Structural Comparison of Diverse HIV-1 Subtypes using Molecular Modelling and Docking Analyses of Integrase Inhibitors

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Study Design
2.3. Nucleic Acid Extraction
2.4. PCR Amplification and Sequencing
2.5. Consensus Sequence Alignment and Mutation Detection
2.6. Protein Modelling
2.7. Protein Preparation
2.8. Model Validation
2.9. INSTI Extractions and Molecular Docking
2.10. Refinement and Energy Minimisation
2.11. Binding Affinity Calculation and Interaction Analysis
2.12. Binding Site Analysis
3. Results
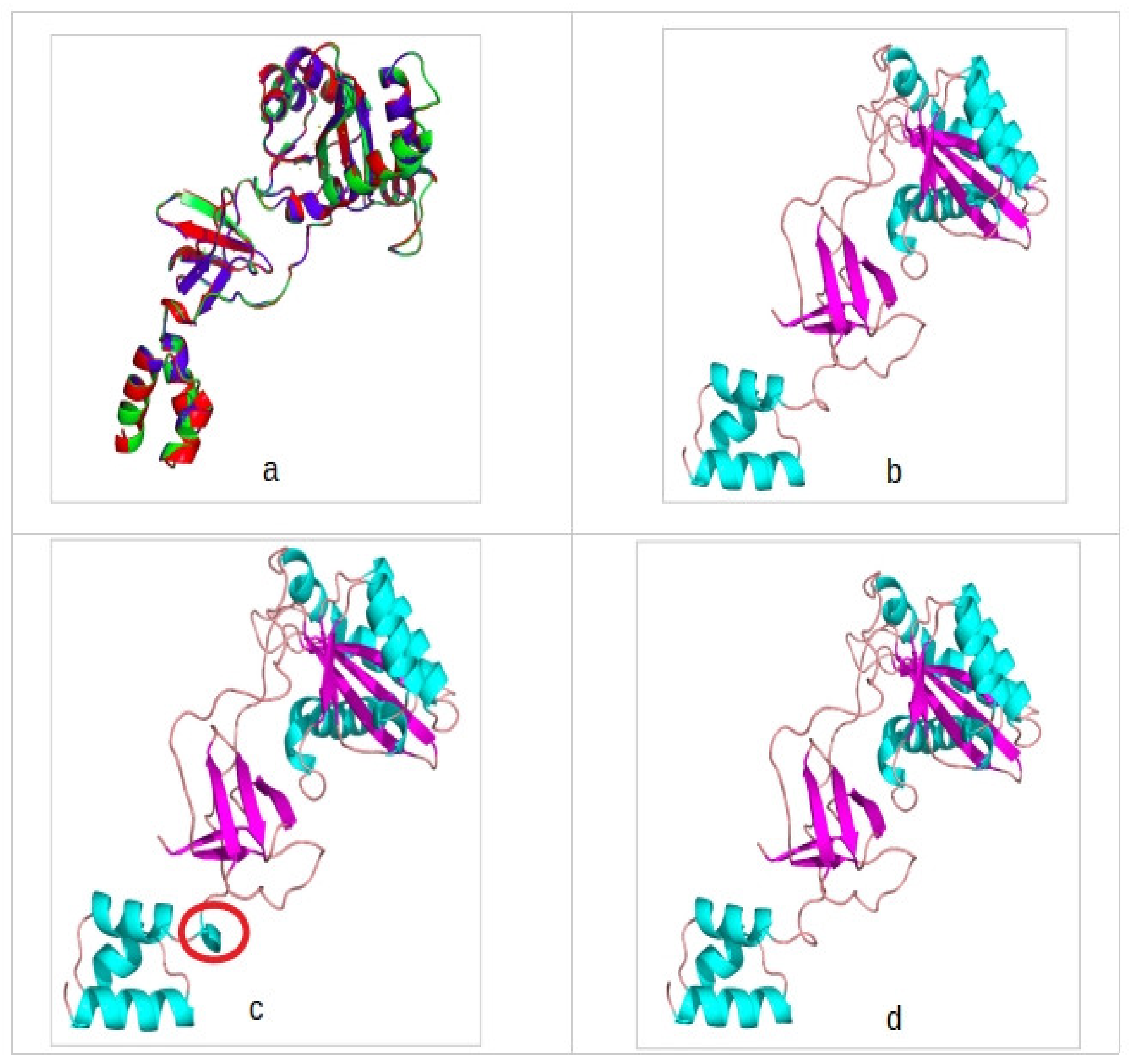
3.1. Sequence Alignments and Protein Structure Assessment
3.2. Molecular Docking and Interaction Analysis
3.3. Binding Site Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Piot, P.; Quinn, T.C. Response to the AIDS Pandemic—A Global Health Model. N. Engl. J. Med. 2013, 368, 2210–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA Integration. Chem. Rev. 2016, 116, 12730–12757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craigie, R. The Molecular Biology of HIV Integrase. Future Virol. 2012, 7, 679–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craigie, R.; Bushman, F.D. HIV DNA Integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grobler, J.A.; Stillmock, K.; Hu, B.; Witmer, M.; Felock, P.; Espeseth, A.S.; Wolfe, A.; Egbertson, M.; Bourgeois, M.; Melamed, J.; et al. Diketo Acid Inhibitor Mechanism and HIV-1 Integrase: Implications for Metal Binding in the Active Site of Phosphotransferase Enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 6661–6666. [Google Scholar] [CrossRef] [Green Version]
- Neamati, N.; Lin, Z.; Karki, R.G.; Orr, A.; Cowansage, K.; Strumberg, D.; Pais, G.C.G.; Voigt, J.H.; Nicklaus, M.C.; Winslow, H.E.; et al. Metal-Dependent Inhibition of HIV-1 Integrase. J. Med. Chem. 2002, 45, 5661–5670. [Google Scholar] [CrossRef]
- Ceccherini-Silberstein, F.; Malet, I.; D’Arrigo, R.; Antinori, A.; Marcelin, A.-G.; Perno, C.-F. Characterization and Structural Analysis of HIV-1 Integrase Conservation. AIDS Rev. 2009, 11, 17–29. [Google Scholar] [PubMed]
- Malet, I.; Soulie, C.; Tchertanov, L.; Derache, A.; Amellal, B.; Traore, O.; Simon, A.; Katlama, C.; Mouscadet, J.-F.; Calvez, V.; et al. Structural Effects of Amino Acid Variations between B and CRF02-AG HIV-1 Integrases. J. Med. Virol. 2008, 80, 754–761. [Google Scholar] [CrossRef]
- Anstett, K.; Brenner, B.; Mesplede, T.; Wainberg, M.A. HIV Drug Resistance against Strand Transfer Integrase Inhibitors. Retrovirology 2017, 14, 36. [Google Scholar] [CrossRef]
- World Health Organisation. Update of Recommendations of First-and Second-Line Antiretroviral Regimens; World Health Organisation: Geneva, Switzerland, 2019. [Google Scholar]
- Rhee, S.-Y.; Grant, P.M.; Tzou, P.L.; Barrow, G.; Harrigan, P.R.; Ioannidis, J.P.A.; Shafer, R.W. A Systematic Review of the Genetic Mechanisms of Dolutegravir Resistance. J. Antimicrob. Chemother. 2019, 74, 3135–3149. [Google Scholar] [CrossRef]
- Santoro, M.M.; Perno, C.F. HIV-1 Genetic Variability and Clinical Implications. ISRN Microbiol. 2013, 2013, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Keyhani, S.; Wang, S.; Hebert, P.; Carpenter, D.; Anderson, G. US Pharmaceutical Innovation in an International Context. Am. J. Public Health 2010, 100, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV Subtype Diversity Worldwide. Curr. Opin. HIV AIDS 2019, 14, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Bar-Magen, T.; Sloan, R.D.; Faltenbacher, V.H.; Donahue, D.A.; Kuhl, B.D.; Oliveira, M.; Xu, H.; Wainberg, M.A. Comparative Biochemical Analysis of HIV-1 Subtype B and C Integrase Enzymes. Retrovirology 2009, 6, 103. [Google Scholar] [CrossRef] [Green Version]
- Lessells, R.; Katzenstein, D.; de Oliveira, T. Are Subtype Differences Important in HIV Drug Resistance? Curr. Opin. Virol. 2012, 2, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Depatureaux, A.; Quashie, P.K.; Mesplède, T.; Han, Y.; Koubi, H.; Plantier, J.-C.; Oliveira, M.; Moisi, D.; Brenner, B.; Wainberg, M.A. HIV-1 Group O Integrase Displays Lower Enzymatic Efficiency and Higher Susceptibility to Raltegravir than HIV-1 Group M Subtype B Integrase. Antimicrob. Agents Chemother. 2014, 58, 7141–7150. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.-S.; Mesplède, T.; Wainberg, M.A. Differences among HIV-1 Subtypes in Drug Resistance against Integrase Inhibitors. Infect. Genet. Evol. 2016, 46, 286–291. [Google Scholar] [CrossRef]
- Llácer Delicado, T.; Torrecilla, E.; Holguín, Á. Deep Analysis of HIV-1 Natural Variability across HIV-1 Variants at Residues Associated with Integrase Inhibitor (INI) Resistance in INI-Naive Individuals. J. Antimicrob. Chemother. 2016, 71, 362–366. [Google Scholar] [CrossRef]
- Obasa, A.E.; Engelbrecht, S.; Jacobs, G.B. Near Full-Length HIV-1 Subtype B Sequences from the Early South African Epidemic, Detecting a BD Unique Recombinant Form (URF) from a Sample in 1985. Sci. Rep. 2019, 9, 6227. [Google Scholar] [CrossRef] [Green Version]
- Courtney, C.R.; Agyingi, L.; Fokou, A.; Christie, S.; Asaah, B.; Meli, J.; Ngai, J.; Hewlett, I.; Nyambi, P.N. Monitoring HIV-1 Group M Subtypes in Yaoundé, Cameroon Reveals Broad Genetic Diversity and a Novel CRF02_AG/F2 Infection. AIDS Res. Hum. Retrovir. 2016, 32, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Abongwa, L.E.; Nyamache, A.K.; Torimiro, J.N.; Okemo, P.; Charles, F. Human Immunodeficiency Virus Type 1 ((HIV-1) Subtypes in the Northwest Region, Cameroon. Virol. J. 2019, 16, 103. [Google Scholar] [CrossRef] [Green Version]
- Obasa, A.E.; Mikasi, S.G.; Brado, D.; Cloete, R.; Singh, K.; Neogi, U.; Jacobs, G.B. Drug Resistance Mutations Against Protease, Reverse Transcriptase and Integrase Inhibitors in People Living With HIV-1 Receiving Boosted Protease Inhibitors in South Africa. Front. Microbiology 2020, 11, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, G.B.; Laten, A.; van Rensburg, E.J.; Bodem, J.; Weissbrich, B.; Rethwilm, A.; Preiser, W.; Engelbrecht, S. Phylogenetic Diversity and Low Level Antiretroviral Resistance Mutations in HIV Type 1 Treatment-Naive Patients from Cape Town, South Africa. AIDS Res. Hum. Retrovir. 2008, 24, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- Brado, D.; Obasa, A.E.; Ikomey, G.M.; Cloete, R.; Singh, K.; Engelbrecht, S.; Neogi, U.; Jacobs, G.B. Analyses of HIV-1 Integrase Sequences Prior to South African National HIV-Treatment Program and Availability of Integrase Inhibitors in Cape Town, South Africa. Sci. Rep. 2018, 8, 4709. [Google Scholar] [CrossRef] [PubMed]
- Mikasi, S.G.; Gichana, J.O.; Van der Walt, C.; Brado, D.; Obasa, A.E.; Njenda, D.; Messembe, M.; Lyonga, E.; Assoumou, O.; Cloete, R.; et al. HIV-1 Integrase Diversity and Resistance-Associated Mutations and Polymorphisms Among Integrase Strand Transfer Inhibitor-Naive HIV-1 Patients from Cameroon. AIDS Res. Hum. Retrovir. 2020, 36, 450–455. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, D.O.; Li, M.; Yang, R.; Rebensburg, S.V.; Ghirlando, R.; Jeon, Y.; Shkriabai, N.; Kvaratskhelia, M.; Craigie, R.; Lyumkis, D. Cryo-EM Structures and Atomic Model of the HIV-1 Strand Transfer Complex Intasome. Science 2017, 355, 89–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Hooft, R.W.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in Protein Structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. [20] VERIFY3D: Assessment of protein models with three-dimensional profiles. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1997; Volume 277, pp. 396–404. [Google Scholar]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from Standard Atomic Volumes as a Quality Measure for Protein Crystal Structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System, version 1.8; Schrodinger LLC.: New York, NY, USA, 2015.
- Hare, S.; Vos, A.M.; Clayton, R.F.; Thuring, J.W.; Cummings, M.D.; Cherepanov, P. Molecular Mechanisms of Retroviral Integrase Inhibition and the Evolution of Viral Resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral Intasome Assembly and Inhibition of DNA Strand Transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef]
- Hare, S.; Smith, S.J.; Métifiot, M.; Jaxa-Chamiec, A.; Pommier, Y.; Hughes, S.H.; Cherepanov, P. Structural and Functional Analyses of the Second-Generation Integrase Strand Transfer Inhibitor Dolutegravir (S/GSK1349572). Mol. Pharm. 2011, 80, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Cook, N.J.; Li, W.; Berta, D.; Badaoui, M.; Ballandras-Colas, A.; Nans, A.; Kotecha, A.; Rosta, E.; Engelman, A.N.; Cherepanov, P. Structural Basis of Second-Generation HIV Integrase Inhibitor Action and Viral Resistance. Science 2020, 367, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Wares, M.; Mesplède, T.; Quashie, P.K.; Osman, N.; Han, Y.; Wainberg, M.A. The M50I Polymorphic Substitution in Association with the R263K Mutation in HIV-1 Subtype B Integrase Increases Drug Resistance but Does Not Restore Viral Replicative Fitness. Retrovirology 2014, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Rogers, L.; Obasa, A.E.; Jacobs, G.B.; Sarafianos, S.G.; Sönnerborg, A.; Neogi, U.; Singh, K. Structural Implications of Genotypic Variations in HIV-1 Integrase From Diverse Subtypes. Front. Microbiol. 2018, 9, 1754. [Google Scholar] [CrossRef] [Green Version]
- Chitongo, R.; Obasa, A.E.; Mikasi, S.G.; Jacobs, G.B.; Cloete, R. Molecular Dynamic Simulations to Investigate the Structural Impact of Known Drug Resistance Mutations on HIV-1C Integrase-Dolutegravir Binding. PLoS ONE 2020, 15, e0223464. [Google Scholar] [CrossRef]
- Lahti, J.L.; Tang, G.W.; Capriotti, E.; Liu, T.; Altman, R.B. Bioinformatics and Variability in Drug Response: A Protein Structural Perspective. J. R. Soc. Interface 2012, 9, 1409–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Nakahara, K.; Seki, T.; Miki, S.; Kawauchi, S.; Suyama, A.; Wakasamorimoto, C.; Kodama, M.; Endoh, T.; Oosugi, E. Selection of Diverse and Clinically Relevant Integrase Inhibitor-Resistant Human Immunodeficiency Virus Type 1 Mutants. Antivir. Res. 2008, 80, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Hatano, H.; Lampiris, H.; Fransen, S.; Gupta, S.; Huang, W.; Hoh, R.; Martin, J.N.; Lalezari, J.; Bangsberg, D.; Petropoulos, C.; et al. Evolution of Integrase Resistance During Failure of Integrase Inhibitor-Based Antiretroviral Therapy. J. Acquir. Immune Defic. Syndr. 2010, 54, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abram, M.E.; Ram, R.R.; Margot, N.A.; Barnes, T.L.; White, K.L.; Callebaut, C.; Miller, M.D. Lack of Impact of Pre-Existing T97A HIV-1 Integrase Mutation on Integrase Strand Transfer Inhibitor Resistance and Treatment Outcome. PLoS ONE 2017, 12, e0172206. [Google Scholar] [CrossRef]
- Ribeiro, A.J.M.; Ramos, M.J.; Fernandes, P.A. The Catalytic Mechanism of HIV-1 Integrase for DNA 3′-End Processing Established by QM/MM Calculations. J. Am. Chem. Soc. 2012, 134, 13436–13447. [Google Scholar] [CrossRef]
- Rogolino, D.; Carcelli, M.; Compari, C.; De Luca, L.; Ferro, S.; Fisicaro, E.; Rispoli, G.; Neamati, N.; Debyser, Z.; Christ, F.; et al. Diketoacid Chelating Ligands as Dual Inhibitors of HIV-1 Integration Process. Eur. J. Med. Chem. 2014, 78, 425–430. [Google Scholar] [CrossRef]
- Musyoka, T.; Tastan Bishop, Ö.; Lobb, K.; Moses, V. The Determination of CHARMM Force Field Parameters for the Mg2+ Containing HIV-1 Integrase. Chem. Phys. Lett. 2018, 711, 1–7. [Google Scholar] [CrossRef]
- Hassounah, S.A.; Alikhani, A.; Oliveira, M.; Bharaj, S.; Ibanescu, R.-I.; Osman, N.; Xu, H.-T.; Brenner, B.G.; Mesplède, T.; Wainberg, M.A. Antiviral Activity of Bictegravir and Cabotegravir against Integrase Inhibitor-Resistant SIVmac239 and HIV-1. Antimicrob. Agents Chemother. 2017, 61, e01695-17. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drug | CRF02_AG (Kcal/Mol) | Subtype B (Kcal/Mol) | Subtype C (Kcal/Mol) | PFV/SIV (Kcal/Mol) |
|---|---|---|---|---|
| Raltegravir | −7.2 | −4.4 | −5.1 | −6.1 |
| Elvitegravir | −3.7 | −3.4 | −3.4 | −4.0 |
| Dolutegravir | −3.0 | −3.4 | −3.0 | −5.4 |
| Bictegravir | −4.0 | −3.5 | −3.5 | −3.9 |
| Cabotagravir | −6.5 | −5.7 | −7.0 | N/A |
| INSTI | HIV-1B (ID:5u1c) | HIV-1C IN | CRF_02_AG IN | PFV/SIV IN | ||||
|---|---|---|---|---|---|---|---|---|
| Hydrogen bonds | Ionic contact | Hydrogen bonds | Ionic contact | Hydrogen bonds | Ionic contact | Hydrogen bonds | Ionic contact | |
| RAL | 2 (CYS10, GLU152) | 2MG | 6 (THY11, GUA22, ASP64, ASP116, TYR143, GLU152) | 2MG | 5 (THY11, GUA22, ASP64, ASP116, GLU152) | 2MG | 4 (ASP128, ASP185, TYR212, GLU221) | 2MG |
| DTG | 5 (THY11, GUA22, ASP64, ASP116, GLU152) | 2MG | 4 (THY11, GUA22, ASP64, GLU152) | 2MG | 4 (ADE21, GUA22, ASP64, ASP116) | 2MG | 3 (ASP128, ASP185, GLU221) | 2MG |
| EVG | 5 (THY11, ADE21, GUA22, ASP116, ARG231) | 1MG | 3 (ADE21, GLU52, ASP64) | 2MG | 6 (THY11, GUA22, ASP64, ASP116, GLU152, ARG231) | 2MG | 1 (GLU221) | 2MG |
| BIC | 4 (THY11, GUA22, ASP64, CYS65) | 1MG | 4 (THY11, GUA22, ASP64, CYS65, ASP116) | 2MG | 5 (THY11, ASP64, CYS65, GLU92, ASP116) | 2MG | 3 (ASP64, ASP116, GLU152) | 2MG |
| CBT | 7 (THY11, ADE21, GUA22, ASP64, THR66, HIS67, ASP116) | 2MG | 8 (THY11, ADE21, GUA22, ASP64, THR66, HIS67, ASP116, GLU152) | 2MG | 6 (THY11, ADE21, GUA22, ASP64, HIS67, ASP116) | 2MG | N/A | N/A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isaacs, D.; Mikasi, S.G.; Obasa, A.E.; Ikomey, G.M.; Shityakov, S.; Cloete, R.; Jacobs, G.B. Structural Comparison of Diverse HIV-1 Subtypes using Molecular Modelling and Docking Analyses of Integrase Inhibitors. Viruses 2020, 12, 936. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090936
Isaacs D, Mikasi SG, Obasa AE, Ikomey GM, Shityakov S, Cloete R, Jacobs GB. Structural Comparison of Diverse HIV-1 Subtypes using Molecular Modelling and Docking Analyses of Integrase Inhibitors. Viruses. 2020; 12(9):936. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090936
Chicago/Turabian StyleIsaacs, Darren, Sello Given Mikasi, Adetayo Emmanuel Obasa, George Mondinde Ikomey, Sergey Shityakov, Ruben Cloete, and Graeme Brendon Jacobs. 2020. "Structural Comparison of Diverse HIV-1 Subtypes using Molecular Modelling and Docking Analyses of Integrase Inhibitors" Viruses 12, no. 9: 936. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090936