A Comparison of Whole Genome Sequencing of SARS-CoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture

 , , , , , ,
, , , , , ,  , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Methods
2.1. Clinical Isolates
2.2. Genome Sequencing
2.3. Genome Assembly
2.4. Assessment of Clinical Diagnostic PCR Primers
2.5. Molecular Epidemiology Analysis
2.6. Data & Software Availability
3. Result
3.1. Clinical Isolates
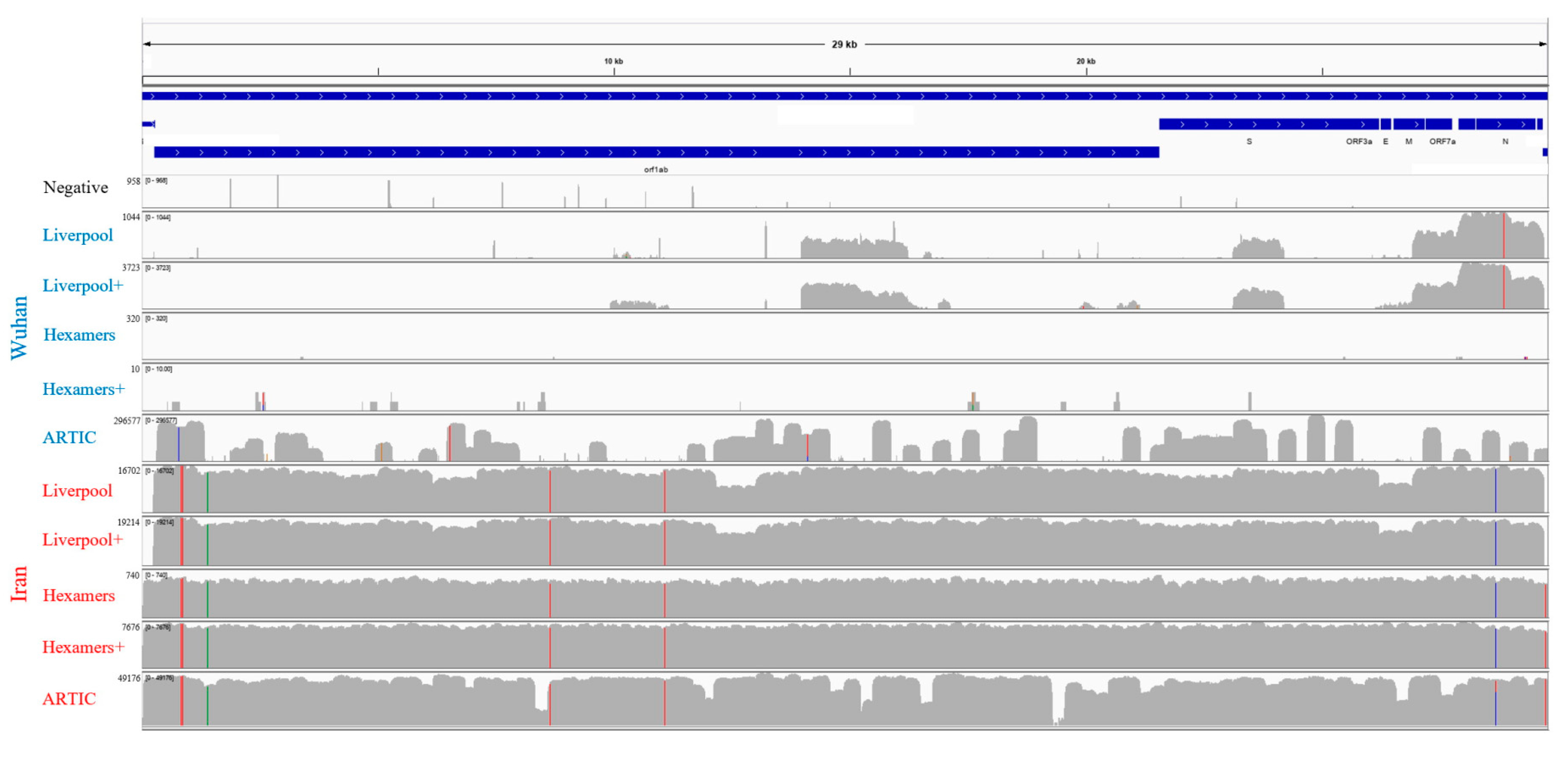
3.2. Genome Sequencing and Assembly
3.3. Assessment of Clinical Diagnostic PCR Primers
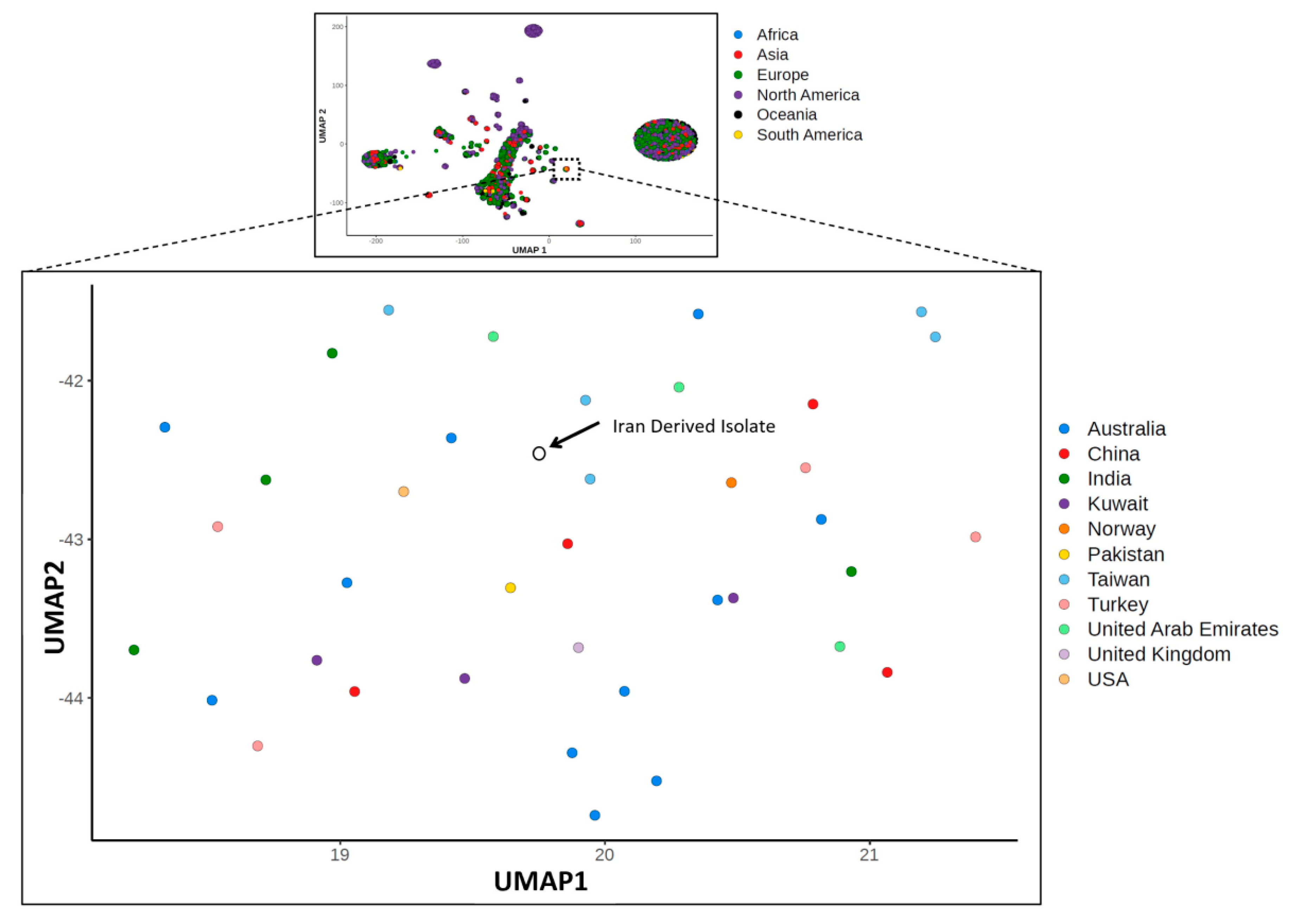
3.4. Molecular Epidemiology Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA 2020, 323, 1843–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Si, H.R.; Zhu, Y.; Yang, X.L.; Anderson, D.E.; Shi, Z.L.; Wang, L.F.; Zhou, P. Discovery of Bat Coronaviruses through Surveillance and Probe Capture-Based Next-Generation Sequencing. mSphere 2020, 5, e00807-19. [Google Scholar] [CrossRef] [Green Version]
- Metsky, H.C.; Siddle, K.J.; Gladden-Young, A.; Qu, J.; Yang, D.K.; Brehio, P.; Goldfarb, A.; Piantadosi, A.; Wohl, S.; Carter, A.; et al. Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat. Biotechnol. 2019, 37, 160–168. [Google Scholar] [CrossRef]
- Depledge, D.P.; Palser, A.L.; Watson, S.J.; Lai, I.Y.C.; Gray, E.R.; Grant, P.; Kanda, R.K.; Leproust, E.; Kellam, P.; Breuer, J. Specific Capture and Whole-Genome Sequencing of Viruses from Clinical Samples. PLoS ONE 2011, 6, e27805. [Google Scholar] [CrossRef]
- Marchand-Senécal, X.; Kozak, R.; Mubareka, S.; Salt, N.; Gubbay, J.B.; Eshaghi, A.; Allen, V.; Li, Y.; Bastien, N.; Gilmour, M.; et al. Diagnosis and Management of First Case of COVID-19 in Canada: Lessons applied from SARS. Clin. Infect. Dis. 2020, ciaa227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBlanc, J.J.; Gubbay, J.B.; Li, Y.; Needle, R.; Arneson, S.R.; Marcino, D.; Charest, H.; Desnoyers, G.; Dust, K.; Fattouh, R.; et al. Real-time PCR-based SARS-CoV-2 detection in Canadian laboratories. J. Clin. Virol. 2020, 128, 104433. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 3 May 2020).
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Deatherage, D.E.; Traverse, C.C.; Wolf, L.N.; Barrick, J.E. Detecting rare structural variation in evolving microbial populations from new sequence junctions using breseq. Front. Genet. 2015, 5, 468. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (Lond. Engl.) 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Vermeiren, C.; Marchand-Senécal, X.; Sheldrake, E.; Bulir, D.; Smieja, M.; Chong, S.; Forbes, J.D.; Katz, K. Comparison of Copan Eswab and FLOQswab for COVID-19 PCR diagnosis: Working around a supply shortage. J. Clin. Microbiol. 2020, 58, e00669-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalla, A.K.; Casto, A.M.; Huang, M.L.W.; Perchetti, G.A.; Sampoleo, R.; Shrestha, L.; Wei, Y.; Zhu, H.; Jerome, K.R.; Greninger, A.L. Comparative Performance of SARS-CoV-2 Detection Assays using Seven Different Primer/Probe Sets and One Assay Kit. J. Clin. Microbiol. 2020, 58, e00557-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, D.K.; Pan, Y.; Cheng, S.M.; Hui, K.P.; Krishnan, P.; Liu, Y.; Ng, D.Y.; Wan, C.K.; Yang, P.; Wang, Q.; et al. Molecular Diagnosis of a Novel Coronavirus (2019-nCoV) Causing an Outbreak of Pneumonia. Clin. Chem. 2020, 66, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Molecular Assays to Diagnose COVID-19: Summary Table of Available Protocols. Available online: https://www.who.int/who-documents-detail/molecular-assays-to-diagnose-covid-19-summary-table-of-available-protocols (accessed on 11 May 2020).
- Uniform Manifold Approximation and Projection for Dimension Reduction. Available online: http://arxiv.org/abs/1802.03426. (accessed on 15 May 2020).
- Kimura, M.A. simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Issues with SARS-CoV-2 Sequencing Data. Virological. 2020. Available online: http://virological.org/t/issues-with-sars-cov-2-sequencing-data/473 (accessed on 11 May 2020).
- Maan, H.; Mbareche, H.; Raphenya, A.R.; Banerjee, A.; Nasir, J.A.; Kozak, R.A.; Knox, N.; Mubareka, S.; McArthur, A.G.; Wang, B. Genotyping SARS-CoV-2 through an interactive web application. The Lancet Digital Health 2020, 2, E340–E341. [Google Scholar] [CrossRef]
- Network, A. hCoV-2019 (nCoV-2019/SARS-CoV-2). Available online: https://artic.network/ncov-2019 (accessed on 24 March 2020).
- Gudbjartsson, D.F.; Helgason, A.; Jonsson, H.; Magnusson, O.T.; Melsted, P.; Norddahl, G.L.; Saemundsdottir, J.; Sigurdsson, A.; Sulem, P.; Agustsdottir, A.B.; et al. Spread of SARS-CoV-2 in the Icelandic Population. N. Engl. J. Med. 2020, 382, 2302–2315. [Google Scholar] [CrossRef]
- Xiao, M.; Liu, X.; Ji, J.; Li, M.; Li, J.; Yang, L.; Sun, W.; Ren, P.; Yang, G.; Zhao, J.; et al. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 2020, 12, 57. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
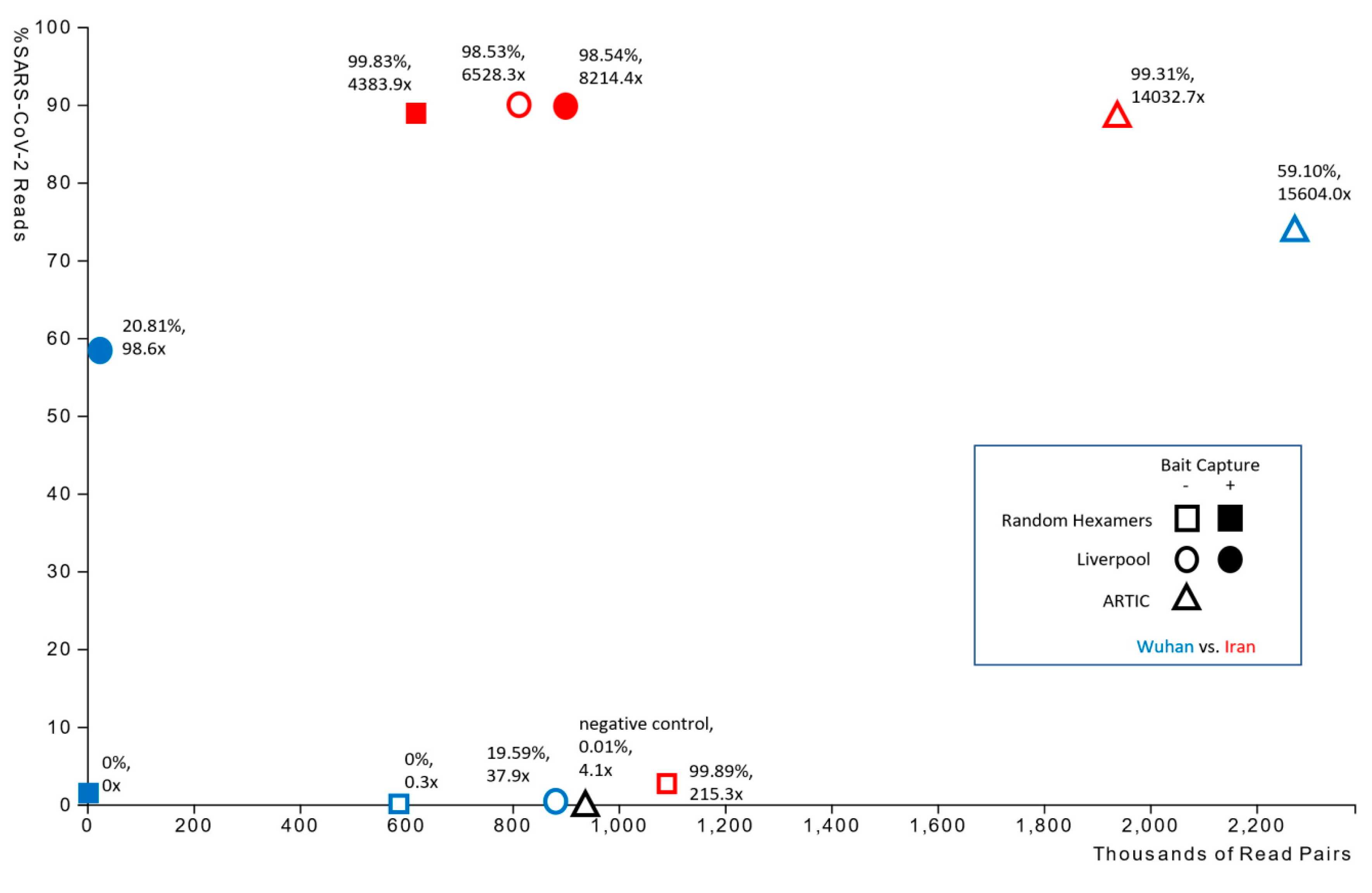
| Sample | Amplification | Enrichment | Number of Paired Reads | Reads from SARS-CoV-2 (%) | SARS-CoV-2 Genome Fraction (%) | Average Depth of Coverage | 0–100x Coverage (%) | 101–1000x Coverage (%) | >1000x Coverage (%) | # iVar Variants |
|---|---|---|---|---|---|---|---|---|---|---|
| Negative | ARTIC | No | 938,693 | 0.01 | 0 | 4.1x | 99.2 | 0.8 | 0.1 | n/a |
| Wuhan | Liverpool | No | 883,212 | 0.52 | 19.587 | 37.9x | 93.88 | 6.08 | 0.04 | 1 |
| Wuhan | Liverpool | Yes | 22,119 | 58.73 | 20.811 | 98.6x | 89.6 | 6.8 | 3.6 | 1 |
| Wuhan | Hexamers | No | 585,396 | 0.01 | 0 | 0.3x | 99.9 | 0.1 | 0.00 | n/a |
| Wuhan | Hexamers | Yes | 1536 | 1.56 | 0 | n/a | n/a | n/a | n/a | n/a |
| Wuhan | ARTIC | No | 2,271,152 | 73.86 | 59.104 | 15,604.0x | 10.6 | 35.5 | 53.9 | 5 |
| Iran | Liverpool | No | 813,975 | 90.13 | 98.53 | 6528.3x | 1.2 | 3.1 | 95.6 | 6 |
| Iran | Liverpool | Yes | 901,124 | 89.76 | 98.54 | 8214.4x | 0.7 | 0.2 | 99.1 | 6 |
| Iran | Hexamers | No | 1,091,011 | 2.77 | 99.89 | 215.3x | 0.43 | 99.56 | 0.00 | 7 |
| Iran | Hexamers | Yes | 619,661 | 89.17 | 99.83 | 4383.9x | 0.2 | 0.3 | 99.6 | 7 |
| Iran | ARTIC | No | 1,935,748 | 88.25 | 99.31 | 14,032.7x | 0.2 | 1.7 | 98.1 | 7 |
| Mutation | Liverpool Alone | Liverpool + Enrichment | Hexamers Alone | Hexamers + Enrichment | ARTIC Amplification | Clinical Diagnostic Primer Mismatch |
|---|---|---|---|---|---|---|
| Unresolved 5′ sequence | 259 bp | 258 bp | 40 bp | 0 bp | 49 bp | |
| Unresolved 3′ sequence | 200 bp | 190 bp | 77 bp | 139 bp | 67 bp | |
| pos. 835 (orf1ab polyprotein) | F190F (TTC→TTT) | F190F (TTC→TTT) | F190F (TTC→TTT) | F190F (TTC→TTT) | F190F (TTC→TTT) | NIID_WH-1_R854 |
| pos. 884 (orf1ab polyprotein) | R207C (CGT→TGT) | R207C (CGT→TGT) | R207C (CGT→TGT) | R207C (CGT→TGT) | R207C (CGT→TGT) | NIID_WH-1_R913 |
| pos. 1397 (orf1ab polyprotein) | V378I (GTA→ATA) | V378I (GTA→ATA) | V378I (GTA→ATA) | V378I (GTA→ATA) | V378I (GTA→ATA) | |
| pos. 8653 (orf1ab polyprotein) | M2796I (ATG→ATT) | M2796I (ATG→ATT) | M2796I (ATG→ATT) | M2796I (ATG→ATT) | M2796I (ATG→ATT) | Spike_F1 |
| pos. 9502 (orf1ab polyprotein) | 5.0% of reads suggest A3079A (GCC→GCT) | Spike_F1 | ||||
| pos. 11,074 (orf1ab polyprotein) | 11.8% of reads suggest a deletion between positions 10,809 and 13,203 | 11.8% of reads suggest a deletion between positions 10,809 and 13,203 | 10.9% of reads suggest a deletion between positions 10,809 and 13,203 | Spike_F1 | ||
| pos. 11,082 (orf1ab polyprotein) | 18.1% of reads suggest a deletion between positions 10,817 and 10,819 | 22.8% of reads suggest a deletion between positions 10,817 and 10,819 | Spike_F1 | |||
| pos. 11,083 (orf1ab polyprotein) | L3606F (TTG→TTT) | L3606F (TTG→TTT) | L3606F (TTG→TTT) | L3606F (TTG→TTT) | L3606F (TTG→TTT) | Spike_F1 |
| pos. 19,285–19,603 (orf1ab polyprotein) | 319 bp coverage gap (no aligned reads); amplicon 64 | |||||
| pos. 27,156 (membrane glycoprotein) | 5.3% of reads suggest S212C (AGT→TGT) | |||||
| pos. 28,688 (nucleocapsid phosphoprotein) | L139L (TTG→CTG) | L139L (TTG→CTG) | L139L (TTG→CTG) | L139L (TTG→CTG) | L139L (TTG→CTG) | 2019-nCoV_N3-F |
| pos. 29,742 (intergenic) | no coverage | no coverage | G→T | G→T | G→T |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasir, J.A.; Kozak, R.A.; Aftanas, P.; Raphenya, A.R.; Smith, K.M.; Maguire, F.; Maan, H.; Alruwaili, M.; Banerjee, A.; Mbareche, H.; et al. A Comparison of Whole Genome Sequencing of SARS-CoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture. Viruses 2020, 12, 895. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080895
Nasir JA, Kozak RA, Aftanas P, Raphenya AR, Smith KM, Maguire F, Maan H, Alruwaili M, Banerjee A, Mbareche H, et al. A Comparison of Whole Genome Sequencing of SARS-CoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture. Viruses. 2020; 12(8):895. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080895
Chicago/Turabian StyleNasir, Jalees A., Robert A. Kozak, Patryk Aftanas, Amogelang R. Raphenya, Kendrick M. Smith, Finlay Maguire, Hassaan Maan, Muhannad Alruwaili, Arinjay Banerjee, Hamza Mbareche, and et al. 2020. "A Comparison of Whole Genome Sequencing of SARS-CoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture" Viruses 12, no. 8: 895. https://0-doi-org.brum.beds.ac.uk/10.3390/v12080895