Viral Metagenomic Profiling of Croatian Bat Population Reveals Sample and Habitat Dependent Diversity

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
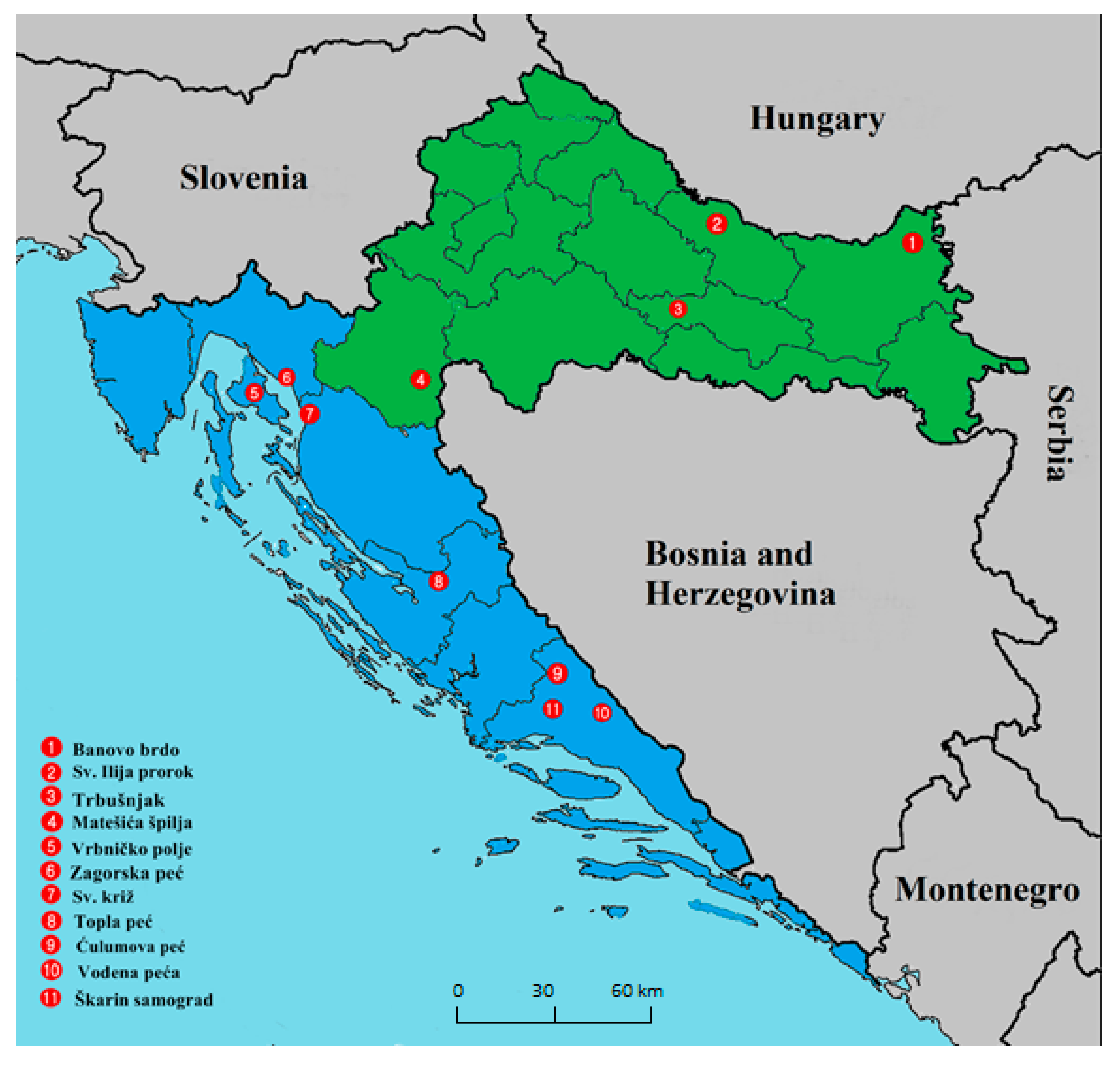
2.1. Bats, Locations, Sampling, and Ethics Statement
2.2. Sample Preparation and Viral Nucleic Acid Extraction
2.3. Library Construction and Nextera XT Illumina Sequencing
2.4. Viral Metagenomic Profiling
2.5. Complete Viral Genome Assembly, Identification and Taxonomic Classification
2.6. Phylogenetic Analyses
2.7. Statistical Analysis
3. Results
3.1. Bats, Locations and Sampling
3.2. Viral Metagenomic Profiling
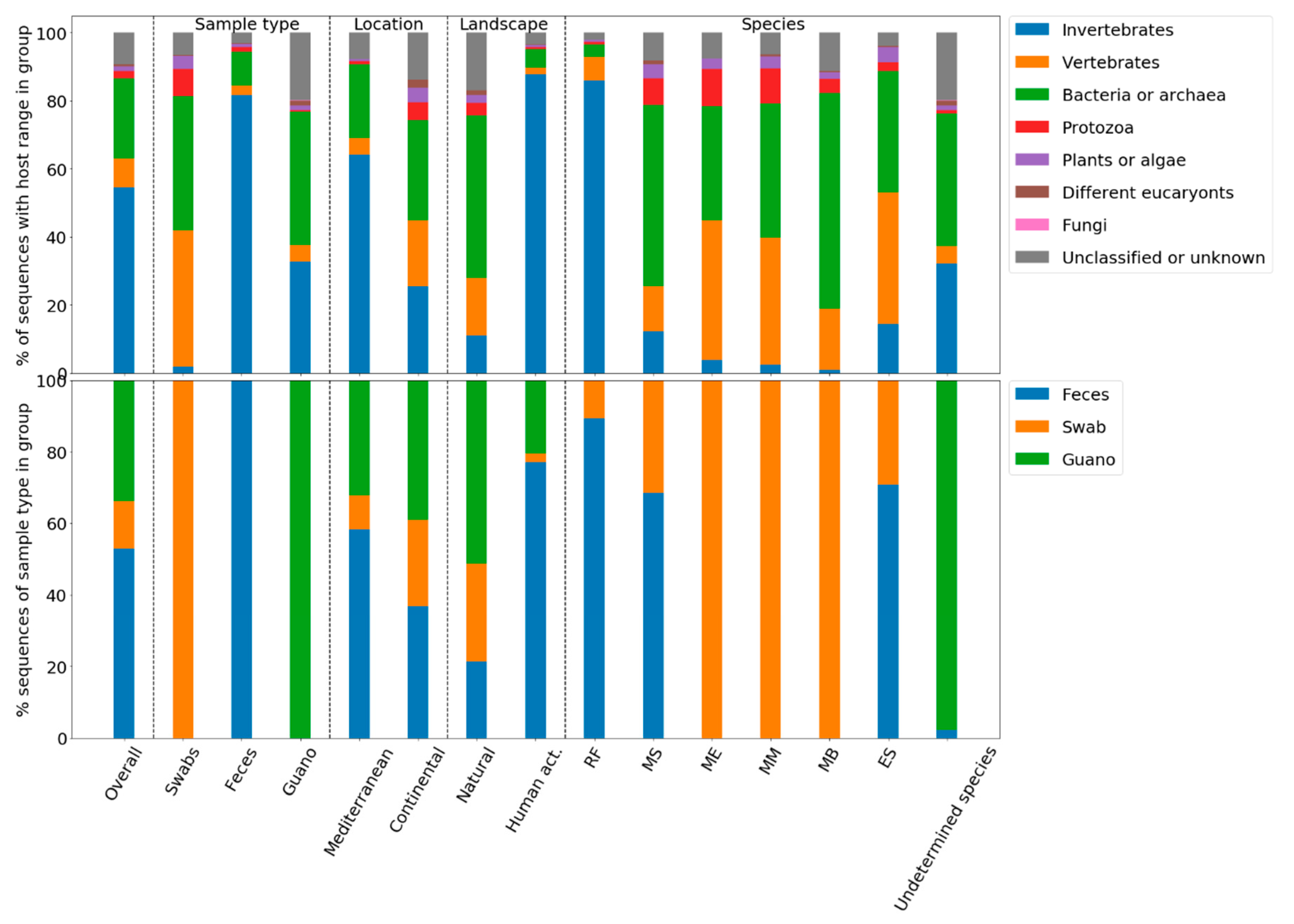
3.3. Eukaryotic Versus Prokaryotic Viruses
3.4. Viral Metagenomic Profiling Revealed Presence of 63 Viral Families; Most Common Were Viruses Infecting Eukaryotes–Insects
3.5. Vertebrate Viruses
Taxonomical Families of Vertebrate Viruses Interesting Due to Zoonotic Potential
3.6. Three Complete and One Nearly Complete Genome Sequences of Novel Viruses Were Identified
3.6.1. Iflavirus
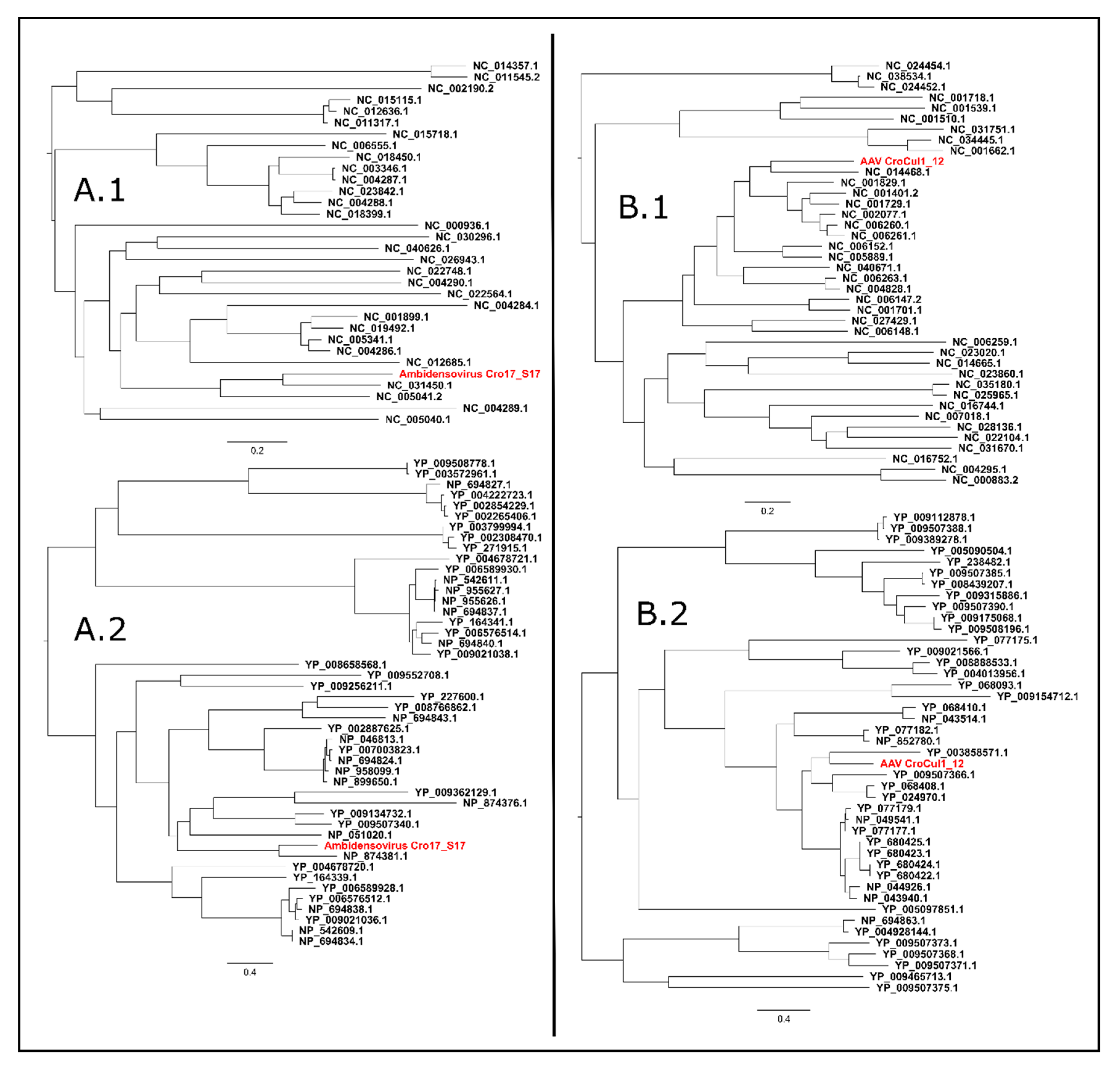
3.6.2. Ambidensovirus
3.6.3. Adeno-Associated Virus (AAV)
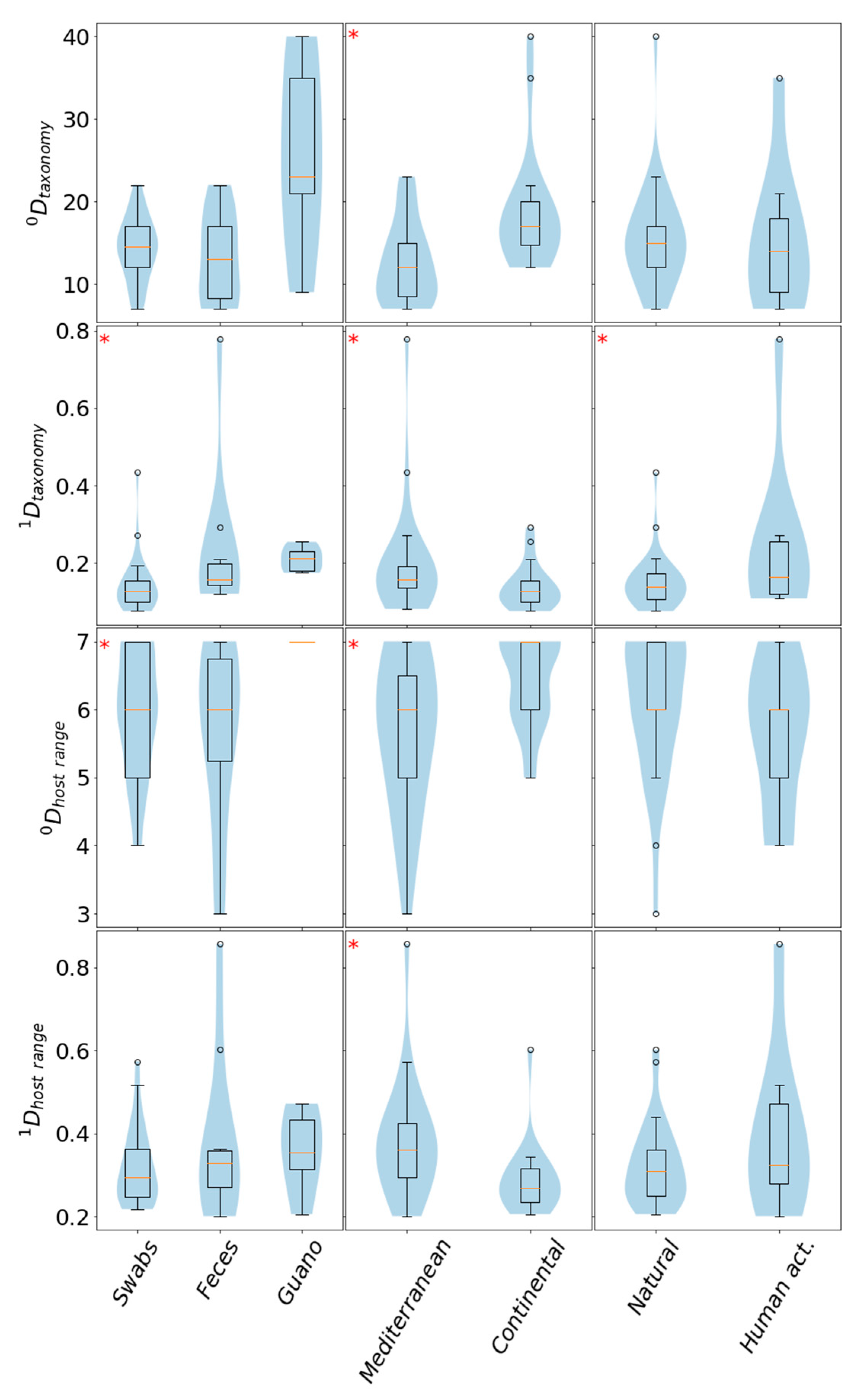
3.7. Quantitative Analysis of Diversity and the Viral Compositions of Sample Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Racey, P.A. The uniqueness of bats. In Bats and Viruses: A New Frontier of Emerging Infectious Diseases; Wang, L.-F., Cowled, C., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Dietz, C.; Kiefer, A. Bats of Britain and Europe; Bloomsbury: London, UK, 2016. [Google Scholar]
- Jones, G.; Jacobs, D.S.; Kunz, T.H.; Wilig, M.R.; Racey, P.A. Carpe noctem: The importance of bats as bioindicators. Endanger. Species Res. 2009, 8, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Lina, P.H.C. Common Names of European Bats; NEP/EUROBATS Secretariat: Bonn, Germany, 2016. [Google Scholar]
- Official Gazette of the Republic of Croatia. Ordinance on Strictly Protected Species. Available online: https://narodne-novine.nn.hr/clanci/sluzbeni/2013_12_144_3086.html (accessed on 14 August 2020).
- Bern Convention. Available online: https://www.coe.int/en/web/bern-convention (accessed on 14 August 2020).
- Ministry of Environmental and Nature Protection of the Republic of Croatia, Nature Protection Directorate & State Institute for Nature Protection. Inf.EUROBATS.MoP7.12—Sixth National Report on the Implementation of the Agreement; Ministry of Environmental and Nature Protection of the Republic of Croatia, Nature Protection Directorate & State Institute for Nature Protection: Zagreb, Croatia, 2014. [Google Scholar]
- Tvrtković, N. The findings of Mehely’s horseshoe bat (Chiroptera) in Croatia in the last century were mistakes in identification. Nat. Croat. 2016, 25, 165–172. [Google Scholar] [CrossRef]
- Jones, K.; Patel, N.; Levy, M.; Storeygard, A.; Balk, D.; Gittleman, J.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 44, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C. Viruses in bats: A historic review. In Bats and Viruses: A New Frontier of Emerging Infectious Diseases; Wang, L.-F., Cowled, C., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Kuzmin, I.V.; Rupprecht, C.E. Bat Lyssaviruses. In Bats and Viruses: A New Frontier of Emerging Infectious Diseases; Wang, L.-F., Cowled, C., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 47–98. [Google Scholar]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlottau, K.; Rissmann, M.; Graaf, A.; Schön, J.; Sehl, J.; Wylezich, C.; Höper, C.; Mettenleiter, D.; Balkema-Buschmann, T.; Harder, A. Experimental Transmission Studies of SARS-CoV-2 in Fruit Bats, Ferrets, Pigs and Chickens. SSRN Electron. J. 2020, 5247. [Google Scholar] [CrossRef]
- Olival, K.; Weekley, C.; Daszak, P. Are bats really “special” as viral reservoirs? What we know and need to know. In Bats and Viruses: A New Frontier of Emerging Infectious Diseases; Wang, L.-F., Cowled, C., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 281–295. [Google Scholar]
- Heneberg, Đ.; Bakić, J.; Heneberg, N.; Nikolić, B.; Agoli, B.; Hronovsky, V.; Dusbabek, F.; Kolman, J.; Blažek, K.; Bakota, M. Ekološko-medicinska ispitivanja pećina dalmatinskog krša. Zb. Vojnomed. Akad. 1968, 43–47. [Google Scholar]
- Pavlinić, I.; Đaković, M.; Lojkić, I. Pseudogymnoascus destructans in Croatia confirmed. Eur. J. Wildl. Res. 2015, 61, 325–328. [Google Scholar] [CrossRef]
- Šimić, I.; Lojkić, I.; Krešić, N.; Cliquet, F.; Picard-Meyer, E.; Wasniewski, M.; Ćukušić, A.; Zrnčić, V.; Bedeković, T. Molecular and serological survey of lyssaviruses in Croatian bat populations. BMC Vet. Res. 2018, 14, 274. [Google Scholar] [CrossRef]
- Pavlinić, I.; Čač, Ž.; Lojkić, I.; Đaković, M.; Bedeković, T.; Lojkić, M. Šišmiši biološki rezervoari i potencijalni prijenosnici lyssavirusa. Vet. Stanica 2009, 40, 297–304. [Google Scholar]
- Croatia Map Blank. Available online: https://hr.wikipedia.org/wiki/Datoteka:Croatia_map_blank.png (accessed on 14 August 2020).
- Banyard, A.C.; Hayman, D.T.; Freuling, C.M.; Muller, T.; Fooks, A.R.; Johnson, N. Bat Rabies. In Rabies: Scientific Basis of the Disease and Its Management; Jackson, A.C., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 215–268. [Google Scholar]
- Lojkić, I.; Bidin, M.; Prpić, J.; Šimić, I.; Krešić, N.; Bedeković, T. Faecal virome of red foxes from peri-urban areas. Comp. Immunol. Microbiol. Infect. Dis. 2016, 45. [Google Scholar] [CrossRef]
- GenBank Non-Redundant Protein Database. Available online: ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz (accessed on 14 August 2020).
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Diamond BLASTx. Available online: https://github.com/bbuchfink/diamond (accessed on 14 August 2020).
- Huson, D.; Mitra, S.; Ruscheweyh, H.; Weber, N.; Schuster, S. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basic Local Alignment Search Tool. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 14 August 2020).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2, Improving the Ultrafast Bootstrap Approximation. Molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.O. Diversity and Evenness: A Unifying Notation and Its Consequences. Ecology 1973, 54, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Kruskal, W.H.; Wallis, W.A. Use of Ranks in One-Criterion Variance Analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Šimić, I.; Zorec, T.M.; Krešić, N.; Poljak, M.; Bedeković, T.; Lojkić, I. Novel Circo-Like Virus Detected in a Croatian Bat Population. Microbiol. Resour. Announc. 2019, 8, 18–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Li, Z.; Yang, F.; Zheng, J.; Feng, Y.; Guo, H.; Li, Y.; Wang, Y.; Su, N.; Zhang, F.; et al. Virome Profiling of Bats from Myanmar by Metagenomic Analysis of Tissue Samples Reveals More Novel Mammalian Viruses. PLoS ONE 2013, 8, 61950. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Salmier, A.; Tirera, S.; De Thoisy, B.; Franc, A.; Darcissac, E.; Donato, D.; Bouchier, C.; Lacoste, V.; Lavergne, A. Virome analysis of two sympatric bat species (Desmodus rotundus and Molossus molossus) in French Guiana. PLoS ONE 2017, 12, e0186943. [Google Scholar] [CrossRef] [Green Version]
- Maceljski, M. Poljoprivredna Entomologija; Zrinski: Čakovec, Croatia, 2002. [Google Scholar]
- Yang, W.T.; Shi, S.H.; Jiang, Y.L.; Zhao, L.; Chen, H.L.; Huang, K.Y.; Yang, G.L.; Wang, C.F. Genetic characterization of a densovirus isolated from great tit (Parus major) in China. Infect. Genet. Evol. 2016, 41, 107–112. [Google Scholar] [CrossRef]
- Ge, X.; Li, Y.; Yang, X.; Zhang, H.; Zhou, P.; Zhang, Y.; Shi, Z. Metagenomic Analysis of Viruses from Bat Fecal Samples Reveals Many Novel Viruses in Insectivorous Bats in China. J. Virol. 2012, 86, 4620–4630. [Google Scholar] [CrossRef] [Green Version]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.M.; Hughes, J.; Mietasch, M.; Modha, S.; Ogliastro, M.; Penzes, J.J.; et al. ICTV virus taxonomy profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef]
- Chen, L.; Liu, B.; Yang, J.; Jin, Q. DBatVir: The database of bat-associated viruses. Database 2014, 2014, bau021. [Google Scholar] [CrossRef] [Green Version]
- Berns, K.; Parrish, C. Parvoviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1768–1792. [Google Scholar]
- Genus: Dependoparvovirus. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/ssdna-viruses/w/parvoviridae/1043/genus-dependoparvovirus (accessed on 14 August 2020).
- Castrignano, S.B.; Nagasse-Sugahara, T.K.; Garrafa, P.; Monezi, T.A.; Barrella, K.M.; Mehnert, D.U. Identification of circo-like virus-Brazil genomic sequences in raw sewage from the metropolitan area of São Paulo: Evidence of circulation two and three years after the first detection. Mem. Do Inst. Oswaldo Cruz 2017, 112, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Castrignano, S.B.; Nagasse-Sugahara, T.K.; Kisielius, J.J.; Ueda-Ito, M.; Brandão, P.E.; Curti, S.P. Two novel circo-like viruses detected in human feces: Complete genome sequencing and electron microscopy analysis. Virus Res. 2013, 178, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat Guano Virome: Predominance of Dietary Viruses from Insects and Plants plus Novel Mammalian Viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic Analysis of the Viromes of Three North American Bat Species: Viral Diversity among Different Bat Species That Share a Common Habitat. J. Virol. 2010, 84, 13004–13018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panonska Nizina. Available online: http://enciklopedija.hr/Natuknica.aspx?ID=46451 (accessed on 14 August 2020).
- Biswas, S.R.; Mallik, A.U. Disturbance effects on species diversity and functional diversity in Riparian and Upland Plant Communities. Ecology 2010, 91, 28–35. [Google Scholar] [CrossRef]
- Victoria, J.G.; Kapoor, A.; Li, L.; Blinkova, O.; Slikas, B.; Wang, C.; Naeem, A.; Zaidi, S.; Delwart, E. Metagenomic Analyses of Viruses in Stool Samples from Children with Acute Flaccid Paralysis. J. Virol. 2009, 83, 4642–4651. [Google Scholar] [CrossRef] [Green Version]
- Tonjes, R.R.; Niebert, M. Relative Age of Proviral Porcine Endogenous Retrovirus Sequences in Sus scrofa Based on the Molecular Clock Hypothesis. J. Virol. 2003, 77, 12363–12368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzian, C.; Pélisson, A.; Bucheton, A. Evolution and phylogeny of insect endogenous retroviruses. BMC Evol. Biol. 2001, 1, 3. [Google Scholar] [CrossRef]
- Belshaw, R.; Pereira, V.; Katzourakis, A.; Talbot, G.; Pačes, J.; Burt, A.; Tristem, M. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2004, 101, 4894–4899. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.N.; Hooley, P.; Roden, D.; Ejtehadi, H.D.; Rylance, P.; Warren, P.; Martin, J.; Murray, P.G. Human endogenous retroviruses: Transposable elements with potential? Clin. Exp. Immunol. 2004, 138, 1–9. [Google Scholar] [CrossRef]
- Garcia-Etxebarria, K.; Garcia-Garcerà, M.; Calafell, F. Consistency of metagenomic assignment programs in simulated and real data. BMC Bioinform. 2014, 15, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | All Sequences | % of Total Sequences | Viral Sequences | % Viral Sequences in Category | % of Total Viral Sequences | No. Samples | % of Samples | 0DTax. Family | 1DTax. Family | 0DHost Range | 1DHost Range |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample type | |||||||||||
| feces | 171,700 | 10.97 | 19,951 | 11.62 | 50.47 | 10 | 18.61 | 30 | 0.457 | 6 | 0.558 |
| guano | 286,222 | 18.28 | 14,377 | 5.02 | 36.37 | 5 | 16.28 | 56 | 0.103 | 7 | 0.346 |
| swab | 1,107,621 | 70.75 | 5199 | 0.47 | 13.15 | 28 | 65.12 | 39 | 0.080 | 6 | 0.312 |
| Location type | |||||||||||
| M | 428,175 | 27.35 | 29,747 | 6.95 | 75.26 | 23 | 53.49 | 45 | 0.243 | 7 | 0.427 |
| C | 1,137,368 | 72.65 | 9780 | 0.86 | 24.74 | 20 | 46.51 | 58 | 0.055 | 6 | 0.227 |
| Landscape type | |||||||||||
| natural | 1,242,052 | 79.33 | 17,066 | 1.37 | 43.18 | 34 | 79.07 | 61 | 0.081 | 7 | 0.309 |
| with human activity | 323,491 | 20.66 | 22,461 | 6.94 | 56.82 | 9 | 20.93 | 45 | 0.428 | 6 | 0.65 |
| Species | |||||||||||
| RF | 449,808 | 28.73 | 19,060 | 4.24 | 48.22 | 11 | 25.58 | 35 | 0.490 | 6 | 0.605 |
| MS | 347,268 | 22.18 | 1634 | 0.47 | 4.13 | 12 | 27.91 | 29 | 0.089 | 6 | 0.317 |
| MM | 267,273 | 17.07 | 955 | 0.36 | 2.42 | 4 | 9.30 | 23 | 0.088 | 6 | 0.294 |
| ME | 35,720 | 2.28 | 236 | 0.66 | 0.60 | 1 | 2.33 | 17 | 0.122 | 5 | 0.292 |
| MB | 36,312 | 2.32 | 380 | 1.05 | 0.96 | 3 | 6.98 | 13 | 0.164 | 6 | 0.444 |
| ES | 79,587 | 5.08 | 452 | 0.57 | 1.14 | 2 | 4.65 | 18 | 0.119 | 6 | 0.279 |
| Undetermined, other | 349,575 | 22.33 | 16,810 | 4.81 | 42.53 | 10 | 23.26 | 58 | 0.093 | 7 | 0.327 |
| Total/Overall | 1,565,543 | 100.00 | 39,527 | 2.52 | 100.00 | 43 | 100.00 | 63 | 0.149 | 7 | 0.342 |
| Families | Host Range | Genome Type | Overall (%) | Sample Type (%) | Geographical Location Group (%) | Landscape Type (%) | Bat Species (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | F | G | M | C | N | H | RF | MS | MM | ME | MB | ES | Undetermined/Mixed | ||||
| Iflaviridae | Invertebrates | (+)ssRNA | 44.663 | 0.058 | 81.424 | 9.780 | 57.391 | 5.951 | 2.385 | 76.786 | 85.236 | 0.061 | 0.000 | 0.424 | 0.000 | 0.000 | 8.364 |
| Siphoviridae | Bacteria or archaea | dsDNA | 9.591 | 12.849 | 2.381 | 18.418 | 10.599 | 6.524 | 20.755 | 1.109 | 1.233 | 19.523 | 7.435 | 5.932 | 27.895 | 12.832 | 17.775 |
| Podoviridae | Bacteria or archaea | dsDNA | 7.352 | 7.828 | 3.198 | 12.944 | 7.971 | 5.470 | 12.604 | 3.361 | 1.884 | 9.364 | 3.770 | 2.119 | 24.211 | 8.407 | 13.224 |
| Parvoviridae | Vertebrates, Unclassified or unknown, Invertebrates | ssDNA | 5.996 | 11.810 | 0.125 | 12.040 | 7.920 | 0.143 | 7.412 | 4.920 | 3.216 | 0.000 | 0.105 | 0.000 | 0.000 | 0.000 | 10.446 |
| Myoviridae | Bacteria or archaea | dsDNA | 4.048 | 9.021 | 2.667 | 4.166 | 2.901 | 7.536 | 8.702 | 0.512 | 0.488 | 12.179 | 10.785 | 15.678 | 8.684 | 5.752 | 6.597 |
| Retroviridae | Vertebrates, Invertebrates | ssRNA-RT | 2.333 | 14.464 | 0.526 | 0.452 | 0.995 | 6.401 | 4.442 | 0.730 | 1.821 | 11.016 | 21.675 | 26.271 | 10.263 | 3.761 | 0.416 |
| Mimiviridae | Protozoa, Unclassified or unknown | dsDNA | 1.728 | 7.540 | 1.178 | 0.390 | 0.635 | 5.051 | 3.475 | 0.401 | 0.624 | 9.058 | 9.634 | 8.475 | 3.421 | 2.434 | 1.666 |
| Ackermannviridae | Bacteria or archaea | dsDNA | 1.477 | 6.040 | 0.396 | 1.329 | 0.054 | 5.808 | 2.514 | 0.690 | 0.687 | 3.611 | 10.471 | 0.424 | 0.263 | 6.858 | 1.553 |
| Iridoviridae | Vertebrates, Invertebrates | dsDNA | 1.126 | 5.905 | 0.326 | 0.508 | 0.561 | 2.843 | 2.156 | 0.343 | 0.818 | 5.263 | 6.911 | 13.136 | 5.263 | 1.106 | 0.482 |
| Poxviridae | Vertebrates, Invertebrates | dsDNA | 1.017 | 3.405 | 0.762 | 0.508 | 0.205 | 3.487 | 2.150 | 0.156 | 0.273 | 3.488 | 5.131 | 0.847 | 2.368 | 29.425 | 0.595 |
| Phycodnaviridae | Plants or algae | dsDNA | 0.878 | 3.443 | 0.652 | 0.264 | 0.286 | 2.679 | 1.799 | 0.178 | 0.310 | 4.162 | 4.188 | 2.542 | 1.842 | 2.655 | 0.922 |
| Microviridae | Bacteria or archaea | ssDNA | 0.620 | 0.385 | 0.296 | 1.155 | 0.740 | 0.256 | 1.025 | 0.312 | 0.178 | 0.612 | 0.628 | 0.000 | 0.263 | 0.221 | 1.148 |
| Reoviridae | Vertebrates, Invertebrates | dsRNA | 0.615 | 0.519 | 0.030 | 1.461 | 0.000 | 2.485 | 1.371 | 0.040 | 0.010 | 0.122 | 2.094 | 0.000 | 0.000 | 1.991 | 1.249 |
| Permutotetraviridae | Unclassified or unknown, Invertebrates | (+)ssRNA | 0.531 | 0.000 | 1.033 | 0.028 | 0.703 | 0.010 | 0.000 | 0.935 | 1.081 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.024 |
| Inoviridae | Bacteria or archaea | ssDNA | 0.443 | 0.692 | 0.015 | 0.946 | 0.545 | 0.133 | 0.949 | 0.058 | 0.000 | 0.796 | 1.047 | 5.508 | 0.000 | 0.221 | 0.821 |
| Herpesviridae | Vertebrates | dsDNA | 0.369 | 1.808 | 0.236 | 0.035 | 0.128 | 1.104 | 0.820 | 0.027 | 0.073 | 2.999 | 2.618 | 0.847 | 0.000 | 1.327 | 0.297 |
| Picobirnaviridae | Vertebrates, Unclassified or unknown | dsRNA | 0.306 | 0.000 | 0.000 | 0.842 | 0.000 | 1.237 | 0.697 | 0.009 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.720 |
| Circoviridae | Vertebrates, Unclassified or unknown, Fungi, Invertebrates | ssDNA | 0.293 | 0.000 | 0.000 | 0.807 | 0.387 | 0.010 | 0.680 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.690 |
| Flaviviridae | Vertebrates | (+)ssRNA | 0.273 | 1.866 | 0.020 | 0.049 | 0.027 | 1.022 | 0.551 | 0.062 | 0.220 | 1.958 | 1.780 | 0.000 | 0.000 | 2.212 | 0.042 |
| Nodaviridae | Vertebrates, Unclassified or unknown, Invertebrates | (+)ssRNA | 0.202 | 0.135 | 0.000 | 0.508 | 0.013 | 0.777 | 0.258 | 0.160 | 0.031 | 0.061 | 0.000 | 0.000 | 0.000 | 0.000 | 0.434 |
| Dicistroviridae | Invertebrates | (+)ssRNA | 0.202 | 0.038 | 0.000 | 0.543 | 0.003 | 0.808 | 0.205 | 0.200 | 0.010 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.464 |
| Partitiviridae | Plants or algae | dsRNA | 0.190 | 0.000 | 0.165 | 0.292 | 0.141 | 0.337 | 0.182 | 0.196 | 0.173 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.250 |
| Totiviridae | Protozoa, Invertebrates | dsRNA | 0.187 | 0.000 | 0.341 | 0.042 | 0.229 | 0.061 | 0.035 | 0.303 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.440 |
| Astroviridae | Vertebrates | (+)ssRNA | 0.175 | 0.327 | 0.000 | 0.362 | 0.003 | 0.695 | 0.398 | 0.004 | 0.000 | 0.979 | 0.105 | 0.000 | 0.000 | 0.000 | 0.309 |
| Coronaviridae | Vertebrates | (+)ssRNA | 0.154 | 0.038 | 0.000 | 0.410 | 0.010 | 0.593 | 0.053 | 0.232 | 0.010 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.351 |
| Picornaviridae | Vertebrates | (+)ssRNA | 0.134 | 0.000 | 0.000 | 0.369 | 0.003 | 0.532 | 0.211 | 0.076 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.315 |
| Solemoviridae | Plants or algae | (+)ssRNA | 0.114 | 0.000 | 0.005 | 0.306 | 0.000 | 0.460 | 0.012 | 0.191 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.221 | 0.262 |
| Polydnaviridae | Invertebrates | dsDNA | 0.089 | 0.654 | 0.000 | 0.007 | 0.064 | 0.164 | 0.152 | 0.040 | 0.026 | 0.367 | 1.257 | 3.390 | 0.789 | 0.000 | 0.006 |
| Tymoviridae | Plants or algae | (+)ssRNA | 0.073 | 0.000 | 0.000 | 0.202 | 0.000 | 0.297 | 0.170 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.173 |
| Pithoviridae | Protozoa, Unclassified or unknown | dsDNA | 0.053 | 0.192 | 0.045 | 0.014 | 0.013 | 0.174 | 0.105 | 0.013 | 0.010 | 0.184 | 0.419 | 0.424 | 0.000 | 0.000 | 0.065 |
| Caulimoviridae | Plants or algae | dsDNA-RT | 0.051 | 0.308 | 0.005 | 0.021 | 0.040 | 0.082 | 0.105 | 0.009 | 0.021 | 0.428 | 0.000 | 0.000 | 0.263 | 1.106 | 0.018 |
| Virgaviridae | Plants or algae | (+)ssRNA | 0.048 | 0.000 | 0.000 | 0.132 | 0.000 | 0.194 | 0.111 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.113 |
| Baculoviridae | Invertebrates | dsDNA | 0.043 | 0.231 | 0.005 | 0.028 | 0.010 | 0.143 | 0.088 | 0.009 | 0.016 | 0.428 | 0.209 | 0.000 | 0.000 | 0.000 | 0.030 |
| Adenoviridae | Vertebrates, Unclassified or unknown | dsDNA | 0.040 | 0.077 | 0.060 | 0.000 | 0.020 | 0.102 | 0.076 | 0.013 | 0.016 | 0.061 | 0.000 | 0.000 | 0.000 | 0.000 | 0.071 |
| Metaviridae | Invertebrates | ssRNA-RT | 0.035 | 0.212 | 0.005 | 0.014 | 0.034 | 0.041 | 0.082 | 0.000 | 0.010 | 0.428 | 0.000 | 0.000 | 0.263 | 0.442 | 0.012 |
| Ascoviridae | Invertebrates | dsDNA | 0.030 | 0.115 | 0.020 | 0.014 | 0.010 | 0.092 | 0.064 | 0.004 | 0.005 | 0.184 | 0.209 | 0.000 | 0.000 | 0.000 | 0.036 |
| Luteoviridae | Plants or algae | (+)ssRNA | 0.025 | 0.000 | 0.010 | 0.056 | 0.000 | 0.102 | 0.018 | 0.031 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.442 | 0.048 |
| Tombusviridae | Plants or algae | (+)ssRNA | 0.025 | 0.000 | 0.000 | 0.070 | 0.000 | 0.102 | 0.006 | 0.040 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.059 |
| Alphatetraviridae | Invertebrates | (+)ssRNA | 0.023 | 0.019 | 0.000 | 0.056 | 0.003 | 0.082 | 0.041 | 0.009 | 0.005 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.048 |
| Peribunyaviridae | Different eucaryonts, Vertebrates | (-)ssRNA | 0.020 | 0.000 | 0.000 | 0.056 | 0.010 | 0.051 | 0.029 | 0.013 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.048 |
| Polycipiviridae | Invertebrates | (+)ssRNA | 0.020 | 0.000 | 0.000 | 0.056 | 0.020 | 0.020 | 0.023 | 0.018 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.048 |
| Bromoviridae | Plants or algae | (+)ssRNA | 0.015 | 0.000 | 0.000 | 0.042 | 0.000 | 0.061 | 0.035 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.036 |
| Phenuiviridae | Vertebrates, Unclassified or unknown, Invertebrates | (-)ssRNA | 0.015 | 0.000 | 0.000 | 0.042 | 0.000 | 0.061 | 0.035 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.036 |
| Alloherpesviridae | Vertebrates | dsDNA | 0.015 | 0.096 | 0.000 | 0.007 | 0.000 | 0.061 | 0.029 | 0.004 | 0.026 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 |
| Leviviridae | Bacteria or archaea | (+)ssRNA | 0.015 | 0.019 | 0.000 | 0.035 | 0.000 | 0.061 | 0.035 | 0.000 | 0.005 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.030 |
| Marseilleviridae | Protozoa, Unclassified or unknown | dsDNA | 0.013 | 0.077 | 0.005 | 0.000 | 0.003 | 0.041 | 0.018 | 0.009 | 0.005 | 0.061 | 0.209 | 0.424 | 0.000 | 0.000 | 0.000 |
| Nudiviridae | Invertebrates | dsDNA | 0.013 | 0.019 | 0.005 | 0.021 | 0.003 | 0.041 | 0.023 | 0.004 | 0.005 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.024 |
| Rhabdoviridae | Invertebrates, Plants or algae, Vertebrates, Unclassified or unknown | (-)ssRNA | 0.013 | 0.000 | 0.000 | 0.035 | 0.000 | 0.051 | 0.029 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.030 |
| Genomoviridae | Unclassified or unknown | ssDNA | 0.010 | 0.019 | 0.000 | 0.021 | 0.010 | 0.010 | 0.012 | 0.009 | 0.005 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.018 |
| Potyviridae | Plants or algae | (+)ssRNA | 0.008 | 0.058 | 0.000 | 0.000 | 0.003 | 0.020 | 0.012 | 0.004 | 0.005 | 0.000 | 0.105 | 0.424 | 0.000 | 0.000 | 0.000 |
| Birnaviridae | Vertebrates, Invertebrates | dsRNA | 0.008 | 0.000 | 0.000 | 0.021 | 0.000 | 0.031 | 0.018 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.018 |
| Malacoherpesviridae | Invertebrates | dsDNA | 0.008 | 0.058 | 0.000 | 0.000 | 0.007 | 0.010 | 0.012 | 0.004 | 0.000 | 0.061 | 0.105 | 0.424 | 0.000 | 0.000 | 0.000 |
| Secoviridae | Plants or algae | (+)ssRNA | 0.005 | 0.000 | 0.000 | 0.014 | 0.003 | 0.010 | 0.012 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.012 |
| Hepeviridae | Vertebrates | (+)ssRNA | 0.005 | 0.019 | 0.000 | 0.007 | 0.000 | 0.020 | 0.006 | 0.004 | 0.000 | 0.061 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 |
| Betaflexiviridae | Plants or algae | (+)ssRNA | 0.005 | 0.019 | 0.000 | 0.007 | 0.000 | 0.020 | 0.012 | 0.000 | 0.005 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 |
| Nanoviridae | Plants or algae | dsDNA | 0.003 | 0.000 | 0.000 | 0.007 | 0.003 | 0.000 | 0.006 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 |
| Spiraviridae | Unclassified or unknown | ssDNA | 0.003 | 0.000 | 0.000 | 0.007 | 0.000 | 0.010 | 0.006 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 |
| Caliciviridae | Vertebrates | (+)ssRNA | 0.003 | 0.000 | 0.000 | 0.007 | 0.000 | 0.010 | 0.006 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 |
| Bicaudaviridae | Bacteria or archaea | dsDNA | 0.003 | 0.000 | 0.005 | 0.000 | 0.000 | 0.010 | 0.006 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 |
| Autolykiviridae | Unclassified or unknown | dsDNA | 0.003 | 0.000 | 0.000 | 0.007 | 0.003 | 0.000 | 0.000 | 0.004 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 |
| Bornaviridae | Vertebrates | (-)ssRNA | 0.003 | 0.019 | 0.000 | 0.000 | 0.003 | 0.000 | 0.006 | 0.000 | 0.000 | 0.061 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Polyomaviridae | Vertebrates | dsDNA | 0.003 | 0.000 | 0.000 | 0.007 | 0.003 | 0.000 | 0.006 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 |
| Asfarviridae | Vertebrates | dsDNA | 0.003 | 0.019 | 0.000 | 0.000 | 0.003 | 0.000 | 0.006 | 0.000 | 0.000 | 0.061 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Unclassified or unknown | Bacteria or archaea, Vertebrates, Different eucaryonts, Plants or algae, Unclassified or unknown, Invertebrates, Protozoa | (-)ssRNA, Unclassified or unknown RNA, ssDNA, (+)ssRNA, dsDNA, dsRNA, Unclassified or unknown | 14.238 | 9.598 | 4.060 | 30.041 | 7.278 | 35.409 | 22.759 | 7.765 | 1.453 | 12.362 | 9.110 | 12.712 | 14.211 | 18.584 | 29.114 |
| No. | Landscape Type | Sample Type | Location | Location Group | Species | All Sequences | Viral Sequences (%) | 0DTax. Family | 1DTax. Family | 0DHost Range | 1DHost Range |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | N | F | 1 | C | MS | 7986 | 645 (8.077) | 22 | 0.210 | 7 | 0.335 |
| 2 | N | S | 1 | C | MS | 76,457 | 170 (0.222) | 15 | 0.090 | 6 | 0.307 |
| 3 | N | S | 1 | C | RF | 107,464 | 210 (0.195) | 17 | 0.109 | 6 | 0.218 |
| 4 | N | S | 9 | M | RF | 9988 | 747 (7.479) | 15 | 0.435 | 5 | 0.574 |
| 5 | N | S | 9 | M | MS | 2560 | 35 (1.367) | 7 | 0.192 | 5 | 0.414 |
| 6 | N | S | 9 | M | MB | 16,903 | 159 (0.941) | 12 | 0.138 | 6 | 0.314 |
| 7 | H | G | 2 | C | / | 78,222 | 1973 (2.522) | 35 | 0.255 | 7 | 0.315 |
| 8 | H | S | 2 | C | MM | 108,367 | 156 (0.144) | 16 | 0.112 | 6 | 0.280 |
| 9 | N | F | 4 | C | RF | 58,257 | 170 (0.292) | 12 | 0.292 | 6 | 0.603 |
| 10 | N | F | 4 | C | ES | 10,238 | 320 (3.126) | 17 | 0.158 | 6 | 0.269 |
| 11 | N | F | 4 | C | MS | 83,747 | 340 (0.406) | 20 | 0.154 | 7 | 0.236 |
| 12 | N | S | 4 | C | RF | 48,398 | 169 (0.349) | 12 | 0.152 | 6 | 0.320 |
| 13 | N | S | 4 | C | RF | 39,251 | 151 (0.385) | 14 | 0.134 | 7 | 0.262 |
| 14 | N | S | 4 | C | MS | 42,389 | 111 (0.262) | 17 | 0.076 | 7 | 0.248 |
| 15 | N | S | 4 | C | RF | 79,074 | 256 (0.324) | 20 | 0.093 | 7 | 0.253 |
| 16 | N | S | 4 | C | ES | 69,349 | 132 (0.19) | 15 | 0.081 | 7 | 0.227 |
| 17 | N | F | 11 | M | MS | 4168 | 15 (0.36) | 7 | 0.159 | 5 | 0.276 |
| 18 | N | F | 11 | M | MS | 6120 | 15 (0.245) | 8 | 0.137 | 3 | 0.364 |
| 19 | N | G | 11 | M | / | 23,648 | 1806 (7.637) | 23 | 0.214 | 7 | 0.435 |
| 20 | N | S | 11 | M | MS, MN | 39,015 | 157 (0.402) | 17 | 0.097 | 6 | 0.261 |
| 21 | N | S | 11 | M | MM | 40,471 | 262 (0.647) | 15 | 0.108 | 7 | 0.248 |
| 22 | N | S | 11 | M | MS | 7833 | 183 (2.336) | 10 | 0.194 | 5 | 0.440 |
| 23 | N | S | 11 | M | RF | 10,063 | 129 (1.282) | 14 | 0.145 | 6 | 0.362 |
| 24 | H | F | 7 | M | RF | 29,564 | 16,998 (57.496) | 7 | 0.779 | 4 | 0.857 |
| 25 | H | G | 7 | M | / | 10,662 | 2644 (24.798) | 21 | 0.232 | 7 | 0.472 |
| 26 | H | S | 7 | M | RF | 38,090 | 114 (0.299) | 8 | 0.272 | 5 | 0.517 |
| 27 | H | S | 7 | M | ME | 35,720 | 236 (0.661) | 18 | 0.109 | 6 | 0.247 |
| 28 | N | S | 8 | M | MB | 2860 | 13 (0.455) | 8 | 0.136 | 4 | 0.336 |
| 29 | N | S | 8 | M | MS | 13,045 | 196 (1.502) | 13 | 0.174 | 6 | 0.416 |
| 30 | N | F | 3 | C | MS | 43,707 | 2129 (4.871) | 17 | 0.140 | 7 | 0.345 |
| 31 | N | G | 3 | C | / | 42,904 | 1853 (4.319) | 40 | 0.175 | 7 | 0.206 |
| 32 | N | S | 3 | C | MS | 42,697 | 277 (0.649) | 19 | 0.095 | 7 | 0.230 |
| 33 | N | S | 3 | C | MM | 55,162 | 302 (0.547) | 20 | 0.102 | 7 | 0.235 |
| 34 | N | S | 3 | C | RF | 63,273 | 235 (0.371) | 16 | 0.106 | 6 | 0.267 |
| 35 | N | S | 3 | C | MS | 53,987 | 101 (0.187) | 14 | 0.125 | 5 | 0.285 |
| 36 | N | S | 3 | C | MM | 26,439 | 80 (0.303) | 14 | 0.130 | 5 | 0.333 |
| 37 | N | S | 10 | M | MB | 16,549 | 208 (1.257) | 11 | 0.156 | 7 | 0.351 |
| 38 | N | S | 10 | M | MS | 7997 | 160 (2.001) | 12 | 0.180 | 6 | 0.414 |
| 39 | H | F | 5 | M | MS, RF | 14,731 | 267 (1.813) | 14 | 0.121 | 6 | 0.202 |
| 40 | H | F | 5 | M | MS, RF | 4915 | 37 (0.753) | 9 | 0.165 | 6 | 0.326 |
| 41 | H | S | 5 | M | RF | 3220 | 36 (1.118) | 9 | 0.157 | 4 | 0.365 |
| 42 | N | G | 6 | M | / | 39,053 | 5116 (13.1) | 9 | 0.181 | 7 | 0.355 |
| 43 | N | S | 6 | M | MS | 51,000 | 214 (0.42) | 22 | 0.082 | 7 | 0.219 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimić, I.; Zorec, T.M.; Lojkić, I.; Krešić, N.; Poljak, M.; Cliquet, F.; Picard-Meyer, E.; Wasniewski, M.; Zrnčić, V.; Ćukušić, A.; et al. Viral Metagenomic Profiling of Croatian Bat Population Reveals Sample and Habitat Dependent Diversity. Viruses 2020, 12, 891. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080891
Šimić I, Zorec TM, Lojkić I, Krešić N, Poljak M, Cliquet F, Picard-Meyer E, Wasniewski M, Zrnčić V, Ćukušić A, et al. Viral Metagenomic Profiling of Croatian Bat Population Reveals Sample and Habitat Dependent Diversity. Viruses. 2020; 12(8):891. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080891
Chicago/Turabian StyleŠimić, Ivana, Tomaž Mark Zorec, Ivana Lojkić, Nina Krešić, Mario Poljak, Florence Cliquet, Evelyne Picard-Meyer, Marine Wasniewski, Vida Zrnčić, Anđela Ćukušić, and et al. 2020. "Viral Metagenomic Profiling of Croatian Bat Population Reveals Sample and Habitat Dependent Diversity" Viruses 12, no. 8: 891. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080891