Spatio-Temporal Mutational Profile Appearances of Swedish SARS-CoV-2 during the Early Pandemic

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sets
2.2. Mutation Sites in Swedish SARS-CoV-2 Sequences
2.3. Recombination Analysis and Selection Pressure
2.4. Evolutionary Dynamics Analysis of Mutational Profiles
2.5. Phylogenetic Inference
3. Results
3.1. Sequence Collection Overview
3.2. Divergence of SARS-CoV-2 in Sweden as Compared to the Prototype Sequence from Wuhan, China
3.3. Mutational Profiles of Swedish SARS-CoV-2
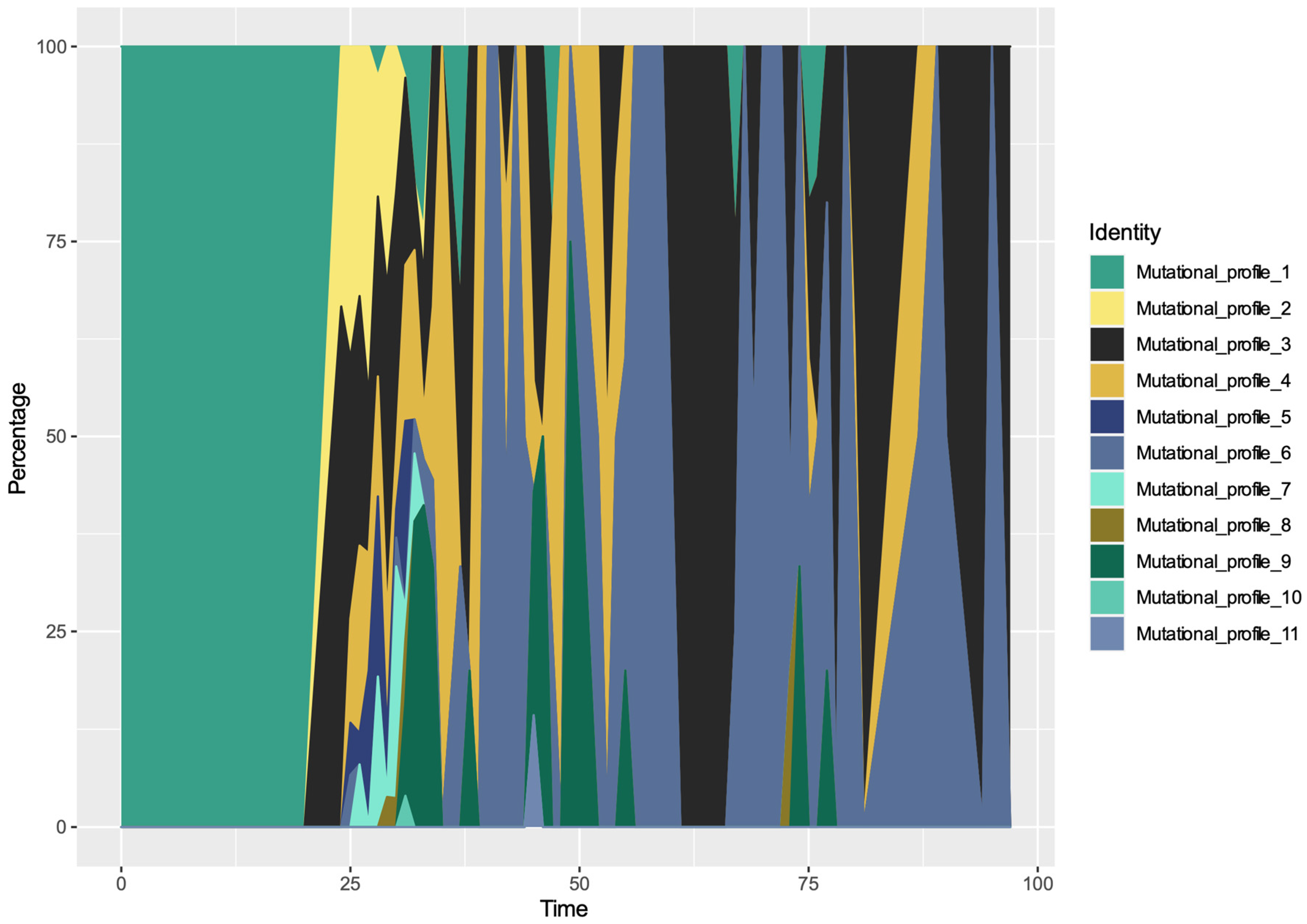
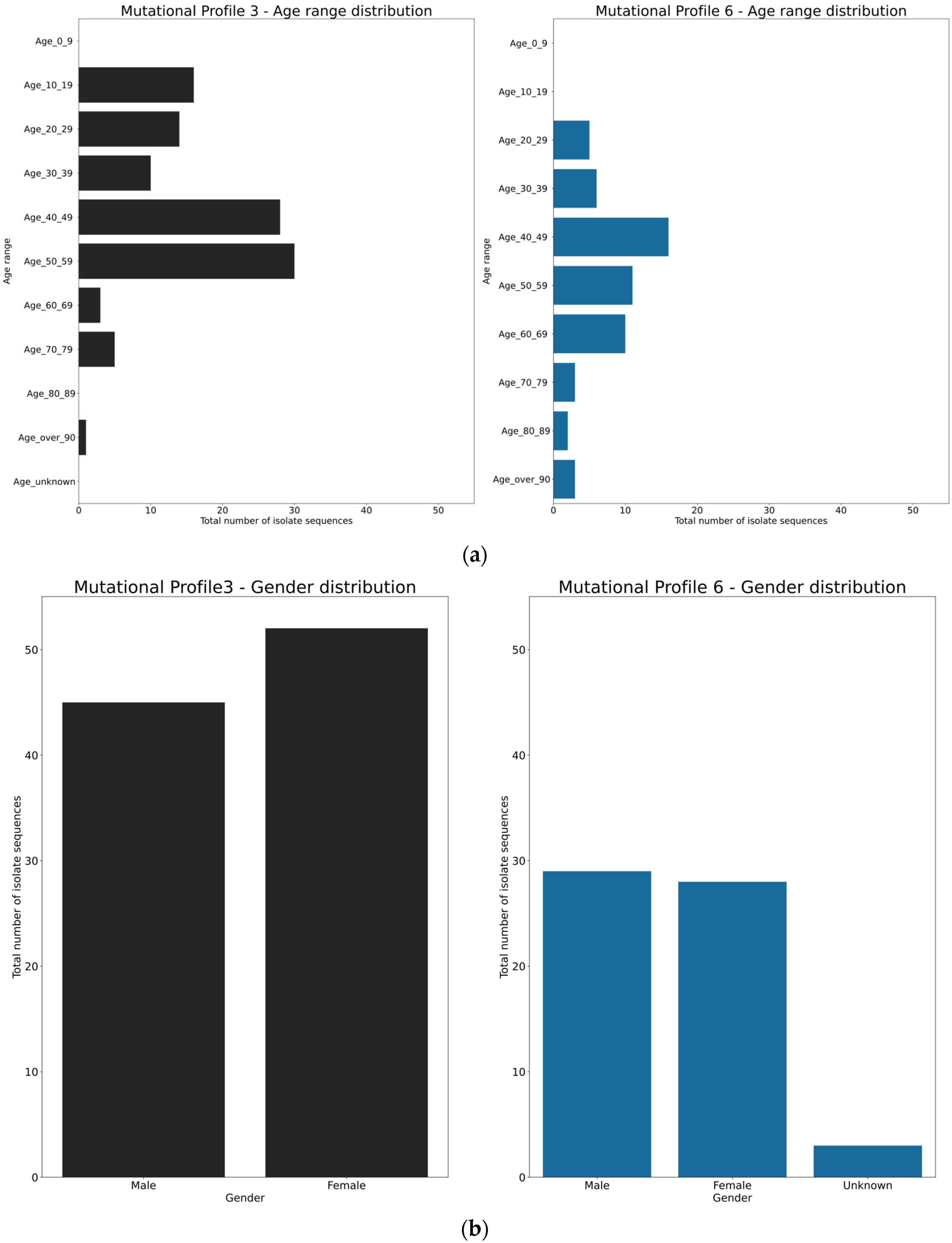
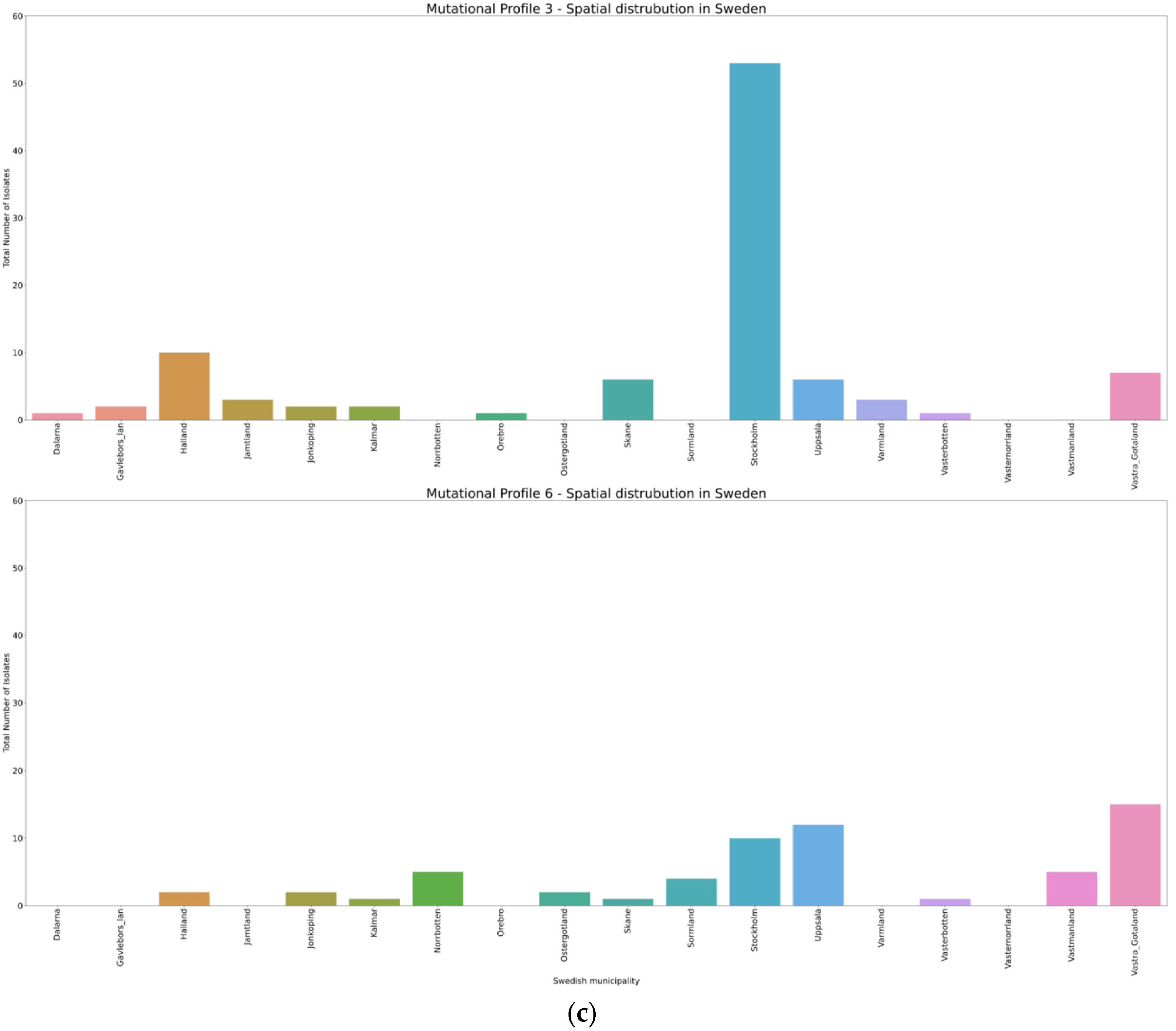
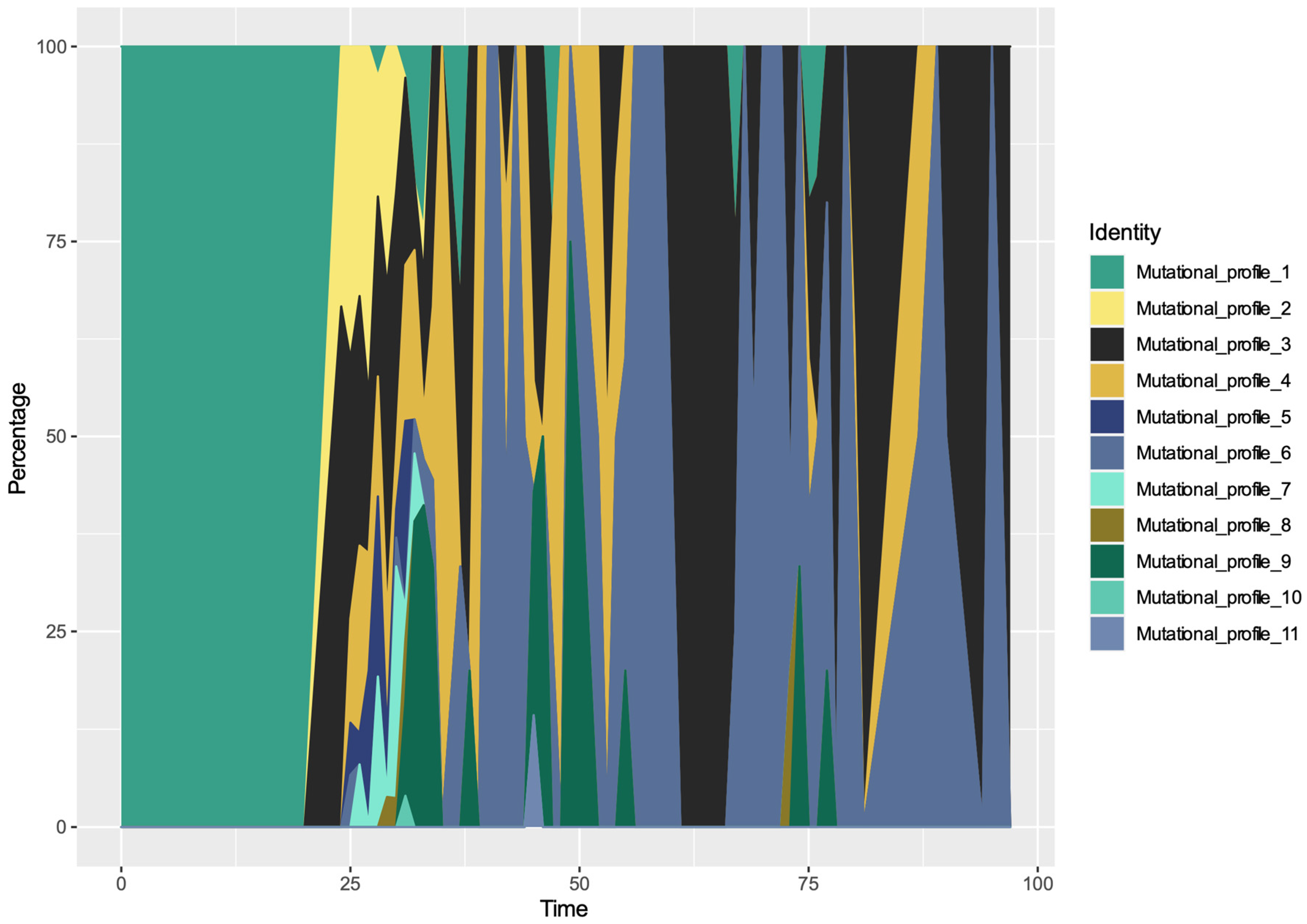
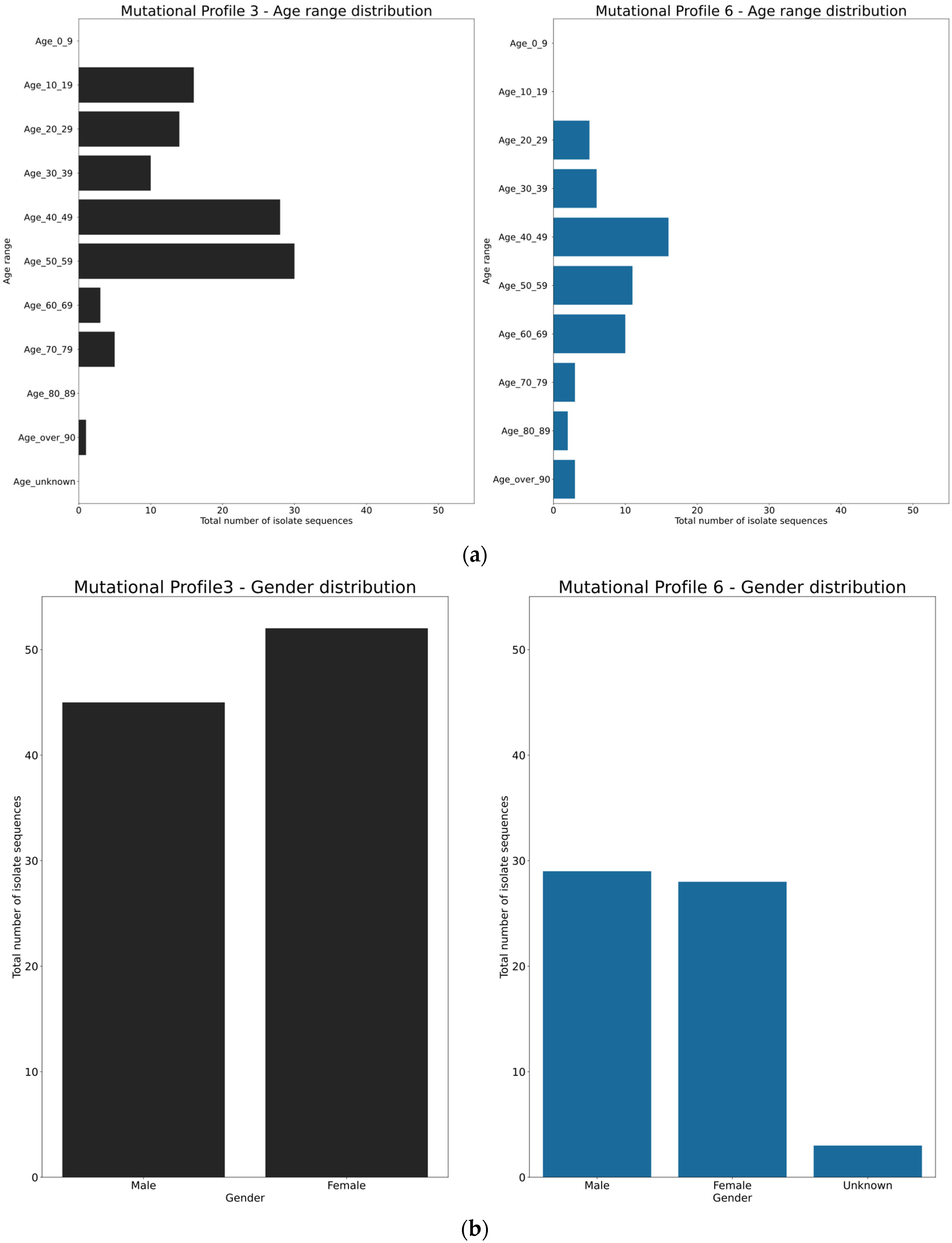
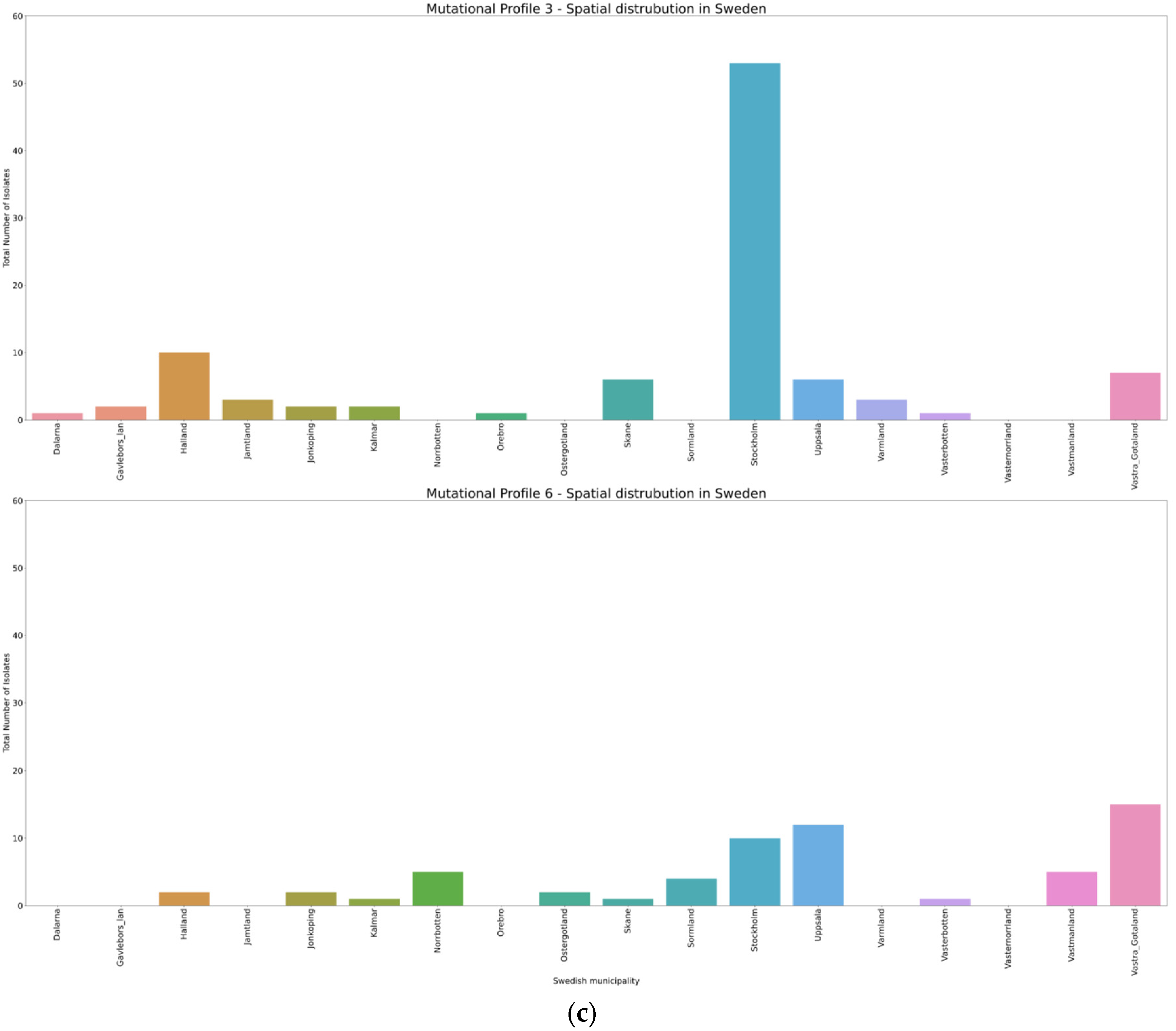
3.4. Spatio-Temporal Mutational Profile Appearances in Swedish SARS-CoV-2 Variants
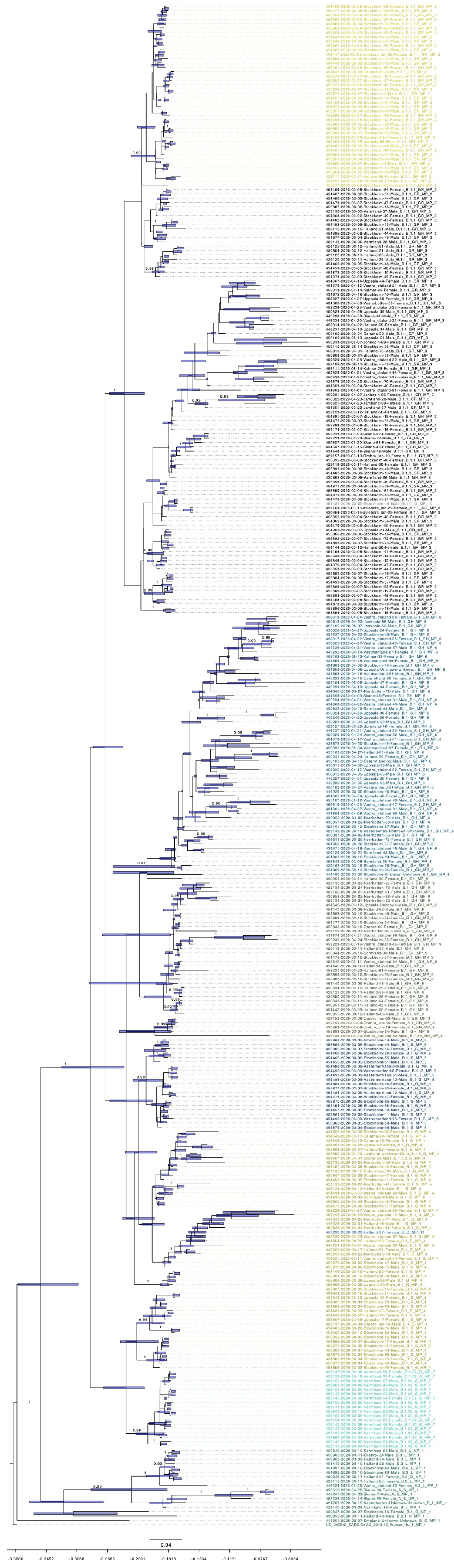
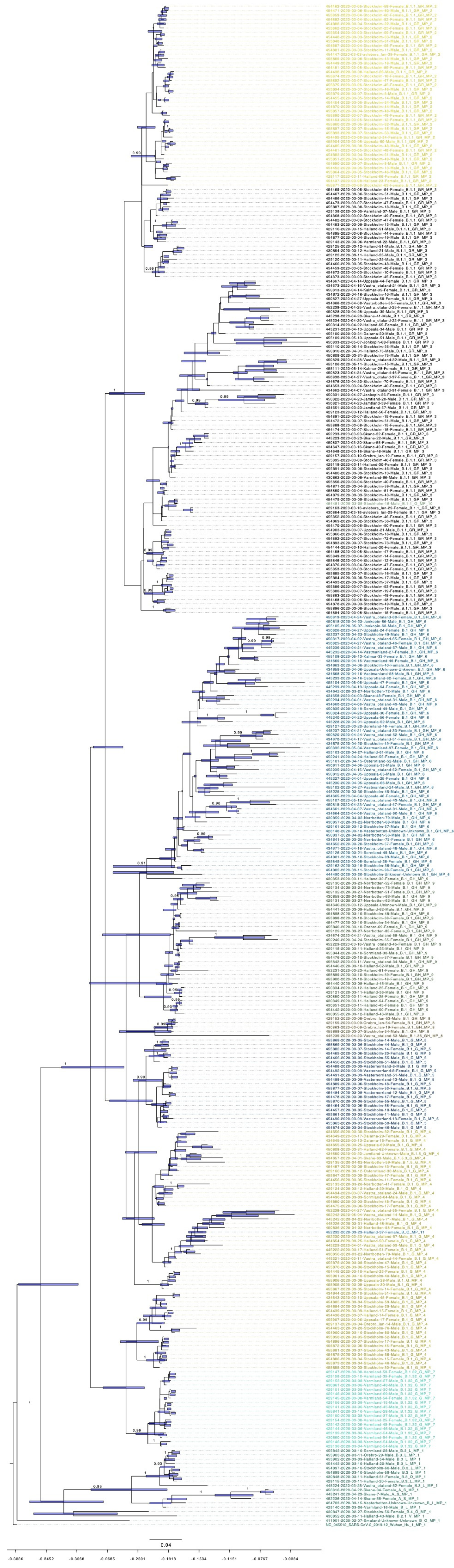
3.5. Bayesian Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guarner, J. Three Emerging Coronaviruses in Two Decades. Am. J. Clin. Pathol. 2020, 153, 420–421. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R.; Peiris, M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antivir. Res. 2013, 100, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Folkhälsomyndigheten. Available online: https://www.folkhalsomyndigheten.se (accessed on 12 September 2020).
- Walker, P.G.T.; Whittaker, C.; Watson, O.J.; Baguelin, M.; Winskill, P.; Hamlet, A.; Djafaara, B.A.; Cucunuba, Z.; Mesa, D.O.; Green, W.; et al. The impact of COVID-19 and strategies for mitigation and suppression in low- and middle-income countries. Science 2020, 369, 413–422. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Kasson, P.M. Managing COVID-19 spread with voluntary public-health measures: Sweden as a case study for pandemic control. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; Package Version 3.6.3.; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: http://www.R-project.org/ (accessed on 12 September 2020).
- Wang, G.T.; Peng, B.; Leal, S.M. Variant association tools for quality control and analysis of large-scale sequence and genotyping array data. Am. J. Hum. Genet. 2014, 94, 770–783. [Google Scholar] [CrossRef] [Green Version]
- Speed, D.; Hemani, G.; Johnson, M.R.; Balding, D.J. Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet. 2012, 91, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Pond, S.L.K. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Folkhälsomyndigheten. Helgenomsekvensering av Svenska SARS-CoV-2 som Orsakar COVID-19. Delrapport 1. 11 June 2020. Available online: https://www.folkhalsomyndigheten.se/publicerat-material/publikationsarkiv/h/helgenomsekvensering-av-svenska-sars-cov-2-som-orsakar-covid-19/ (accessed on 12 September 2020).
- Folkhälsomyndigheten. Helgenomsekvensering av Svenska SARS-CoV-2 som Orsakar COVID-19. Delrapport 2. 7 July 2020. Available online: https://www.folkhalsomyndigheten.se/publicerat-material/publikationsarkiv/h/helgenomsekvensering-av-svenska-sars-cov-2-som-orsakar-covid-19-del2/ (accessed on 12 September 2020).
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Yuan, L.; Huang, X.Y.; Liu, Z.Y.; Zhang, F.; Zhu, X.L.; Yu, J.Y.; Ji, X.; Xu, Y.P.; Li, G.; Li, C.; et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef] [Green Version]
- Urbanowicz, R.A.; McClure, C.P.; Sakuntabhai, A.; Sall, A.A.; Kobinger, G.; Muller, M.A.; Holmes, E.C.; Rey, F.A.; Simon-Loriere, E.; Ball, J.K. Human Adaptation of Ebola Virus during the West African Outbreak. Cell 2016, 167, 1079–1087.e1075. [Google Scholar] [CrossRef] [Green Version]
- Cyranoski, D. Profile of a killer: The complex biology powering the coronavirus pandemic. Nature 2020, 581, 22–26. [Google Scholar] [CrossRef]
- Korber, B.F.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Tang, X.C.; Agnihothram, S.S.; Jiao, Y.; Stanhope, J.; Graham, R.L.; Peterson, E.C.; Avnir, Y.; Tallarico, A.S.; Sheehan, J.; Zhu, Q.; et al. Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc. Natl. Acad. Sci. USA 2014, 111, E2018–E2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, J.; Li, W.; Roberts, A.; Matthews, L.J.; Murakami, A.; Vogel, L.; Wong, S.K.; Subbarao, K.; Farzan, M.; Marasco, W.A. Evaluation of human monoclonal antibody 80R for immunoprophylaxis of severe acute respiratory syndrome by an animal study, epitope mapping, and analysis of spike variants. J. Virol. 2005, 79, 5900–5906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Zhang, Y.; Li, J.; Xiao, Y.; Zhang, J.; Wang, Y.; Chen, L.; Paranhos-Baccala, G.; Wang, J. Genetic drift of human coronavirus OC43 spike gene during adaptive evolution. Sci. Rep. 2015, 5, 11451. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Keyaerts, E.; Lemey, P.; Moes, E.; Li, S.; Vandamme, A.M.; Van Ranst, M. Circulation of genetically distinct contemporary human coronavirus OC43 strains. Virology 2005, 337, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Chibo, D.; Birch, C. Analysis of human coronavirus 229E spike and nucleoprotein genes demonstrates genetic drift between chronologically distinct strains. J. Gen. Virol. 2006, 87, 1203–1208. [Google Scholar] [CrossRef]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, L.; Oliva, R. D936Y and Other Mutations in the Fusion Core of the SARS-Cov-2 Spike Protein Heptad Repeat 1 Undermine the Post-Fusion Assembly. bioRxiv 2020. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Petrone, M.E.; Holmes, E.C. We shouldn’t worry when a virus mutates during disease outbreaks. Nat. Microbiol. 2020, 5, 529–530. [Google Scholar] [CrossRef] [Green Version]
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Daniel, P.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Membrebe, J.V.; Suchard, M.A.; Rambaut, A.; Baele, G.; Lemey, P. Bayesian Inference of Evolutionary Histories under Time-Dependent Substitution Rates. Mol. Biol. Evol. 2019, 36, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Warnock, R.C.; Yang, Z.; Donoghue, P.C. Exploring uncertainty in the calibration of the molecular clock. Biol. Lett. 2012, 8, 156–159. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Caucci, S.; Clementi, N.; Vicenti, I.; Dragoni, F.; Cattelan, A.M.; Menzo, S.; Pan, A.; Callegaro, A.; et al. Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic. Viruses 2020, 12, 798. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site (NC, 045512) | Ref (NC, 045512) | Variant Change | Wild Type Frequency (%) | Variant Frequency (%) | aa Change | Type | Region | Amino Acid Property Change |
|---|---|---|---|---|---|---|---|---|
| 241 | C | T | 5.2 | 94.8 | transition | 5′UTR gene | ||
| 1059 | C | T | 73.6 | 26.4 | T265I | transition | ORF1ab/nsp2 | Polar, uncharged R group to Non-polar aliphatic R group |
| 3037 | C | T | 5.7 | 94.3 | F924F | transition | ORF1ab/nsp3 | |
| 12790 | A | G | 94.8 | 5.2 | T4175T | transition | ORF1ab/nsp9 | |
| 13568 | C | T | 94.8 | 5.2 | A43V | transition | RNA-dependent RNA polymerase | Both are non-polar aliphatic R groups |
| 14408 | C | T | 6.0 | 94.0 | P323L | transition | RNA-dependent RNA polymerase | Polar, uncharged R group to Non-polar aliphatic R group |
| 22583 | G | A | 94.8 | 5.2 | V341I | transition | S | Both are non-polar aliphatic R groups |
| 23403 | A | G | 5.2 | 94.8 | D614G | transition | S | Negatively charged R group to Non-polar aliphatic R group |
| 24368 | G | T | 82.8 | 17.2 | D936Y | transversion | S | Negatively charged R group to Polar aliphatic R group |
| 25563 | G | T | 72.2 | 27.8 | Q57H | transversion | ORF3a | Negatively charged R group to Positively charged R group |
| 27046 | C | T | 88.0 | 12.0 | T175M | transition | M | Polar, uncharged R group to Non-polar aliphatic R group |
| 28881 | G | A | 59.9 | 40.1 | R203K | transition | N | Both are positive R groups |
| 28882 | G | A | 59.9 | 40.1 | R203K | transition | N | Both are positive R groups |
| 28883 | G | C | 59.9 | 40.1 | G204R | transversion | N | Non-polar aliphatic R groups to Positively charged R group |
| Profiles | PANGOLIN Lineage | GISAID Lineage | Frequency (%) | Mutational Profiling |
|---|---|---|---|---|
| Mutational Profile 1 | B | O | 4.89 | WT |
| B.4 | O | WT | ||
| B | L | WT | ||
| B.3 | L | WT | ||
| B.3 | O | WT | ||
| B.2.1 | V | WT | ||
| A | S | WT | ||
| Mutational Profile 2 | B.1.1 | GR | 12.1 | 241C > T, 3037C > T, 14408C > T, 23403A > G, 27046C > T, 28881G > A, 28882G > A, 28883G > C |
| Mutational profile 3 | B.1.1 | GR | 27.9 | 241C > T, 3037C > T, 14408C > T, 23403A > G, 28881G > A, 28882G > A, 28883G > C |
| B.1.1.1 | GR | 241C > T, 3037C > T, 14408C > T, 23403A > G, 28881G > A, 28882G > A, 28883G > C | ||
| Mutational profile 4 | B.1 | G | 15.5 | 241C > T, 3037C > T, 14408C > T, 23403A > G |
| B.1.5 | G | 241C > T, 3037C > T, 14408C > T, 23403A > G | ||
| B.1.5.3 | G | 241C > T, 3037C > T, 14408C > T, 23403A > G | ||
| Mutational Profile 5 | B.1 | G | 6.03 | 241C > T, 3037C > T, 14408C > T, 22583G > A, 23403A > G |
| Mutational Profile, 6 | B.1 | GH | 17.2 | 241C > T, 1059C > T, 3037C > T, 14408C > T, 23403A > G, 24368G > T, 25563G > T |
| Mutational Profile 7 | B.1.32 | G | 5.17 | 241C > T, 3037C > T, 12790A > G, 14408C > T, 14408C > T, 23403A > G |
| Mutational Profile 8 | B.1 | GH | 1.43 | 241C > T, 3037C > T, 14408C > T, 23403A > G, 25563G > T |
| Mutational Profile, 8 | B.1.36 | GH | 241C > T, 3037C > T, 14408C > T, 23403A > G, 25563G > T | |
| Mutational Profile 9 | B.1 | GH | 9.20 | 241C > T, 1059C > T, 3037C > T, 14408C > T, 23403A > G, 25563G > T |
| Mutational Profile 10 | B.1.1 | O | 0.287 | 241C > T, 14408C > T, 23403A > G, 28881G > A, 28882G > A, 28883G > C |
| Mutational Profile 11 | B | O | 0.287 | 241C > T, 14408C > T, 23403A > G |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, J.; Hickman, R.A.; Li, J.; Lu, X.; Lindahl, J.F.; Lundkvist, Å.; Järhult, J.D. Spatio-Temporal Mutational Profile Appearances of Swedish SARS-CoV-2 during the Early Pandemic. Viruses 2020, 12, 1026. https://0-doi-org.brum.beds.ac.uk/10.3390/v12091026
Ling J, Hickman RA, Li J, Lu X, Lindahl JF, Lundkvist Å, Järhult JD. Spatio-Temporal Mutational Profile Appearances of Swedish SARS-CoV-2 during the Early Pandemic. Viruses. 2020; 12(9):1026. https://0-doi-org.brum.beds.ac.uk/10.3390/v12091026
Chicago/Turabian StyleLing, Jiaxin, Rachel A. Hickman, Jinlin Li, Xi Lu, Johanna F. Lindahl, Åke Lundkvist, and Josef D. Järhult. 2020. "Spatio-Temporal Mutational Profile Appearances of Swedish SARS-CoV-2 during the Early Pandemic" Viruses 12, no. 9: 1026. https://0-doi-org.brum.beds.ac.uk/10.3390/v12091026