruses 2017, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A system of cytokines encapsulated in extracellular vesicles. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, E.; Narayanan, A.; van Duyne, R.; Shabbeer-Meyering, S.; Iordanskiy, S.; Saifuddin, M.; Das, R.; Afonso, P.V.; Sampey, G.C.; Chung, M.; et al. Human T-lymphotropic virus type 1-infected cells secrete exosomes that contain tax protein. J. Biol. Chem. 2014, 289, 22284–22305. [Google Scholar] [CrossRef] [Green Version]
- Barclay, R.A.; Schwab, A.; DeMarino, C.; Akpamagbo, Y.; Lepene, B.; Kassaye, S.; Iordanskiy, S.; Kashanchi, F. Exosomes from uninfected cells activate transcription of latent HIV-1. J. Biol. Chem. 2017, 292, 14764. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, W.; Gomez-Lopez, N.; Erez, O.; Romero, R.; Margolis, L. Extracellular vesicles generated by placental tissues ex vivo: A transport system for immune mediators and growth factors. Am. J. Reprod. Immunol. 2018, 80, e12860. [Google Scholar] [CrossRef]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, E.; Saifuddin, M.; Sampey, G.; Shafagati, N.; van Duyne, R.; Iordanskiy, S.; Kehn-Hall, K.; Liotta, L.; Petricoin, E.; Young, M.; et al. The use of nanotrap particles technology in capturing HIV-1 virions and viral proteins from infected cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- DeMarino, C.; Barclay, R.A.; Pleet, M.L.; Pinto, D.O.; Branscome, H.; Paul, S.; Lepene, B.; El-Hage, N.; Kashanchi, F. Purification of high yield extracellular vesicle preparations away from virus. J. Vis. Exp. 2019, 151, e59876. [Google Scholar] [CrossRef]
- Evans, M.K.; Lepkowski, J.M.; Powe, N.R.; LaVeist, T.; Kuczmarski, M.F.; Zonderman, A.B. Healthy aging in neighborhoods of diversity across the life span (HANDLS): Overcoming barriers to implementing a longitudinal, epidemiologic, urban study of health, race, and socioeconomic status. Ethn. Dis. 2010, 20, 267–275. [Google Scholar]
- Noren Hooten, N.; Abdelmohsen, K.; Gorospe, M.; Ejiogu, N.; Zonderman, A.B.; Evans, M.K. MicroRNA expression patterns reveal differential expression of target genes with age. PLoS ONE 2010, 5, e10724. [Google Scholar] [CrossRef] [Green Version]
- Gale, M.; Tan, S.L.; Katze, M.G. Translational control of viral gene expression in eukaryotes. Microbiol. Mol. Biol. Rev. 2000, 64, 239–280. [Google Scholar] [CrossRef] [Green Version]
- Buono, R.; Longo, V.D. Starvation, stress resistance, and cancer. Trends Endocrinol. Metab. 2018, 29, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Brandhorst, S.; Harputlugil, E.; Mitchell, J.R.; Longo, V.D. Protective effects of short-term dietary restriction in surgical stress and chemotherapy. Ageing. Res. Rev. 2017, 39, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. MTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.C.; Schuldiner, O.; Neufeld, T.P. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 2004, 7, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochaba, J.; Powers, A.F.; Tremble, K.A.; Greenlee, S.; Post, N.M.; Matson, J.E.; MacLeod, A.R.; Guo, S.; Aghajan, M. A novel and translational role for autophagy in antisense oligonucleotide trafficking and activity. Nucl. Acids Res. 2019, 47, 11284–11303. [Google Scholar] [CrossRef]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK signaling pathway in renal fibrosis. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, E.V.; Hosokawa, H.; Ungerbäck, J. Mechanisms of action of hematopoietic transcription factor PU.1 in initiation of T-cell development. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Glimcher, L.H.; Singh, H. Transcription factors in lymphocyte development—T and B cells get together. Cell 1999, 96, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Awad, R.M.; de Vlaeminck, Y.; Maebe, J.; Goyvaerts, C.; Breckpot, K. Turn back the TIMe: Targeting tumor infiltrating myeloid cells to revert cancer progression. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Ren, J.P.; Zhao, J.; Dai, J.; Griffin, J.W.D.; Wang, L.; Wu, X.Y.; Morrison, Z.D.; Li, G.Y.; Gazzar, M.E.; Ning, S.B.; et al. Hepatitis C virus-induced myeloid-derived suppressor cells regulate T-cell differentiation and function via the signal transducer and activator of transcription 3 pathway. Immunology 2016, 148, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Pleet, M.L.; Erickson, J.; DeMarino, C.; Barclay, R.A.; Cowen, M.; Lepene, B.; Liang, J.; Kuhn, J.H.; Prugar, L.; Stonier, S.W.; et al. Ebola Virus VP40 Modulates Cell Cycle and Biogenesis of Extracellular Vesicles. J. Infect. Dis. 2018. [Google Scholar] [CrossRef]
- Sadaie, M.R.; Tschachler, E.; Valerie, K.; Rosenberg, M.; Felber, B.K.; Pavlakis, G.N.; Klotman, M.E.; Wong-Staal, F. Activation of tat-defective human immunodeficiency virus by ultraviolet light. New Biol. 1990, 2, 479–486. [Google Scholar]
- Sadale, M.R.; Hager, G.L. Induction of Developmentally Programmed Cell Death and Activation of HIV by Sodium Butyrate. Virology 1994, 202, 513–518. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Qian, H. Biological effects of radiation on cancer cells. Mil. Med. Res. 2018, 5. [Google Scholar] [CrossRef]
- Ghiglione, Y.; Turk, G. Nef performance in macrophages: The master orchestrator of viral persistence and spread. Curr. HIV Res. 2011, 9, 505–513. [Google Scholar] [CrossRef]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 Latency in Monocytes/Macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-Y.; Zheng, Y.-H.; He, Y.; Chen, Z.; He, B. The role of autophagy in THP-1 macrophages resistance to HIV- vpr-induced apoptosis. Exp. Cell Res. 2017, 351, 68–73. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [Green Version]
- El-Naseery, N.I.; Mousa, H.S.E.; Noreldin, A.E.; El-Far, A.H.; Elewa, Y.H.A. Aging-associated immunosenescence via alterations in splenic immune cell populations in rat. Life Sci. 2020, 241, 117168. [Google Scholar] [CrossRef]
- Zhou, F.; Shen, Q.; Claret, F.X. Novel roles of reactive oxygen species in the pathogenesis of acute myeloid leukemia. J. Leukoc. Biol. 2013, 94, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; MacEwan, D.J. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood 2008, 111, 3793–3801. [Google Scholar] [CrossRef] [Green Version]
- Sillar, J.R.; Germon, Z.P.; De Iuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Raja, R.; Farooqui, S.R.; Ahmad, S.; Banerjea, A.C. USP7 deubiquitinase controls HIV-1 production by stabilizing Tat protein. Biochem. J. 2017, 474, 1653–1668. [Google Scholar] [CrossRef]
- Deng, L.; de la Fuente, C.; Fu, P.; Wang, L.; Donnelly, R.; Wade, J.D.; Lambert, P.; Li, H.; Lee, C.G.; Kashanchi, F. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology 2000, 277, 278–295. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.E.; Banks, C.A.S.; Shilatifard, A.; Conaway, J.W.; Conaway, R.C. ELL-associated factors 1 and 2 are positive regulators of RNA polymerase II elongation factor ELL. Proc. Natl. Acad. Sci. USA 2005, 102, 10094–10098. [Google Scholar] [CrossRef] [Green Version]
- Nolte-’t Hoen, E.; Cremer, T.; Gallo, R.C.; Margolis, L.B. Extracellular vesicles and viruses: Are they close relatives? Proc. Natl. Acad. Sci. USA 2016, 113, 9155–9161. [Google Scholar] [CrossRef] [Green Version]
- Altman, J.K.; Sassano, A.; Kaur, S.; Glaser, H.; Kroczynska, B.; Redig, A.J.; Russo, S.; Barr, S.; Platanias, L.C. Dual mTORC2/mTORC1 Targeting Results in Potent Suppressive Effects on Acute Myeloid Leukemia (AML) Progenitors. Clin. Cancer Res. 2011, 17, 4378–4388. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.Y.; Sykes, D.B.; Ameri, S.; Kalaitzidis, D.; Charles, J.F.; Nelson-Maney, N.; Wei, K.; Cunin, P.; Morris, A.; Cardona, A.E.; et al. The metabolic regulator mTORC1 controls terminal myeloid differentiation. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]
- Willems, L.; Chapuis, N.; Puissant, A.; Maciel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202. [Google Scholar] [CrossRef] [Green Version]
- Garibaldi, C.; Jereczek-Fossa, B.A.; Marvaso, G.; Dicuonzo, S.; Rojas, D.P.; Cattani, F.; Starzyńska, A.; Ciardo, D.; Surgo, A.; Leonardi, M.C.; et al. Recent advances in radiation oncology. Ecancermedicalscience 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Bentzen, S.M. Preventing or reducing late side effects of radiation therapy: Radiobiology meets molecular pathology. Nat. Rev. Cancer 2006, 6, 702–713. [Google Scholar] [CrossRef]
- Falcke, S.E.; Rühle, P.F.; Deloch, L.; Fietkau, R.; Frey, B.; Gaipl, U.S. Clinically Relevant Radiation Exposure Differentially Impacts Forms of Cell Death in Human Cells of the Innate and Adaptive Immune System. Int. J. Mol. Sci. 2018, 19, 3574. [Google Scholar] [CrossRef] [Green Version]
- Jaffray, D.A.; Gospodarowicz, M.K. Radiation Therapy for Cancer. In Cancer: Disease Control Priorities, 3rd ed.Gelband, H., Jha, P., Sankaranarayanan, R., Horton, S., Eds.; The International Bank for Reconstruction and Development/The World Bank: Washington, DC, USA, 2015; Volume 3, ISBN 978-1-4648-0349-9. [Google Scholar]
- Singh, V.K.; Romaine, P.L.P.; Newman, V.L. Biologics as countermeasures for acute radiation syndrome: Where are we now? Expert Opin. Biol. Ther. 2015, 15, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Tezuka, K.; Xun, R.; Tei, M.; Ueno, T.; Tanaka, M.; Takenouchi, N.; Fujisawa, J. An animal model of adult T-cell leukemia: Humanized mice with HTLV-1-specific immunity. Blood 2014, 123, 346–355. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | Sample Type | HIV-1 | cART; Years | Hepatitis B | Hepatitis C | Collection Date |
|---|---|---|---|---|---|---|---|
| 1 | Female | PBMC | +*2 | +; n/a | - | - | 13 September 2017 |
| 2 | Male | PBMC | +#8 | +; n/a | - | - | 20 September 2017 |
| 3 | Female | PBMC | +#5 | +; 11.7 | - | + | 4 October2017 |
| 4 | Female | PBMC | + | +; 14.9 | - | - | 8 November2017 |
| 5 | Female | PBMC | + | +; 9.4 | - | - | 10 January 2018 |
| 6 | Male | PBMC | +#14 | +; 12 | - | - | 12 February 2018 |
| 7 | Male | PBMC | + | +; n/a | - | + | 1 March2018 |
| 8 | Female | PBMC | +#3 | +; 10.3 | - | + | 1 March 2018 |
| ID | Lot Number | Sex | Age | Ethnicity | Sample Type | Collection Date |
|---|---|---|---|---|---|---|
| 9 | 2010113879 | Male | 26 | African American | PBMC | 4 September 2019 |
| 10 | 2010113875 | Male | 26 | Hispanic/Latino | PBMC | 28 August 2019 |
| 11 | 2010113854 | Female | 42 | Caucasian | PBMC | 26 July 2019 |
| 12 | 2010113876 | Female | 43 | Caucasian | PBMC | 27 August 2019 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, D.O.; DeMarino, C.; Vo, T.T.; Cowen, M.; Kim, Y.; Pleet, M.L.; Barclay, R.A.; Noren Hooten, N.; Evans, M.K.; Heredia, A.; et al. Low-Level Ionizing Radiation Induces Selective Killing of HIV-1-Infected Cells with Reversal of Cytokine Induction Using mTOR Inhibitors. Viruses 2020, 12, 885. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080885
Pinto DO, DeMarino C, Vo TT, Cowen M, Kim Y, Pleet ML, Barclay RA, Noren Hooten N, Evans MK, Heredia A, et al. Low-Level Ionizing Radiation Induces Selective Killing of HIV-1-Infected Cells with Reversal of Cytokine Induction Using mTOR Inhibitors. Viruses. 2020; 12(8):885. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080885
Chicago/Turabian StylePinto, Daniel O., Catherine DeMarino, Thy T. Vo, Maria Cowen, Yuriy Kim, Michelle L. Pleet, Robert A. Barclay, Nicole Noren Hooten, Michele K. Evans, Alonso Heredia, and et al. 2020. "Low-Level Ionizing Radiation Induces Selective Killing of HIV-1-Infected Cells with Reversal of Cytokine Induction Using mTOR Inhibitors" Viruses 12, no. 8: 885. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080885