Exploring the Remarkable Diversity of Culturable Escherichia coli Phages in the Danish Wastewater Environment


1
Department of Environmental Science, Aarhus University, Frederiksborgvej 399, 4000 Roskilde, Denmark
2
Department of Plant and Environmental Sciences, University of Copenhagen, Thorvaldsensvej 40, 1871 Frederiksberg C, Denmark
*
Authors to whom correspondence should be addressed.
Viruses 2020, 12(9), 986; https://0-doi-org.brum.beds.ac.uk/10.3390/v12090986
Submission received: 17 July 2020
/
Revised: 24 August 2020
/
Accepted: 31 August 2020
/
Published: 4 September 2020
(This article belongs to the Special Issue Viruses of Microbes 2020: The Latest Conquests on Viruses of Microbes)
Abstract
:Phages drive bacterial diversity, profoundly influencing microbial communities, from microbiomes to the drivers of global biogeochemical cycling. Aiming to broaden our understanding of Escherichia coli (MG1655, K-12) phages, we screened 188 Danish wastewater samples and isolated 136 phages. Ninety-two of these have genomic sequences with less than 95% similarity to known phages, while most map to existing genera several represent novel lineages. The isolated phages are highly diverse, estimated to represent roughly one-third of the true diversity of culturable virulent dsDNA Escherichia phages in Danish wastewater, yet almost half (40%) are not represented in metagenomic databases, emphasising the importance of isolating phages to uncover diversity. Seven viral families, Myoviridae, Siphoviridae, Podoviridae, Drexlerviridae, Chaseviridae, Autographviridae, and Microviridae, are represented in the dataset. Their genomes vary drastically in length from 5.3 kb to 170.8 kb, with a guanine and cytosine (GC) content ranging from 35.3% to 60.0%. Hence, even for a model host bacterium, substantial diversity remains to be uncovered. These results expand and underline the range of coliphage diversity and demonstrate how far we are from fully disclosing phage diversity and ecology.
1. Introduction
Phages are important ecological contributors, renewing organic matter supplies in nutrient cycles and driving bacterial diversity by enabling co-existence of competing bacteria by “Killing the winner” and by serving as genomic reservoirs and transport units [1,2]. Phage genomes are known to contain auxiliary metabolism genes (AMGs), toxins, and virulence factors [3,4,5,6,7]. Through lysogeny and transduction, they can transfer metabolic traits including antibiotic resistance to their hosts and can confer immunity against homologous phages [1].
Still, despite their ecological role, potential as antimicrobials and the fact that they carry a multitude of unknown genes with great potential for biotechnological applications, phages are vastly understudied. Around 10,000 phage genomes have now been published [8]. Though the number increases rapidly, we may have merely scratched the surface of the expected diversity. It is estimated that at least one billion bacterial species exist [9]. Hence, only phages targeting a tiny fraction of potential hosts have been reported. Efforts to disclose the range and diversity of phages targeting a single host have revealed a stunning display of diversity. The most scrutinized phage host is the Mycobacterium smegmatis, for which the Science Education Alliance Phage Hunters program has isolated more than 4700 phages and fully sequenced 680. These represent 30 distinct phage clusters [10,11]. This endeavour has provided a unique insight into viral and host diversity, evolution, and genetics [12,13,14,15]. No other phage host has been equally targeted, but numerous Escherichia coli phages (coliphages) have been isolated. The International Committee on Taxonomy of Viruses (ICTV) currently recognises 276 phage species originally isolated from Escherichia species [16], while 733 Escherichia phage assemblies are listed in the European Nucleotide Archive (ENA) [17].
As phages are expected to have evolutionary potential to migrate across microbial populations, host species may not be an ideal indicator of relatedness, but it serves as an excellent starting point to explore phage diversity. The hierarchical classification of phages is complicated by the high degree of horizontal gene transfer. Consequently, several classification systems have been proposed [18,19,20]. We may not yet have reached a point where it is reasonable to establish the criteria for a universal system [20]. Nonetheless, a system enabling a mutual understanding and exchange of knowledge is needed. Accordingly, we have classified our phages as per the ICTV guidelines [21].
Here, we aim to expand our understanding of coliphage diversity, by screening for coliphages targeting a single strain of E. coli. Earlier studies on coliphage diversity have explored other aspects. Grose & Casjens (2014) did a comprehensive in silico study on 337 genomes of tailed phages infecting Enterobacteriaceae to characterise the known diversity [8]. Jurczak-Kurek et al. (2016) isolated 60 coliphages from sewage samples on a single E. coli strain and thoroughly assessed the physiological diversity but did not sequence any of the coliphages [22]. Korf et al. (2019) used 29 individual E. coli strains from various sources to isolate, characterise, and sequence 50 diverse tailed coliphages including representatives of novel phage lineages, verifying that there is still something to discover [23]. Mathieu et. al., (2020) explored the presence of virulent and temperate coliphages in 648 faecal samples from 1-year-old children, revealing interesting compositional trends likely to impact gut microbiota dynamics [24].
In this study we applied the High Throughput Screening (HiTS) screening method on 188 wastewater samples using the K-12 MG1655 strain as host. This approach favours virulent, culturable dsDNA phages with a large burst size and a short latency period [25]. We hypothesised that using a non-pathogenic lab-strain like MG1655 would provide a broad diversity of phages as opposed to wildtype strains which are likely better equipped for avoiding a wide array of successful phage infections. Hence, we expected the screening to yield coliphages that were easy to work with but also distinct enough to expand the number of known coliphages.
2. Materials and Methods
The screening for coliphages was performed with the HiTS method as described in [25], though instead of direct plaque sequencing [26], lysates of wells giving rise to plaques were sequenced. In short, an overnight enrichment (37 °C) was performed in microplates with E. coli, media, and wastewater (0.5 mL/well); the next day, the enrichments were filtrated (0.45 µm), re-inoculated (∼1 μL), and re-incubated overnight (37 °C); on the third day, a second filtration (0.45 µm) and a spot-test (soft-agar overlay) were performed to indicate positive wells.
2.1. Sample Bacteria and Media
Inlet wastewater samples (188) were collected (40–50 mL) at two- to four time-points during July and August 2017 from 48 Danish wastewater treatment plants (WWTPs) geographically distributed in Denmark. The samples were centrifuged (9000× g, 4 °C, 10 min) and the supernatant filtered (0.45 μm) before storage in aliquots (−20 °C) until screening. The host bacterium was E. coli (MG1655, K-12), and the medium, Lysogeny Broth (LB), was amended with CaCl2 and MgCl2 (final concentration 10 mM).
2.2. Sequencing and Genome Characterisation
DNA extractions, clean-up (ZR-96 Clean and Concentrator kit, Zymo Research, Irvine, CA, USA), and sequencing libraries (Nextera® XT DNA kit, Illumina, San Diego, CA, USA) were performed according to manufacturer’s protocols with minor modifications as described in Kot et al. (2014) [26]. The libraries were sequenced as paired-end reads on an Illumina NextSeq platform with the Mid Output Kit v2 (300 cycles). The obtained reads were trimmed and assembled in CLC Genomics Workbench version 10.1.1. (CLC BIO, Aarhus, Denmark). Overlapping reads were merged with the following settings: mismatch cost: 2, minimum score: 15, gap cost: 3 and maximum unaligned end mismatches: 0, and then assembled de novo. Additional assemblies were constructed using SPAdes version 3.12.0 [27]. Gene prediction and annotation were performed using a customized RASTtk version 2.0 [28] workflow with GeneMark [29], with manual curation and verification using BLASTP [30], HHpred [31], and Pfam version 32.0 [32], or de novo annotation was performed using VIGA version 0.11.0 [33] based on DIAMOND searches (RefSeq Viral protein database) and HMMer searches (pVOG HMM database). All genomes were assessed for antibiotic resistance genes, bacterial virulence genes, restriction-modification genes, and auxiliary metabolism genes (AMGs) using ResFinder 3.1 [34,35], VirulenceFinder 2.0 [36], Restriction-ModificationFinder 1.1 (REBASE) [37], and VIBRANT version 1.0.1 [38], respectively.
2.3. Bioinformatics
Nucleotide (NT) and amino acid (AA) similarities were calculated using tools recommended by the ICTV [21], i.e., BLAST [30] for identification of the closest relative (BLASTn when possible, discontinuous megaBLAST (word size 16) for larger genomes), and Gegenees version 2.2.1 [39] for assessing phylogenetic NT (BLASTn) and AA (tBLASTx) distances of multiple genomes, with fragment size 200 bp and step size 100 bp. Intergenomic nucleotide sequence similarity and aligned genome fractions between all isolated phage species were plotted with VIRIDIC [40]. NT similarity was determined as percentage query cover multiplied by percentage identity. Novel phages were categorised according to ICTV taxonomy. The criterion of 95% DNA sequence similarity for demarcation of species was applied to identify novel species representatives and to determine uniqueness within the dataset. Evolutionary analyses for phylogenomic trees were conducted in MEGA7 version 2.1 (default settings) [41]. These were based on the large terminase subunit (Caudovirales), a gene commonly applied for phylogenetic analysis [42,43] and on the DNA replication gene (gpA) (Microviridae). The NT sequences were aligned by MUSCLE [44] and the evolutionary history inferred by the Maximum Likelihood method based on the Tamura-Nei model [45]. The trees with the highest log-likelihood and are shown. Pairwise whole genome comparisons were performed with Easyfig 2.2.2 [46] (BLASTn), curated by adding color-codes and identifiers in Inkscape version 0.92.2. The R package iNEXT [47,48] in R studio version 1.1.456 [49] was used for rarefaction, species diversity (q = 0, datatype: incidence_raw), extrapolation thereof (estimadeD), and estimation of sample coverage. The visualisation of genome sizes and GC contents was prepared in Excel version 16.31. Blast+ 2.9.0 [50] was used to perform a NT search of the coliphages (queries) against a database with the IMG/VR v2.0 database sequences [51] and the human gut virome database (GVD) v 1.7 [52]. Reads from metagenomes and metaviromes were mapped using bbmap 38.22 [53]. Genome breadth and depth coverage was calculated using genomecov from BEDtools 2.28.0 [54] and BamM 1.7.3 [55], respectively.
3. Results and Discussion
3.1. Wastewater Coliphages Are Remarkably Diverse
The sequenced coliphages were analysed strictly in silico, focusing on their relatedness to known phages, taxonomy, and distinctive characteristics. The genome assemblies had a coverage of ×20-12122 with an average of ×390.5 (Table S1). The genome screening algorithms identified no homologs of known virulence or antibiotic resistance genes. Though not a definitive exclusion, this is interpreted as a reduced risk of presence, a preferable trait for phage therapy. The majority of genes identified when screening for AMGs code for phage DNA modification pathways (Table S2).
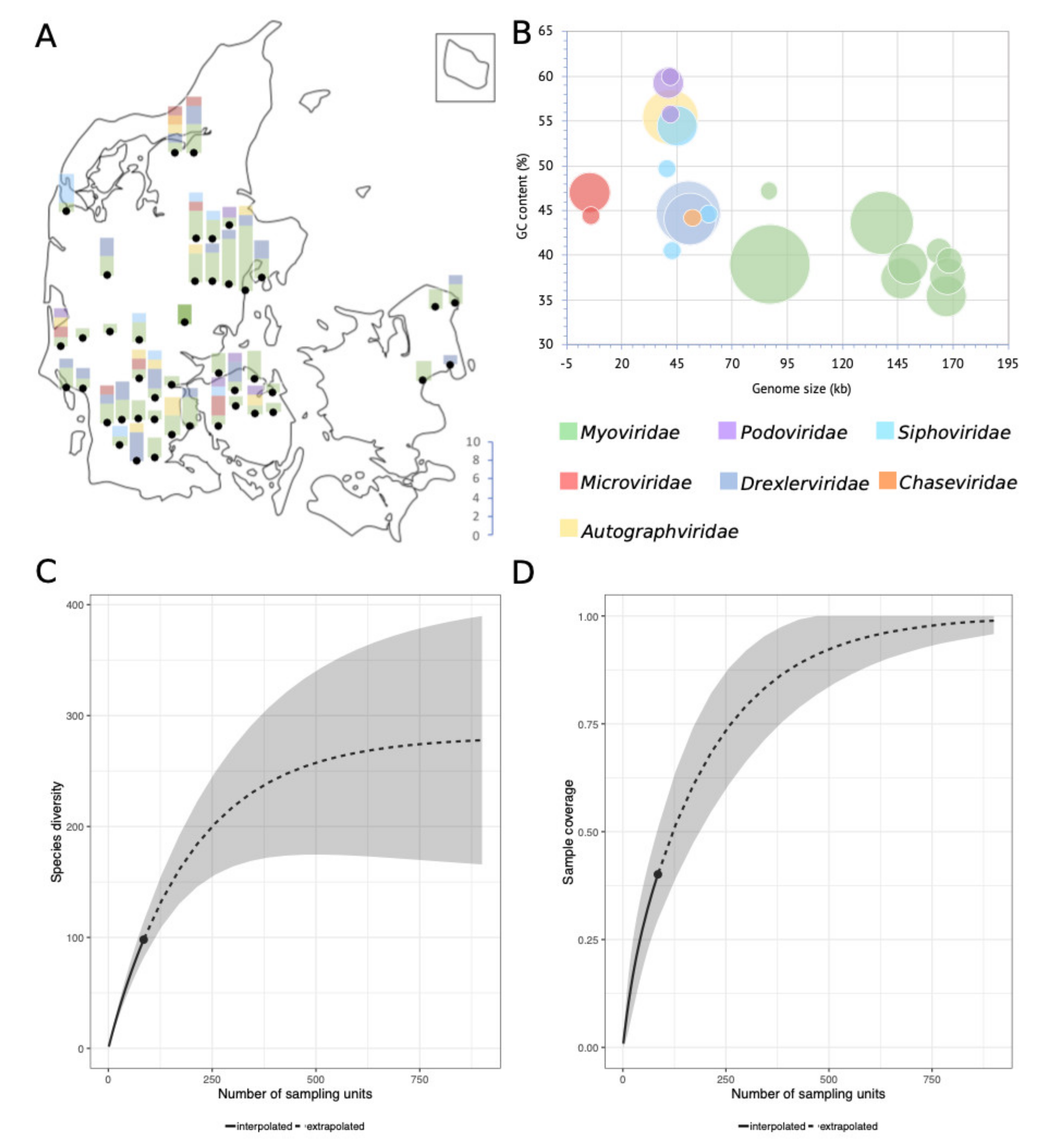
The isolation method (HiTS) favours easily culturable plaque-forming virulent phages [25]. Still, even though we screened wastewater samples, which is a commonly used source for isolation of coliphages, we identified 136 coliphages of which 92 differed by ≥5% from published phage genomes and some with nucleotide (NT) similarities as low as 29% (Table 1). Based on Blastn analyses and the 95% nucleotide similarity demarcation, 104 of the coliphages are unique phage species (Table 1, Figure 1). Based on DNA homology and phylogeny, the 104 unique coliphages group into 14 distinct clusters and 7 single phages (Figure 1 and Figure S1). Coliphages were identified in samples from 44 of the 48 WWTPs (Table S1). There was no substantial difference in phage diversity distribution between samples of urban or rural origin (Figure S2). Samples without coliphages likely reflect the crude nature of the screening method and in some cases sequence or assembly issues and not actual absence. From the majority of positive samples (n = 58) a single phage was sequenced, though some lysates held more than one phage (28 lysates: 2 phages, 6 lysates: 3 phages, 1 lysate: 4 phages, Table S1).
The 95% nucleotide identity demarcation of species is an arbitrary delimitation. It does not consider the biological importance of the non-identical sequence parts and imposes a discrepancy between the demarcation of species depending on genome size. However, it provides a means to quantify and compare relatedness enabling estimations of, e.g., culturable virulent coliphage species richness in the Danish wastewater environment (Figure 2. An extrapolation of species richness (q = 0) predicts a total of 311 distinct species (requiring a sample size of ∼900 phages) (Table S3). The relatively small sample size in this study (n = 136) may subject the estimation to a large prediction bias. The sampling-method also introduces bias by selecting for abundance, latency, and burst size, thereby potentially underestimating diversity. Sequencing and assembly methods as well as the choice of a host further reduce the number of detected phage genomes. Nonetheless, the results indicate the minimal diversity of culturable virulent dsDNA coliphages (MG1655, K-12) in Danish wastewater, estimated to be as a minimum in the range of 183 to 350 unique phage species (Figure 2, Table S3).
The diversity of tailed dsDNA coliphages is well documented [8,22,23] and it is to be expected that a screening of nearly 200 wastewater samples would yield hitherto unknown phages. However, considering the use of only a single host strain and a crude isolation method ensuring that only a single or the few most successful phage(s) from each sample were sequenced, then the degree of novelty and diversity revealed is remarkable and verifies our hypothesis, as well as the efficiency of the HiTS method for exploring diverse phages of a single host [25].
3.2. Taxonomy of the 104 Novel Coliphages
Based on the confirmed morphology of closely related phages, six different families of the order Caudovirales are represented; Myoviridae (57.4%), Drexlerviridae former Siphoviridae (18.4%), Autographviridae former Podoviridae (7.4%), Siphoviridae (6.6%), Podoviridae (3.7%) and Chaseviridae (0.7%), as well as Microviridae (5.9%), order Petitvirales (Figure 1). A similar distribution of coliphages from surface water, manure, sewage, and animal faeces was found by Korf et al. (2019); 70% Myoviridae, 22% Siphoviridae, and 8% Podoviridae [23]. Grose & Casjens (2014) also identified more clusters belonging to the Myoviridae, than the Siphoviridae and the fewest for the Podoviridae when analysing genomes of Caudovirales infecting Enterobacteriaceae [8]. Jurczak-Kurek et al. (2016) found more Siphoviridae than Myoviridae, but also found the Podoviridae to be the least abundant coliphages in sewage [22]. However, these distributions likely reflect abundance distributions of culturable phages and not necessarily natural abundances.
3.2.1. Fifty-Five Myoviridae Species
The 55 Myoviridae phage species represent the greatest span in genome sizes, from the Suspvirus mistaenkt (86.7 kb) to the Dhakavirus dhaeg (170.8 kb) (Figure 2B), and all, except the Krischvirus, code for tRNAs (Table 1). The Myoviridae group into eight distinct clusters and one single phage (mistaenkt), representing three subfamilies; Tevenvirinae, Vequintavirinae and Ounavirinae, in addition to the Phapecoctaviruses and a cluster of six unclassified Myoviridae (Figure 1, Table 1). The Tevenvirinae represent four genera, two krischviruses, five tequatrovirus, two dhakaviruses and two mosigviruses notable for their ability to perform arabinosylation of hmC [56].
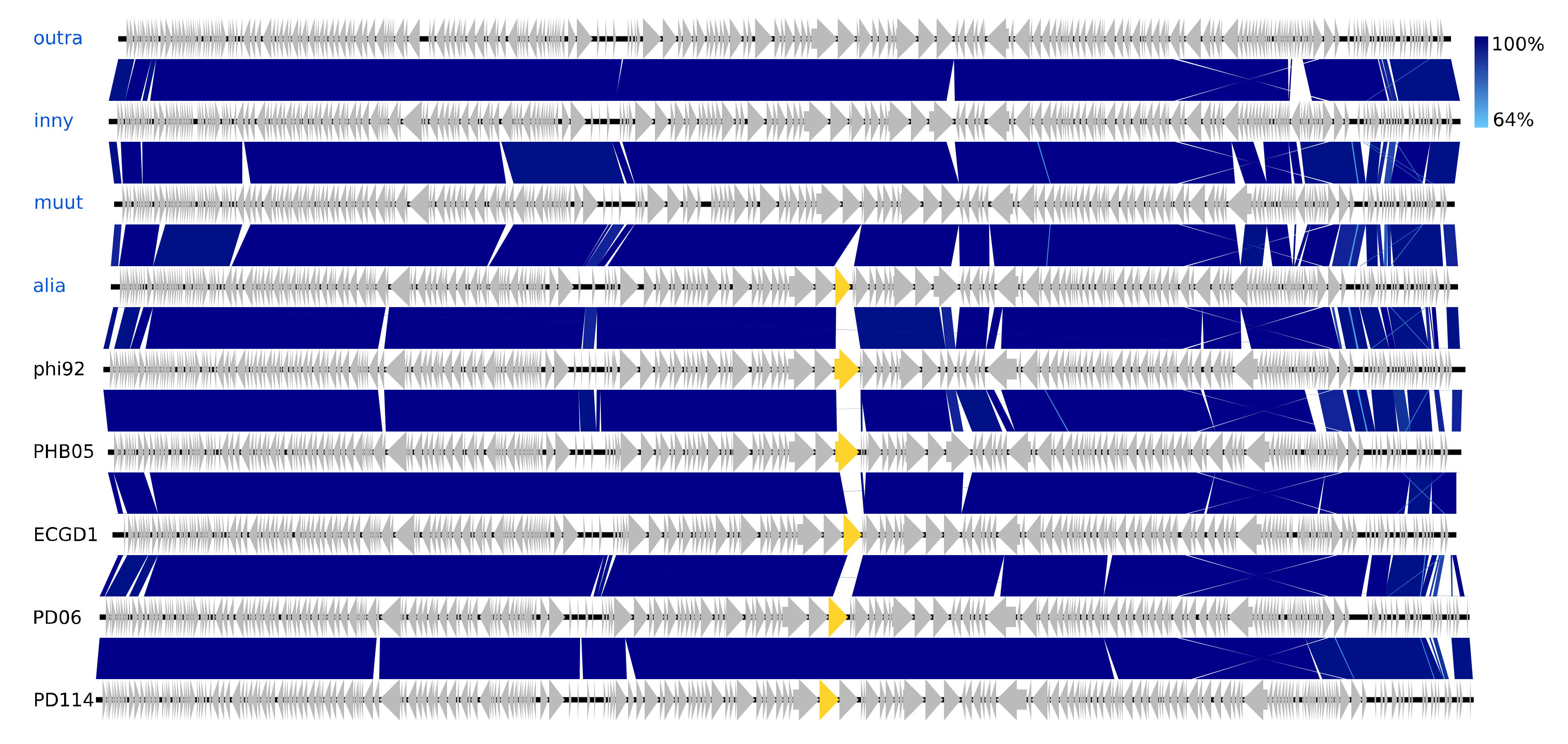
The isolated Vequintavirinae are all vequintaviruses closely related (91.1–93.8%, BLAST) to classified species and were identified in samples from 12 of the 48 WWTPs. All of the Ounavirinae but the Suspvirus mistaenkt are felixounaviruses (89.7–93.9%, BLAST). The Felixounavirus is a relatively large genus with 16 recognized species isolated from Escherichia and Salmonella. In this study, felixounaviruses were identified 33 times in samples from no less than 23 WWTPs, indicating that they are ubiquitous in the Danish wastewater environment and that they are easily cultivated, though the method prevents assessment of relative preponderance. Felixounaviruses often have broad within-genus host ranges, and isolates have been shown to be able to rapidly expand their host range when challenged, co-coinciding with mutations in the long tail gene [57,58]. Five of the Myoviridae are members of the newly announced genus Phapecoctavirus with substantial similarity (86–90%, BLAST) to the type species Escherichia phage phAPEC8 (JX561091) [23,59]. The five phages in the last of the Myoviridae clusters are an even more homogeneous group than the phapecoctaviruses (Figure 3 and Figure S1). All five are closely related (92–95%, BLAST) to the same five unclassified Enterobacteriaceae phages vB_Ecom_PHB05 (MF805809), vB_vPM_PD06 (MH816848), ECGD1 (KU522583), phi92 (NC_023693), and vB_vPM_PD114 (MH675927) [60,61]; this group of nine phages is distinct (<44% NT similarity, BLAST) from all other described phages and thus represents a yet to be classified genus, presumably with the first sequenced phage phi92 as type species. Phi92 was isolated in 1982 and has been thoroughly characterised; it has a broad across-genus (Salmonella, Escherichia) host range enabled by multiple divergent tail fibres and can infect both non-capsulated and encapsulated hosts as it has a unique endosialidase tailspike encoded by gene 143 [60,61]. Interestingly, this gene appears to be unique for phi92, though other versions of a putative tailspike are present at the same position in the genomes of alia, PHB05, ECGD1, and the two PD06 and PD114 phages (Figure 3). Both the phapecoctaviruses and the unclassified Myoviridae genomes code for a complete dTDP-rhamnose biosynthesis pathway. The presence of a dTDP-rhamnose biosynthesis pathway in the DNA metabolism region of phage genomes is peculiar; one possible explanation is that these phages utilize rhamnose for glycosylation of hydroxy-methylated NTs in the same manner as the T4-generated glucosyl-hmC [56].
3.2.2. A New Addition to the Small Family Chaseviridae
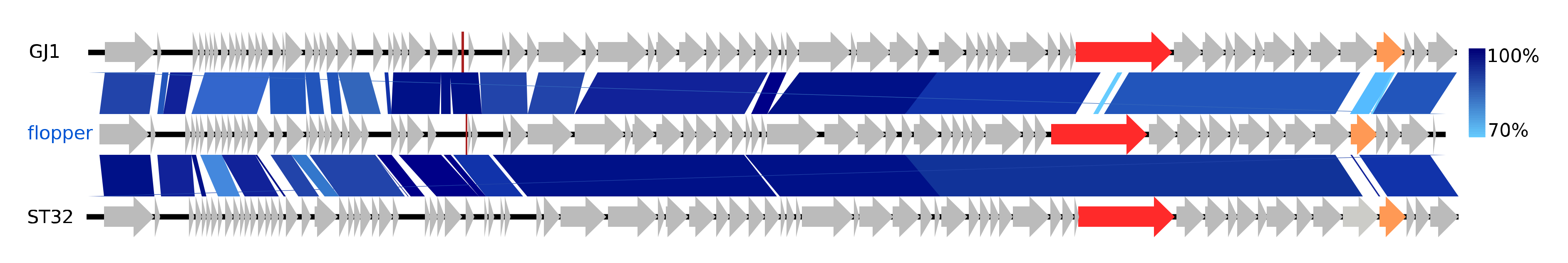
The distinctive phage flopper only shares NT similarity (38.5–87%, BLAST) with ten other phages; it belongs to the newly established Carltongylesvirus (80.8–87% NT similarity, BLAST) of the new family Chaseviridae. This family currently has only nine species and the Carltongylesvirus only two species, Escherichia phage phiEcoM_GJ1 (EF460875) and Escherichia phage ST32 (MF044458). Both type species GJ1 and ST32 have broad within-genus host ranges [62,63]. NT similarity between flopper and GJ1 is partially low in the gene for the putative tail tape measure and also low between all three phage genomes in a tail fiber gene (Figure 4). The carltongylesviruses are unique in having characteristic Myoviridae morphology, i.e., icosahedral head, neck, and a contractile tail with tail fibres and also code for RNA polymerases, a feature otherwise characteristic to the T7-like phages of the Autographiviridae family [62,64,65].
3.2.3. Six Microviridae of Two Genera
The single phage Lilleven and the five gequatroviruses belong to the subfamily Bullavirinae, family Microviridae, order Petitvirales, characterised by ssDNA non-enveloped icosahedral phages (Table 1). Lilleven is a novel species of the genus Alphatrevirus, closely related to (93.9% NT similarity, BLAST, 89–90% AA similarity, Gegenees) the Alphatrevirus Enterobacteria phage St1 (NC_012868) (Figure S3). The five gequatroviruses only differ from one another by single NT polymorphisms and in non-coding regions (Figure S3). They cluster and share genomic organisation and extensive NT similarity (92.6–94.5%, BLAST) with the unclassified Microviridae Escherichia phage SECphi17 (LT960607), but only have 59.1–67.9% NT similarity (BLAST) with recognised Gequatrovirus species, with which they have almost no sequence similarity in the region coding for the major spike protein (gpG), a distinctive marker of the subfamily Bullavirinae involved in host attachment (Figure S3, Table 1) [66]. However, considering the pronounced gene synteny between their relatively small genomes and a conserved AA similarity (62–64%, Gegenees), they are considered gequatroviruses.
The sequencing of the Microviridae is peculiar, as library preparation with the Nextera® XT DNA kit applies transposons targeting dsDNA. However, during Microviridae infection, the host polymerase converts the viral ssDNA into an intermediate state of covalently closed dsDNA, which is then replicated in a rolling circle by viral replication proteins transcribed by the host RNA polymerase [67]. This intermediate state may have enabled the library preparation. The presence of host DNA (2.8–39.1% of reads) in the sequence results of these samples indicates an insufficient initial DNase I treatment (Table S4), which can be attributed to chemical inhibition or inactivation of the enzyme by adhesion to the sides of wells. Hence, it is reasonable to assume that the extracted microvirus DNA was captured as free dsDNA inside host cells during ongoing infections.
3.2.4. Twenty Drexlerviridae Phages Including a New Linage Representative
The 20 species of the new family Drexlerviridae represent a considerable expansion of the new subfamily Tempevirinae [68]. Eight of the Drexlerviridae belong to the new genus Warwickvirus (five species) with Escherichia virus swan01 as type species (LT841308), as they have ≥84.9% NT similarity (BLASTS) to recognised species thereof. The other eight are of the genus Hanrivervirus (NT: 86–90%, BLAST and AA: 77–85% Gegenees, Figure S4), currently consisting of only the type species Shigella virus pSf-1 (NC_021331) isolated from the Han River in Korea [69]. The warwickviruses and hanriverviruses isolated in this study all have comparable genome sizes, GC contents, and gene organisation with the respective type species (Figure 5, Table 1). During their differentiation, many deletions and insertions of small hypothetical genes have occurred; most notable is a unique version of a putative tail-spike protein in seven of the new Hanrivervirus species and all of the new Warwickvirus species, indicating a variety of divergent host ranges (Figure 5). All the hanriverviruses code for (putative) dam, and Psf-1 is resistant against at least six restriction endonucleases [69], suggesting that these phages employ DNA methylation as a defence strategy.
The last Drexlerviridae is Jahat. The warwickviruses and hanriverviruses form a monophyletic clade together with Jahat (Figure 1 and Figure S1). Even though Jahat has its own branch, this phage has gene synteny, slightly higher but comparable GC content, and shares an equal degree of NT similarity ≤68.7% with phages of both the Hanrivervirus and Warwickvirus (Figure 5). Hence, Jahat cannot with confidence be assigned to either genus but falls in between, barely different enough to represent its own genus—an indicator of the genetic continuum of phages challenging taxonomic delimitations.
3.2.5. Eight Siphoviridae Species and a Novel Genus Representative
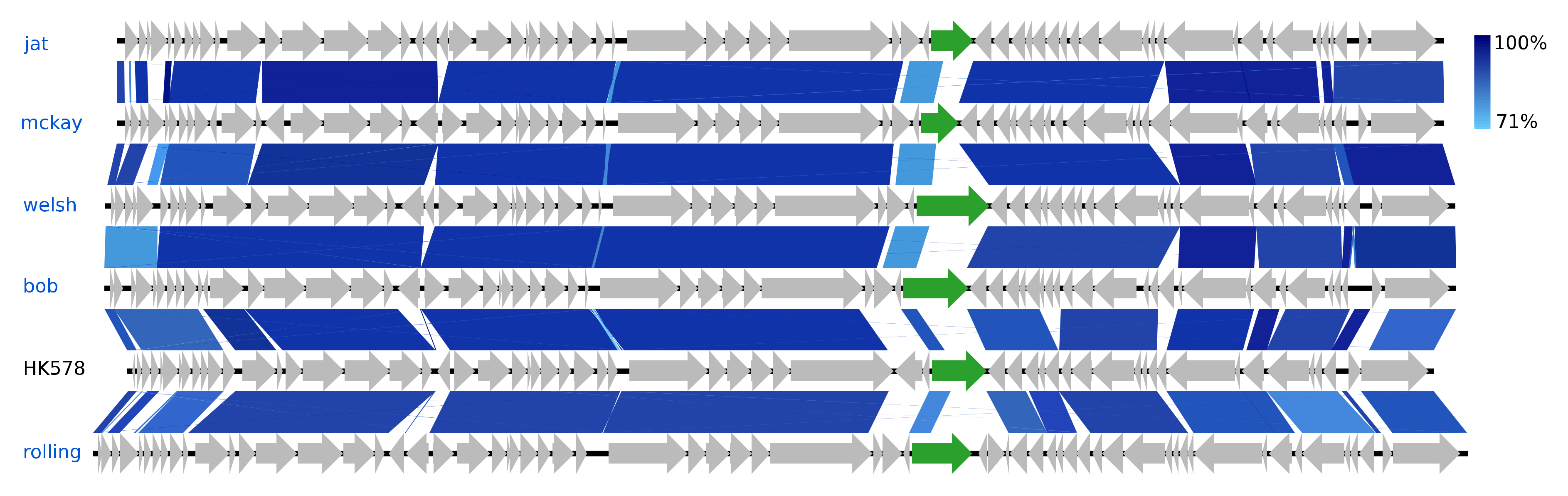
The eight Siphoviridae species vary greatly in GC content, ranging from 44.6% (Skure) to 54.6% (welsh), but are quite similar in genome sizes, 49.7–54.6 kb (Figure 2). Five of these phages are of the genus Dhillonvirus as they have substantial NT similarity (77–80%, BLAST) and pronounced gene synteny with the type species Escherichia virus HK578. As with the hanriverviruses and warwickviruses, their genomes only differ in minor hypothetical genes and have limited NT similarity in a gene of highly variable length coding for a tail fiber (gp26 in HK578) (Figure 6), a phenomenon also observed in the dhillonviruses isolated by Korf et al. (2019), which correspondingly had divergent host ranges [23]. Each of the three remaining Siphoviridae represents a different genus. Based on NT similarity and the presence of the canonical 7-deazaguanine operon, Skure is of the 13-species genus Seuratvirus, while buks is assigned to the two-species genus Jerseyvirus, subfamily Guernseyvirinae.
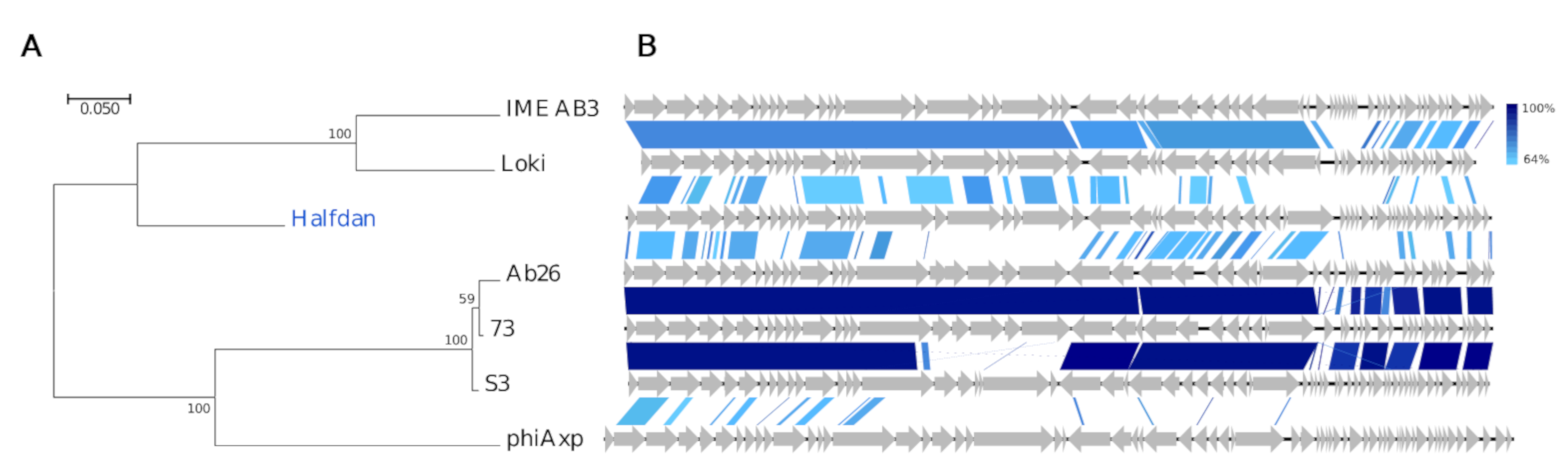
Interestingly, the Siphoviridae Halfdan has only minuscule similarity with described phages (12–29%, BLAST). These entail two Pseudomonas phages vB_PaeS_SCUT-S3 (MK165657) and Ab26 (HG962376) [70], both Septimatreviruses, two Acinetobacter phages of the Lokivirus IMEAB3 (KF811200) and type species Acinetobacter virus Loki [71], and to a lesser degree the unclassified Achromobacter phage phiAxp-1 (KP313532) [72]. They have a common gene organization, yet their intra-Gegenees scores are low (≤1% BLASTn, <43% BLASTx, Figure S5), and NT similarity is negligent in roughly one-third of Halfdan’s 57 CDSs (Figure 7). The TerL-based phylogeny and AA similarity also indicate a distant relation, although grouping Halfdan closer with the lokiviruses (40–43%, Gegenees BLATSx) than the septimatreviruses (33–34%, Gegenees BLATSx) (Figure 7, Figure S5). Clearly Halfdan is distinct from all other described phages and hence the first phage sequenced of a new Siphoviridae genus.
3.2.6. Nine Autographiviridae Species
The nine Autographiviridae all have the hallmarks of this new family, i.e., unidirectionally encoded genes and RNA polymerases [65,68,73]. They belong to the genus Bonnellvirus, as they have conserved gene organisation, a similar GC content, and also share considerable NT similarity (69–93%, BLAST) with the type species Enterobacteria phage J8-65 (NC_025445) (Figure S6). The genomes of the nine new bonnellviruses and J8-65 are highly similar with differences primarily in small hypothetical genes, though Lidtsur codes for a unique version of tailspike colanidase (Figure S6). Lidtsur was deposited to the NCBI GenBank before the others and is currently the only one which is an ICTV-approved species representative.
3.2.7. Four Podoviridae Species Including Two Novel Genus Representatives
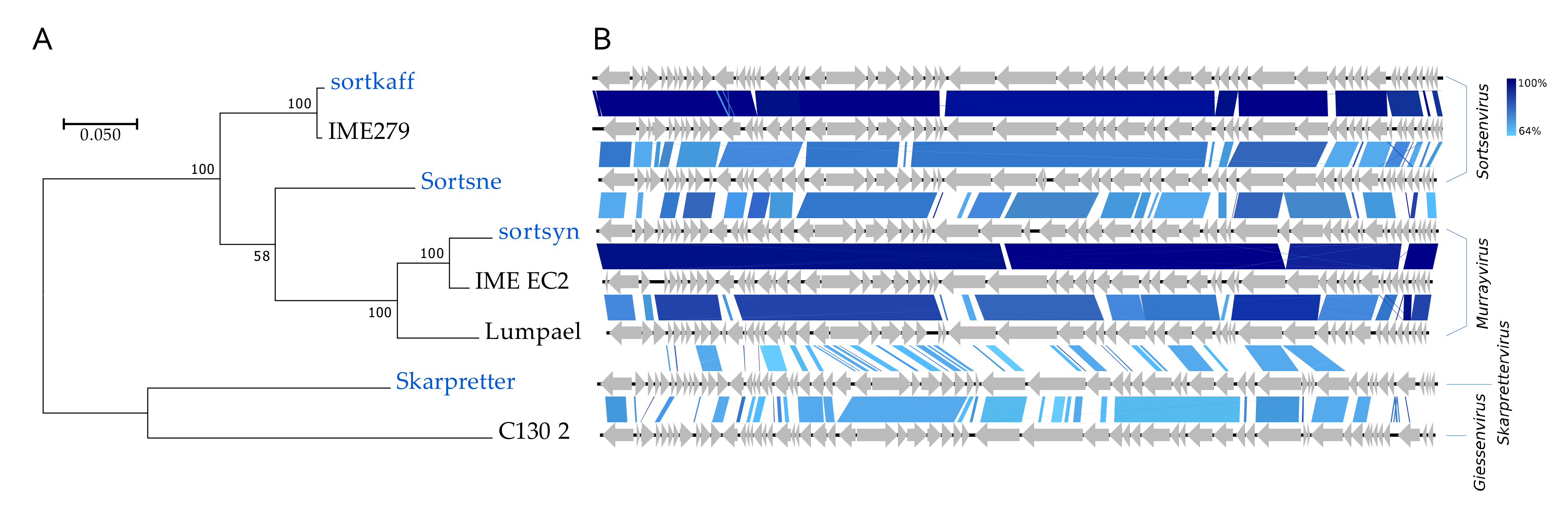
The four Podoviridae all have high (>59%) GC contents and represent no less than three distinct genera (Figure 2, Figure 8). Skarpretter is the type and only species of the genus Skarprettervirus [74]. Skarpretter is distinct from all described phages sharing only 38% NT similarity (BLAST) with the Giessenvirus Escherichia phage C130_2 (MH363708) isolated from cheese [75] (Figure 8 and Figure S7). Sortsne is the type species of the genus Sortsnevirus [74], currently consisting of only Sortsne and Klebsiella phage vB_KpnS_IME279 (MF614100); however, based on high NT similarity and conserved gene organization with IME279 (89.8%, BLAST), we suggest that sortkaff also belongs in Sortsnevirus (Figure 8 and Figure S7). The last Podoviridae sortsyn is of the new 2-species genus Murrayvius [76], as it shares a high degree of NT similarity and has conserved gene organization with the type species Enterobacteria phage IME_EC2 (KF591601) isolated from hospital sewage [77] (Figure 8).
3.3. The Wastewater Coliphages Are Largely Absent in Metaviromes
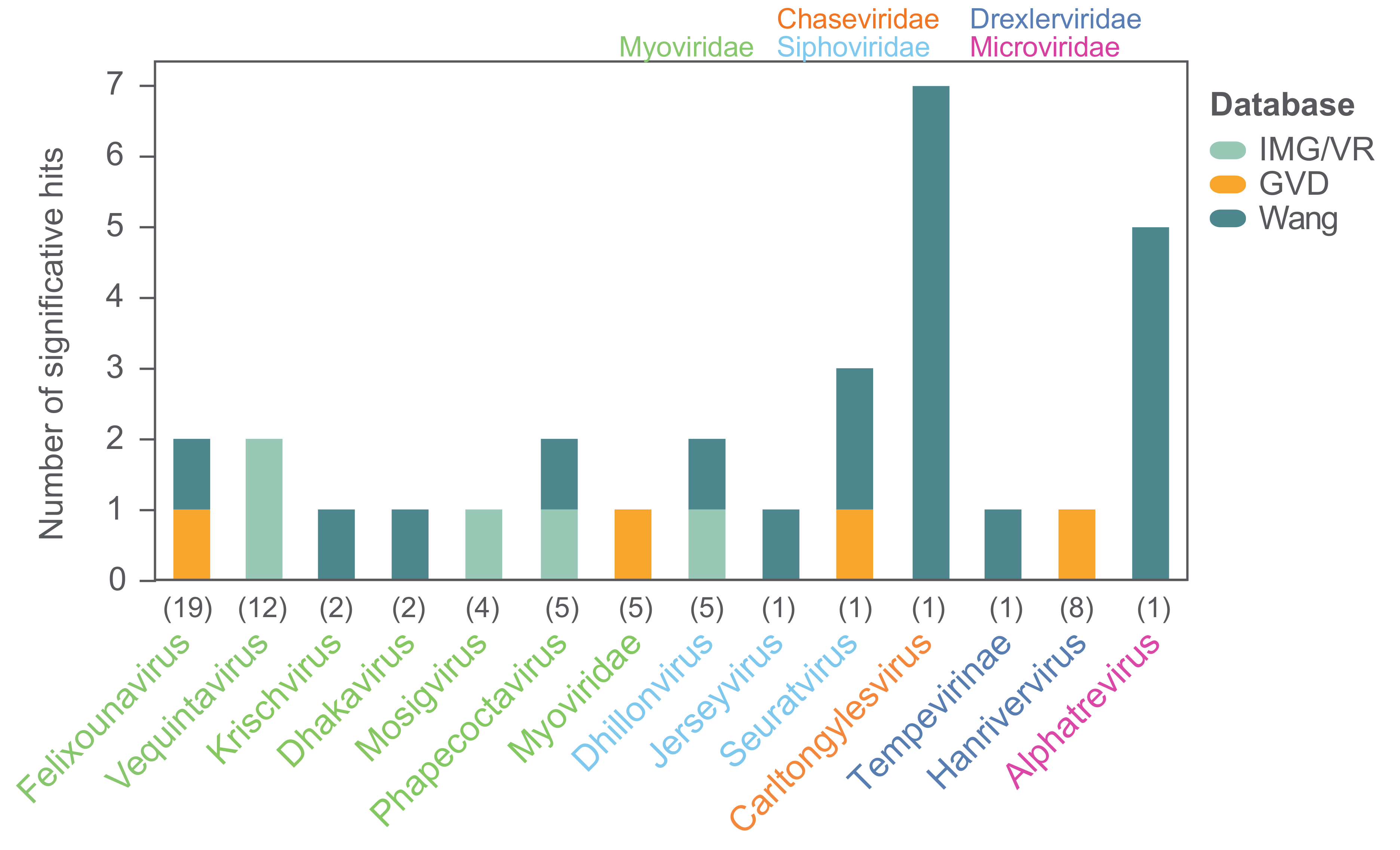
In order to investigate the prevalence of the 104 coliphage species in different environments we mapped the reads of 510 metagenomes from studies of primarily Danish wastewater, pig, and human gut samples (Table S5) [78,79]. The threshold for significant hits was set as mapped reads covering ≥70% of a coliphage genome, and the distribution of the mapped reads was assessed to verify that this threshold ensured identification of closely related phages (Figure S8). No hits were found for any of the coliphages. This is likely a consequence of sequencing depth and sample preparation, as prior to sequencing, these metagenome samples were concentrated by centrifugation as a pellet or by CsCl gradient and the supernatant was either discarded or stored for future studies, and as a result, a large proportion of potential phage reads was omitted. Subsequently, we also searched for the coliphages in hundreds of metavirome datasets (Table S5) from Irish and Chinese faecal, human, animal, and water samples using the same read mapping method (Table S6). There were no hits to the human faecal contigs from Ireland [80], while 22 of the 104 coliphage genomes (21%) representing ten genera were covered by >70% by reads from 10 (mammals and birds) of the 38 (26%) Chinese Wang study libraries (Figure 9) [80]. For most phage genera, only reads from a single sample matched, though reads from five metaviromes (pet dog, pig, yak, and flamingo faeces) matched (>70% read coverage) with the Alphatrevirus Lilleven, and reads from seven metaviromes (dog, red panda, giant panda, non-human primate, masked civet, pig, and chicken faeces) matched (>70% read coverage) with the Carltongylesvirus flopper (Table S6). Finally, the genome sequences of the 104 coliphages (queries) were blasted against a database of 735,106 uncultured viral genomes (UVIGs) from the Integrated Microbial Genomes/Virus (IMG/VR) database, derived from a wide range of sample types including marine, freshwater, terrestrial, and hosts [51], as well as 13,203 UVIGs from human gut samples retrieved from the human gut virome (GVD) database (Table S7) [52]. The coliphage genomes were also blasted against the 8392 isolated virus genomes (iVGs) of the IMG/VR database and based on the observed alignment coverage distribution (Figure S9), significant matches were defined as those covering >80% of coliphage genomes. With this threshold, there were significant matches for 23 of the 104 (22%) coliphage genomes to four of the 735,106 (0.0005%) IMG/VR UVIG sequences (Figure 9, Table S7).
Only phages from 14 of the 24 taxonomic groups of coliphages from this study had matches in the virome databases assessed. For only 14 of the 62 coliphages with matches, a closely related phage could be identified in more than one virome. Although the coliphages are omnipresent and culturable in Danish wastewater, they are for a large part not represented in metagenomic data, and therefore these coliphage genomes provide valuable information. A lack of representation in metagenomic data could be caused by low natural abundance as this would result in insufficient sequencing depth for genome assembly within metagenomes/-viromes. The Siphoviridae Halfdan, the Myoviridae mistaenkt, and the phages of the Drexlerviridae genus Warwickvirus, the Autographviridae genus Bonnellvirus, all the Podoviridae genera, Sortsnevirus, Murrayvirus and Skarprettervirus, as well as the Microviridae genus Gequatrovirus did not match any UVIG sequences, nor did the reads from any virome cover ≥70% of their genomes. Surprisingly, the gequatroviruses were not detected in any of the gut-viromes, even though that apart from temperate and crAss-like phages, Microviridae dominates in human, mammal, and bird gut-microbiomes and that 860 Microviridae genomes were assembled from the assorted Wang et al. (2019) metaviromes [80,81]. However, due to the relatively small size of the Microviridae genomes, substitution of a single gene is enough to warrant a ∼5–33% difference in NT similarity, putting them below the set threshold for identification. The fact that both Halfdan and the bonnellviruses and all the Podoviridae represent novel genera with very few close relatives suggests that these lineages are under-sampled and not sufficiently abundant in the environments explored by metagenomic sequencing and assessed in this study. These findings underline the importance of isolating and sequencing individual phages in order to uncover diversity. It is plausible that phages selected for by plating techniques are not those that are naturally abundant; however, this cannot be concluded based on these results. Future studies should compare the diversity obtained by isolating to metagenomic sequencing of metaviromes of identical samples in order to establish the degree of discrepancy between these two methods.
4. Conclusions
By screening 188 wastewater samples, we identified 104 coliphages species (MG1655–K12), enabling us to predict the species richness of culturable virulent dsDNA coliphages in Danish wastewater, which is predicted to be at least 183–350 and expected to fluctuate drastically over time. The true species richness is likely even higher as the isolation, DNA extraction, library construction, and genome assembly method as well as the choice of a host all are liable to reduce the number of phages detected. Ninety-two of the newly isolated coliphages represent novel species of seven families; Myoviridae, Siphoviridae, Podoviridae, Drexlerviridae, Chaseviridae, and Microviridae. Though most of them distribute into 18 established genera, the diversity of these many phages isolated from a single strain is notable. They vary greatly in genome size and have a broad GC content range.
Apart from the analyses applied, the main difference between this and the comprehensive Korf et al., study from 2019 [23] is the isolation approach. Korf et al., isolated 50 phages from various sample types over several years from a wide collection of clinical E. coli isolates, whereas the wastewater sample collection and phage isolation in this study were performed in a matter of weeks on a single strain of E. coli. Still, the distribution of phage types including many of the same genera and the discovery of a handful of phages with limited similarity to known phages are in many aspects comparable, suggesting that the method of isolation (plaque purification) is perhaps the key limiting factor for uncovering the diversity of coliphages. However, fewer than 60% of the 104 coliphages are represented in the assessed metaviromes, emphasising the importance of cultivating phages to uncover the true diversity.
These findings add to our understanding of phage ecology and diversity, and through classification of these many phages we come yet another step closer to a more refined taxonomic understanding of phages. Furthermore, the numerous and diverse phages isolated in this study, all lytic to the same single strain, serve as an excellent opportunity to learn important phage-host interactions in future studies. These include, but are not limited to, lysogen-induced phage immunity, host-range, and anti-RE systems.
Finally, the first genus representative for at least three novel genera was sequenced in this study. Skarprettervirus and Sortsnevirus of the Podoviridae have been accepted by the ICTV. We propose that Halfdan is the type species of a new Siphoviridae genus, that the four novel Myoviridae species muut, alia, outra, and inny together with five unclassified Myoviridae also represent a new genus, and as the Drexlerviridae Jahat cannot with confidence be assigned to any described genera, Jahat may also represent its own lineage. In conclusion, this study shows that uncharted territory remains for even well-studied phage hosts and that cultivation approaches uncover vital genomes that seem absent from metagenomic studies.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/1999-4915/12/9/986/s1, Figure S1: VIRIDIC Plot of intergenomic sequence similarity, aligned genome fraction and genome length ratio for the 104 coliphage species, Figure S2: Distribution of the 136 coliphage species per WWTP location (urban vs rural), Figure S3: Phylogenetic and genomic analyses of the six Microviridae coliphages, Figure S4: Phylogenomic nucleotide distances (Gegenees, BLASTn) for the Hanrivervirus coliphages, Figure S5: Phylogenomic nucleotide distances (Gegenees, BLASTn) for phage Halfdan, Figure S6: Phylogenetic and genomic analyses of the Bonnellvirus coliphages, Figure S7: Phylogenomic nucleotide distances (Gegenees, BLASTn) for the Podoviridae coliphages, Figure S8: Mapped reads from the Wang et al. (2019) study to the genomes of flopper and Lilleven, Figure S9: Alignment coverage distribution of IMG/VR iVGs to the 104 coliphage genomes and significant hits to these, Table S1: List of phages per wastewater sample and corresponding sequence result metrics, Table S2: AMG screening results, Table S3: Species richness estimations, Table S4: Read mapping results for samples with Microviridae, Table S5: List of sequence data assessed for matches with the 104 coliphage genomes, Table S6: Mapped reads from the Wang et al. (2019) study and the IMG/VR microbiomes, Table S7: IMG/VR and GVD blast results.
Author Contributions
Conceptualization, N.S.O., W.K. and L.H.H.; Data curation, N.S.O. and L.F.-J.; Formal analysis, N.S.O. and L.F.-J.; Funding acquisition, W.K. and L.H.H.; Investigation, N.S.O.; Methodology, N.S.O., W.K. and L.H.H.; Project administration, N.S.O., W.K. and L.H.H.; Resources, N.S.O., W.K. and L.H.H.; Supervision, W.K. and L.H.H.; Validation, N.S.O. and W.K.; Visualization, N.S.O.; Writing–original draft, N.S.O.; Writing–review & editing, N.S.O., L.F.-J., W.K. and L.H.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Human Frontier Science Program Grant RGP0024/2018, Villum Experiment Grant 17595, and Aarhus University Research Foundation AUFF Grant E-2015-FLS-7-28.
Acknowledgments
The authors thank the many members of the Danish Water and Wastewater Association (DANVA) who kindly supplied us with time-series of wastewater samples from their WWTPs. A special thank you is extended to the Billund and Grindsted WWTP of Billund Vand & Energi, Lynetten, Avedøre and Damhusåen WWTP of BIOFOS, Kolding WWTP of BlueKolding, Esbjerg Øst, Esbjerg Vest, Ribe, Varde og Skovlund WWTPs of DIN Forsyning, Drøsbro, Hadsten, Hammel, Hinnerup and Voldum WWTPs of Favrskov Forsyning, Haerning WWTP of Herning Vand, Hillerød WWTP of Hillerød Forsyning, Lemvig WWTP of Lemvig Vand og Spildevand, Bevtoft, Gram, Haderslev, Halk, Jegerup, Nustrup, Over Jerstal, Skovby, Skrydstrup, Sommersted, Vojens and Årøsund WWTPs of Provas, Marselisborg, Egå and Viby WWTPs of Aarhus vand, Hedensted, Juelsminde and Tørring WWTPs of Hedensted Spildevand, Ejby Mølle, Bogense, Hofmansgave, Hårslev, Nordvest, Nordøst, Otterup and Søndersø WWTPs of VandCenterSyd, Øst and Vest WWTPs of Aalborg Forsyning and finally Helsingør WWTP of Forsyning Helsingør.
Conflicts of Interest
The authors declare no competing interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Weinbauer, M.G.; Rassoulzadegan, F. Are viruses driving microbial diversification and diversity? Environ. Microbiol. 2003, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.S.; Wilhelm, S.W. Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000 Biol. Rep. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crummett, L.T.; Puxty, R.J.; Weihe, C.; Marston, M.F.; Martiny, J.B. The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology 2016, 499, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.F. Bacteriophage-Encoded Bacterial Virulence Factors and Phage–Pathogenicity Island Interactions. Adv. Virus Res. 2012, 82, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Fortier, L.-C.; Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 2013, 4, 354–365. [Google Scholar] [CrossRef]
- Volkova, V.V.; Lu, Z.; Besser, T.; Gröhn, Y.T. Modeling the infection dynamics of bacteriophages in enteric Escherichia coli: Estimating the contribution of transduction to antimicrobial gene spread. Appl. Environ. Microbiol. 2014, 80, 4350–4362. [Google Scholar] [CrossRef] [Green Version]
- Bearson, B.L.; Allen, H.K.; Brunelle, B.W.; Lee, I.S.; Casjens, S.R.; Stanton, T.B. The agricultural antibiotic carbadox induces phage-mediated gene transfer in Salmonella. Front. Microbiol. 2014, 5, 52. [Google Scholar] [CrossRef] [Green Version]
- Grose, J.H.; Casjens, S.R. Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae. Virology 2014, 468, 421–443. [Google Scholar] [CrossRef] [Green Version]
- Dykhuizen, D. Species Numbers in Bacteria. Proc. Calif. Acad. Sci. 2005, 56, 62. [Google Scholar]
- Hatfull, G.F.; Pedulla, M.L.; Jacobs-Sera, D.; Cichon, P.M.; Foley, A.; Ford, M.E.; Gonda, R.M.; Houtz, J.M.; Hryckowian, A.J.; Kelchner, V.A.; et al. Exploring the Mycobacteriophage Metaproteome: Phage Genomics as an Educational Platform. PLoS Genet. 2006, 2, e92. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F. Mycobacteriophages: Windows into Tuberculosis. PLoS Pathog. 2014, 10, e1003953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfull, G.F.; Jacobs-Sera, D.; Lawrence, J.G.; Pope, W.H.; Russell, D.A.; Ko, C.C.; Weber, R.J.; Patel, M.C.; Germane, K.L.; Edgar, R.H.; et al. Comparative Genomic Analysis of 60 Mycobacteriophage Genomes: Genome Clustering, Gene Acquisition, and Gene Size. J. Mol. Biol. 2010, 397, 119–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedrick, R.M.; Jacobs-Sera, D.; Bustamante, C.A.; Garlena, R.A.; Mavrich, T.N.; Pope, W.H.; Reyes, J.C.; Russell, D.A.; Adair, T.; Alvey, R.; et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2017, 2, 16251. [Google Scholar] [CrossRef] [PubMed]
- Jacobs-Sera, D.; Marinelli, L.J.; Bowman, C.; Broussard, G.W.; Bustamante, C.G.; Boyle, M.M.; Petrova, Z.O.; Dedrick, R.M.; Pope, W.H.; Advancing, S.E.; et al. On the nature of mycobacteriophage diversity and host preference. Virology 2012, 434, 187–201. [Google Scholar] [CrossRef]
- Hatfull, G.F. The Secret Lives of Mycobacteriophages. Adv. Virus Res. 2012, 82, 179–288. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic. Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [Green Version]
- European Nucleotide Archive ENA Browser. Available online: https://www.ebi.ac.uk/ena/browser/text-search?query=escherichiaphage (accessed on 29 May 2020).
- Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Imbroglios of viral taxonomy: Genetic exchange and failings of phenetic approaches. J. Bacteriol. 2002, 184, 4891–4905. [Google Scholar] [CrossRef] [Green Version]
- Aiewsakun, P.; Adriaenssens, E.M.; Lavigne, R.; Kropinski, A.M.; Simmonds, P. Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: Steps towards a unified taxonomy. J. Gen. Virol. 2018, 99, 1331–1343. [Google Scholar] [CrossRef]
- Nelson, D. Phage taxonomy: We agree to disagree. J. Bacteriol. 2004, 186, 7029–7031. [Google Scholar] [CrossRef] [Green Version]
- Adriaenssens, E.; Brister, J.R. How to Name and Classify Your Phage: An Informal Guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Jurczak-Kurek, A.; Gąsior, T.; Nejman-Faleńczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Necel, A.; Jakubowska-Deredas, M.; Narajczyk, M.; Richert, M.; et al. Biodiversity of bacteriophages: Morphological and biological properties of a large group of phages isolated from urban sewage. Sci. Rep. 2016, 6, 34338. [Google Scholar] [CrossRef] [PubMed]
- Korf, I.H.; Meier-Kolthoff, J.P.; Adriaenssens, E.M.; Kropinski, A.M.; Nimtz, M.; Rohde, M.; Van Raaij, M.J.; Wittmann, J. Still Something to Discover: Novel Insights into Escherichia coli Phage Diversity and Taxonomy. Viruses 2019, 11, 454. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, A.; Dion, M.; Deng, L.; Tremblay, D.; Moncaut, E.; Shah, S.A.; Stokholm, J.; Krogfelt, K.A.; Schjørring, S.; Bisgaard, H.; et al. Virulent coliphages in 1-year-old children fecal samples are fewer, but more infectious than temperate coliphages. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Olsen, N.S.; Hendriksen, N.B.; Hansen, L.H.; Kot, W. A New High-throughput Screening (HiTS) Method for Phages—Enabling Crude Isolation and Fast Identification of Diverse Phages with Therapeutic Potential. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kot, W.; Vogensen, F.K.; Sørensen, S.J.; Hansen, L.H. DPS—A rapid method for genome sequencing of DNA-containing bacteriophages directly from a single plaque. J. Virol. Methods 2014, 196, 152–156. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic. Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic. Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic. Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- González-Tortuero, E.; Sutton, T.D.; Velayudhan, V.; Shkoporov, A.N.; Draper, L.A.; Stockdale, S.R.; Ross, R.P.; Hill, C. VIGA: A sensitive, precise and automatic de novo VIral Genome Annotator. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Kleinheinz, K.A.; Joensen, K.G.; Larsen, M.V. Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 2014, 4, e27943. [Google Scholar] [CrossRef] [Green Version]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection of Verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE-a database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic. Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of virome function from genomic sequences. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Ågren, J.; Sundström, A.; Håfström, T.; Segerman, B. Gegenees: Fragmented Alignment of Multiple Genomes for Determining Phylogenomic Distances and Genetic Signatures Unique for Specified Target Groups. PLoS ONE 2012, 7, e39107. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC-a Novel Tool to Calculate the Intergenomic 1 Similarities of Prokaryote-Infecting Viruses 2; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2020. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Tavares, P.; Petit, M.A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [Green Version]
- Mercanti, D.J.; Rousseau, G.M.; Capra, M.L.; Quiberoni, A.; Tremblay, D.M.; Labrie, S.J.; Moineau, S. Genomic Diversity of Phages Infecting Probiotic Strains of Lactobacillus paracasei. Appl. Environ. Microbiol. 2016, 82, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic. Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.C.; Ma, K.H.; Chao, A. iNEXT: An R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- Chao, A.; Gotelli, N.J.; Hsieh, T.C.; Sander, E.L.; Ma, K.H.; Colwell, R.K.; Ellison, A.M. Rarefaction and extrapolation with Hill numbers: A framework for sampling and estimation in species diversity studies. Ecol. Monogr. 2014, 84, 45–67. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. RStudio: Integrated Development for R; RStudio Inc.: Boston, MA, USA, 2016. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Paez-Espino, D.; Chen, I.M.; Palaniappan, K.; Ratner, A.; Chu, K.; Szeto, E.; Pillay, M.; Huang, J.; Markowitz, V.M.; Nielsen, T.; et al. IMG/VR: A database of cultured and uncultured DNA viruses and retroviruses. Nucleic. Acids Res. 2017, 45, D457–D465. [Google Scholar] [CrossRef]
- Gregory, A.C.; Zablocki, O.; Howell, A.; Bolduc, B.; Sullivan, M.B. The human gut virome database. bioRxiv 2019. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Download|SourceForge.net. 2013. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 16 June 2020).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- GitHub—Ecogenomics/BamM: Metagenomics-Focused BAM File Manipulation. 2020. Available online: https://github.com/ecogenomics/BamM (accessed on 24 August 2020).
- Thomas, J.A.; Orwenyo, J.; Wang, L.X.; Black, L.W. The odd “RB” phage—Identification of arabinosylation as a new epigenetic modification of DNA in T4-like phage RB69. Viruses 2018, 10, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropinski, A.M.; Sulakvelidze, A.; Konczy, P.; Poppe, C. Salmonella phages and prophages—Genomics and practical aspects. Methods Mol. Biol. 2007, 394, 133–175. [Google Scholar]
- Rivera, D.; Hudson, L.K.; Denes, T.G.; Hamilton-West, C.; Pezoa, D.; Moreno-Switt, A.I. Two phages of the genera felixunavirus subjected to 12 h challenge on salmonella infantis showed distinct genotypic and phenotypic changes. Viruses 2019, 11, 586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsonos, J.; Adriaenssens, E.M.; Klumpp, J.; Hernalsteens, J.P.; Lavigne, R.; De Greve, H. Complete genome sequence of the novel Escherichia coli phage phAPEC8. J. Virol. 2012, 86, 13117–13118. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, D.; Buettner, F.F.; Browning, C.; Nazarov, S.; Rabsch, W.; Bethe, A.; Oberbeck, A.; Bowman, V.D.; Stummeyer, K.; Mühlenhoff, M.; et al. A Multivalent Adsorption Apparatus Explains the Broad Host Range of Phage phi92: A Comprehensive Genomic and Structural Analysis. J. Virol. 2012, 86, 10384–10398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzer, D.; Browning, C.; Stummeyer, K.; Oberbeck, A.; Mühlenhoff, M.; Gerardy-Schahn, R.; Leiman, P.G. Structure and biochemical characterization of bacteriophage phi92 endosialidase. Virology 2015, 477, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Jamalludeen, N.; Kropinski, A.M.; Johnson, R.P.; Lingohr, E.; Harel, J.; Gyles, C.L. Complete genomic sequence of bacteriophage phiEcoM-GJ1, a novel phage that has myovirus morphology and a podovirus-like RNA polymerase. Appl. Environ. Microbiol. 2008, 74, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Geagea, H.; Rousseau, G.M.; Labrie, S.J.; Tremblay, D.M.; Liu, X.; Moineau, S. Characterization of the Escherichia coli Virulent Myophage ST32. Viruses 2018, 10, 616. [Google Scholar] [CrossRef] [Green Version]
- Kropinski, A.M.; Anany, H.; Adriaenssens, E.M. Create One New Family (Chaseviridae) Including Six New Genera in the Order Caudovirales 2019.047B. Available online: https://ictv.global/ICTV/proposals/2019.047B.zip (accessed on 17 May 2020).
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Labrie, S.J.; Dupuis, M.È.; Tremblay, D.M.; Plante, P.L.; Corbeil, J.; Moineau, S. A new Microviridae phage isolated from a failed biotechnological process driven by Escherichia coli. Appl. Environ. Microbiol. 2014, 80, 6992–7000. [Google Scholar] [CrossRef] [Green Version]
- Doore, S.M.; Fane, B.A. The microviridae: Diversity, assembly, and experimental evolution. Virology 2016, 491, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018-2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.W.; Kim, J.H.; Shin, S.P.; Han, J.E.; Chai, J.Y.; Park, S.C. Characterization and complete genome sequence of the Shigella bacteriophage pSf-1. Res. Microbiol. 2013, 164, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Essoh, C.; Latino, L.; Midoux, C.; Blouin, Y.; Loukou, G.; Nguetta, S.P.; Lathro, S.; Cablanmian, A.; Kouassi, A.K.; Vergnaud, G.; et al. Investigation of a Large Collection of Pseudomonas aeruginosa Bacteriophages Collected from a Single Environmental Source in Abidjan, Côte d’Ivoire. PLoS ONE 2015, 10, e0130548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, D.; Wand, M.E.; Briers, Y.; Lavigne, R.; Sutton, J.M.; Reynolds, D.M. Characterisation and genome sequence of the lytic Acinetobacter baumannii bacteriophage vB_AbaS_Loki. PLoS ONE 2017, 12, e0172303. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, E.; Qi, Z.; Li, H.; Wei, X.; Lin, W.; Zhao, R.; Jiang, A.; Yang, H.; Yin, Z.; et al. Isolation and molecular characterisation of Achromobacter phage phiAxp-3, an N4-like bacteriophage. Sci. Rep. 2016, 6, 24776. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.; Kropinski, A.M.; Alfernas-Zerbini, P.; Buttimer, C.; Lavigne, R.; Bister, J.R.; Tolstoy, I.; Morozova, V.V.; Babkin, I.V.; Kozlova, Y.N.; et al. Create one new family (Autographiviridae) including nine subfamilies and one hundred and thirty-two genera in the order Caudovirales. Microb. Biotechnol. 2019, 13, 1428–1445. [Google Scholar]
- Kropinski, A.M.; Adriaenssens, E.M. Create Four New Genera Including Six New Species in the Family Podoviridae 2019.076B. Available online: https://ictv.global/ICTV/proposals/2019.076B.zip (accessed on 17 May 2020).
- Sváb, D.; Falgenhauer, L.; Rohde, M.; Chakraborty, T.; Tóth, I. Complete genome sequence of C130_2, a novel myovirus infecting pathogenic Escherichia coli and Shigella strains. Arch. Virol. 2019, 164, 321–324. [Google Scholar] [CrossRef] [Green Version]
- Kropinski, A.M.; Adriaenssens, E.M. Create One New Genus (Murrayvirus) Including of Two New Species in the Family Siphoviridae 2019.069B. Available online: https://ictv.global/ICTV/proposals/2019.069B.zip (accessed on 17 May 2020).
- Hua, Y.; An, X.; Pei, G.; Li, S.; Wang, W.; Xu, X.; Fan, H.; Huang, Y.; Zhang, Z.; Mi, Z.; et al. Characterization of the morphology and genome of an Escherichia coli podovirus. Arch. Virol. 2014, 159, 3249–3256. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Munk, P.; Njage, P.; Van Bunnik, B.; McNally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J.; et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Xiao, L.; Estellé, J.; Kiilerich, P.; Ramayo-Caldas, Y.; Xia, Z.; Feng, Q.; Liang, S.; Pedersen, A.Ø.; Kjeldsen, N.J.; Liu, C.; et al. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol. 2016, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ling, Y.; Shan, T.; Yang, S.; Xu, H.; Deng, X.; Delwart, E.; Zhang, W. Gut virome of mammals and birds reveals high genetic diversity of the family Microviridae. Virus Evol. 2019, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host. Microbe. 2019, 26, 527–541.e5. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Phylogenetic trees, bootstrap: 100, scalebar: substitutions per site. (A) Caudovirales based on large terminase subunit terL (Maximum log Likelihood: −1801.27). (B) Microviridae based on the DNA replication protein gene gpA (Maximum log Likelihood: −3922.55). (C) Taxonomic distribution of phages identified in 94 Danish wastewater samples, based on similarity to closest related and the ICTV Master Species list. 1 ≤95% similarity to other phages in the dataset. 2 ≤95% similarity to other phages in the dataset and the NCBI GenBank. 3 The Murrayvirus genus has by mistake been classified as Siphoviridae, but a proposed move to Podoviridae will be included in the 2021 ICTV ratification.
Figure 1.
Phylogenetic trees, bootstrap: 100, scalebar: substitutions per site. (A) Caudovirales based on large terminase subunit terL (Maximum log Likelihood: −1801.27). (B) Microviridae based on the DNA replication protein gene gpA (Maximum log Likelihood: −3922.55). (C) Taxonomic distribution of phages identified in 94 Danish wastewater samples, based on similarity to closest related and the ICTV Master Species list. 1 ≤95% similarity to other phages in the dataset. 2 ≤95% similarity to other phages in the dataset and the NCBI GenBank. 3 The Murrayvirus genus has by mistake been classified as Siphoviridae, but a proposed move to Podoviridae will be included in the 2021 ICTV ratification.

Figure 2.
(A) Number of phages and taxonomic family distribution per WWTP, as well as approximate geographical location thereof. (B) Bubble-diagram of the 104 unique coliphage species displaying genome size and GC content distribution; the area of the bubbles indicates the number of phages. (C) Sample completeness curve with confidence intervals (0.95). (D) Sample-size-based rarefaction and extrapolation curve with confidence intervals (0.95). Sampling units in figures C and D denote isolated phages (n = 136).
Figure 2.
(A) Number of phages and taxonomic family distribution per WWTP, as well as approximate geographical location thereof. (B) Bubble-diagram of the 104 unique coliphage species displaying genome size and GC content distribution; the area of the bubbles indicates the number of phages. (C) Sample completeness curve with confidence intervals (0.95). (D) Sample-size-based rarefaction and extrapolation curve with confidence intervals (0.95). Sampling units in figures C and D denote isolated phages (n = 136).

Figure 3.
Pairwise alignment of the unclassified Myoviridae phage species from this study (blue text) and their closest relatives (black text), the color bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). The genes marked with yellow code for a tailspike (gene 143 in phi92).
Figure 3.
Pairwise alignment of the unclassified Myoviridae phage species from this study (blue text) and their closest relatives (black text), the color bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). The genes marked with yellow code for a tailspike (gene 143 in phi92).

Figure 4.
Pairwise alignment of the new Carltongylesvirus phage flopper (blue) and the Carltongylesvirus phages GJ1 and ST32; the colour bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points. Genes coding for the putative tail tape measure protein (red) and a tail fiber (orange) are colorised.
Figure 4.
Pairwise alignment of the new Carltongylesvirus phage flopper (blue) and the Carltongylesvirus phages GJ1 and ST32; the colour bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points. Genes coding for the putative tail tape measure protein (red) and a tail fiber (orange) are colorised.

Figure 5.
Pairwise alignment of the new Hanrivervirus phages (green text) and type species pSf-1 (black text), the new Warwickvirus phages (blue text), and type species swan01 (black text) and Jahat (brown text); the color bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points. Coloured genes (purple) code for a putative tail fiber.
Figure 5.
Pairwise alignment of the new Hanrivervirus phages (green text) and type species pSf-1 (black text), the new Warwickvirus phages (blue text), and type species swan01 (black text) and Jahat (brown text); the color bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points. Coloured genes (purple) code for a putative tail fiber.

Figure 6.
Pairwise alignment of the new Dhillonvirus phage species (blue text) and the type species HK578 (black text); the colour bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points. Highlighted genes (green) code for a putative tail fiber.
Figure 6.
Pairwise alignment of the new Dhillonvirus phage species (blue text) and the type species HK578 (black text); the colour bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points. Highlighted genes (green) code for a putative tail fiber.

Figure 7.
Comparisons of the new phage lineage representative Halfdan (blue text) and closest relatives (black text). (A) Phylogenetic tree, Maximum log Likelihood: −7678.71, bootstrap 100, large terminase subunit TerL, scalebar: substitutions per site. (B) Pairwise alignment of phage genomes; color bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points.
Figure 7.
Comparisons of the new phage lineage representative Halfdan (blue text) and closest relatives (black text). (A) Phylogenetic tree, Maximum log Likelihood: −7678.71, bootstrap 100, large terminase subunit TerL, scalebar: substitutions per site. (B) Pairwise alignment of phage genomes; color bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points.

Figure 8.
Comparisons of the Podoviridae phage species from this study (blue text) with closest relatives (black text). (A) Phylogenetic tree, Maximum log Likelihood: −8023.43, bootstrap 100, large terminase subunit TerL, scalebar: substitutions per site. (B) Pairwise alignment of phage genomes; color bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points.
Figure 8.
Comparisons of the Podoviridae phage species from this study (blue text) with closest relatives (black text). (A) Phylogenetic tree, Maximum log Likelihood: −8023.43, bootstrap 100, large terminase subunit TerL, scalebar: substitutions per site. (B) Pairwise alignment of phage genomes; color bars between genomes indicate percent pairwise similarity (Easyfig, BlASTn). Genomes have been modified to have similar starting points.

Figure 9.
Significant hits for finding close relatives of the phages isolated in this study in three databases. Hits are defined as hits ≥80% genome coverage when blasting the coliphage genomes against the IMG/VR and GVD databases, and as mapped reads covering ≥70% of individual coliphage genomes when mapping reads from the Wang study. The coliphages are grouped according to genera, and numbers in parentheses denote the number of coliphage species representing each genus; only genera with significant hits are shown. Color-codes of genera denote taxonomic family.
Figure 9.
Significant hits for finding close relatives of the phages isolated in this study in three databases. Hits are defined as hits ≥80% genome coverage when blasting the coliphage genomes against the IMG/VR and GVD databases, and as mapped reads covering ≥70% of individual coliphage genomes when mapping reads from the Wang study. The coliphages are grouped according to genera, and numbers in parentheses denote the number of coliphage species representing each genus; only genera with significant hits are shown. Color-codes of genera denote taxonomic family.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
List of 104 unique Escherichia phage species identified in 94 Danish wastewater samples. Phages (n) denotes the number of phages isolated with more than 95% nucleotide sequence similarity. Similarity is sequence identity (%) times sequence coverage (%) to closest relative (Blastn). Taxonomy is based on similarity (BLASTn) to closest related.
Table 1.
List of 104 unique Escherichia phage species identified in 94 Danish wastewater samples. Phages (n) denotes the number of phages isolated with more than 95% nucleotide sequence similarity. Similarity is sequence identity (%) times sequence coverage (%) to closest relative (Blastn). Taxonomy is based on similarity (BLASTn) to closest related.
| Phage | Accession | Phages (n) | Genome (bp) | ORFs | tRNAs | GC (%) | Family; Genus | Similarity (%) | Closest Relative | Accession |
|---|---|---|---|---|---|---|---|---|---|---|
| tootiki | MN850647 | 1 | 88,257 | 128 | 22 | 39 | Myoviridae; Felixounavirus | 90.2 | Escherichia phage vB_EcoM_Alf5 | NC_031082.1 |
| mio | MN850631 | 1 | 83,431 | 121 | 18 | 39.1 | Myoviridae; Felixounavirus | 89.7 | Salmonella virus VSe11 | MG251391.1 |
| allfine | MN850633 | 1 | 86,963 | 125 | 20 | 39 | Myoviridae; Felixounavirus | 91.2 | Escherichia phage vB_EcoM-AYO145A | NC_028825.1 |
| bumzen | MN850635 | 3 | 87,360 | 126 | 20 | 39.1 | Myoviridae; Felixounavirus | 92.5 | Escherichia phage vB_EcoM_Alf5 | NC_031082.1 |
| dune | MN850636 | 1 | 88,511 | 129 | 20 | 39 | Myoviridae; Felixounavirus | 91.5 | Escherichia phage vB_EcoM_VpaE1 | NC_027337.1 |
| warpig | MN850637 | 1 | 86,106 | 127 | 17 | 39 | Myoviridae; Felixounavirus | 93 | Escherichia phage vB_EcoM_VpaE1 | NC_027337.1 |
| radambza | MN850639 | 1 | 86,702 | 127 | 19 | 38.9 | Myoviridae; Felixounavirus | 91.6 | Escherichia phage vB_EcoM_VpaE1 | NC_027337.1 |
| ekra | MN850644 | 1 | 87,282 | 128 | 20 | 38.9 | Myoviridae; Felixounavirus | 92.9 | Escherichia phage vB_EcoM_Alf5 | NC_031082.1 |
| humlepung | MN850564 | 3 | 85,311 | 119 | 19 | 39.1 | Myoviridae; Felixounavirus | 92.1 | Escherichia phage vB_EcoM_VpaE1 | NC_027337.1 |
| finno | MN850619 | 1 | 87,554 | 129 | 20 | 38.9 | Myoviridae; Felixounavirus | 89.7 | Escherichia phage vB_EcoM-AYO145A | NC_028825.1 |
| garuso | MN850566 | 2 | 85,798 | 130 | 20 | 38.9 | Myoviridae; Felixounavirus | 90.9 | Escherichia phage vB_EcoM-AYO145A | NC_028825.1 |
| momo | MN850580 | 1 | 88,168 | 130 | 20 | 39 | Myoviridae; Felixounavirus | 90.7 | Escherichia phage vB_EcoM-AYO145A | NC_028825.1 |
| heid | MN850577 | 6 | 87,590 | 126 | 20 | 39 | Myoviridae; Felixounavirus | 91.2 | Escherichia phage vB_EcoM_Alf5 | NC_031082.1 |
| skuden | MN850585 | 1 | 87,263 | 131 | 20 | 38.9 | Myoviridae; Felixounavirus | 91.1 | Escherichia phage vB_EcoM_VpaE1 | NC_027337.1 |
| pinkbiff | MN850603 | 1 | 88,814 | 129 | 20 | 39 | Myoviridae; Felixounavirus | 93.9 | Escherichia phage vB_EcoM_Alf5 | NC_031082.1 |
| fjerdesal | MN850605 | 3 | 87,715 | 128 | 21 | 39 | Myoviridae; Felixounavirus | 90.6 | Escherichia phage vB_EcoM_AYO145A | NC_028825.1 |
| andreotti | MN850610 | 1 | 83,391 | 117 | 20 | 39.2 | Myoviridae; Felixounavirus | 91.9 | Escherichia phage vB_EcoM_VpaE1 | NC_027337.1 |
| nataliec | MN850611 | 2 | 89,137 | 134 | 20 | 39 | Myoviridae; Felixounavirus | 90.3 | Escherichia phage vB_EcoM_AYO145A | NC_028825.1 |
| adrianh | MN850614 | 2 | 88,226 | 128 | 19 | 38.9 | Myoviridae; Felixounavirus | 91.1 | Escherichia phage vB_EcoM_Alf5 | NC_031082.1 |
| mistaenkt | MN850587 | 1 | 86,664 | 128 | 22 | 47.2 | Myoviridae; Suspvirus | 91.1 | Escherichia phage SUSP2 | NC_028935.2 |
| nimi | MN850626 | 1 | 137,039 | 213 | 5 | 43.7 | Myoviridae; Vequintavirus | 93.3 | Escherichia phage LL12 | MH491969.1 |
| navn | MN850642 | 1 | 141,707 | 224 | 4 | 43.6 | Myoviridae; Vequintavirus | 91.1 | Escherichia coli O157 typing phage 4 | KP869102.1 |
| nomine | MN850649 | 1 | 137,991 | 220 | 5 | 43.6 | Myoviridae; Vequintavirus | 91.5 | Escherichia phage LL12 | MH491969.1 |
| naswa | MN850595 | 1 | 138,583 | 222 | 5 | 43.6 | Myoviridae; Vequintavirus | 93.1 | Escherichia phage LL12 | MH491969.1 |
| naam | MN850630 | 1 | 137,129 | 215 | 5 | 43.7 | Myoviridae; Vequintavirus | 94.5 | Escherichia coli O157 typing phage 4 | KP869102.1 |
| ime | MN850576 | 2 | 137,114 | 217 | 5 | 43.6 | Myoviridae; Vequintavirus | 93.1 | Escherichia phage LL12 | MH491969.1 |
| magaca | MN850612 | 1 | 135,826 | 217 | 5 | 43.6 | Myoviridae; Vequintavirus | 96 | Escherichia phage slur12 | LN881735.1 |
| nom | MN850646 | 1 | 136,114 | 213 | 5 | 43.6 | Myoviridae; Vequintavirus | 92.6 | Escherichia phage LL12 | MH491969.1 |
| isim | MN850597 | 1 | 138,289 | 219 | 5 | 43.6 | Myoviridae; Vequintavirus | 93.8 | Escherichia phage LL12 | MH491969.1 |
| nomo | MN850578 | 1 | 137,702 | 218 | 5 | 43.7 | Myoviridae; Vequintavirus | 93.3 | Escherichia phage APCEc02 | NC_041869.1 |
| inoa | MN850593 | 1 | 138,710 | 220 | 5 | 43,6 | Myoviridae; Vequintavirus | 92 | Escherichia phage APCEc02 | NC_041869.1 |
| pangalan | MN850627 | 3 | 136,944 | 215 | 5 | 43.7 | Myoviridae; Vequintavirus | 94.8 | Escherichia phage vB_EcoM_FFH2 | NC_024134.1 |
| tuntematon | MN850618 | 2 | 150,473 | 279 | 11 | 39.1 | Myoviridae; Phapecoctavirus | 89.6 | Escherichia phage phAPEC8 | NC_020079.1 |
| anhysbys | MN850648 | 1 | 149,335 | 271 | 11 | 39.1 | Myoviridae; Phapecoctavirus | 91.5 | Escherichia phage phAPEC8 | NC_020079.1 |
| ukendt | MN850565 | 1 | 150,947 | 266 | 11 | 39 | Myoviridae; Phapecoctavirus | 88.7 | Escherichia phage phAPEC8 | NC_020079.1 |
| nepoznato | MN850571 | 4 | 151,514 | 265 | 10 | 38.9 | Myoviridae; Phapecoctavirus | 85.6 | Escherichia phage phAPEC8 | NC_020079.1 |
| nieznany | MN850598 | 1 | 144,998 | 254 | 11 | 39.1 | Myoviridae; Phapecoctavirus | 88.9 | Escherichia phage phAPEC8 | NC_020079.1 |
| muut | MN850573 | 1 | 146,307 | 243 | 13 | 37.4 | Myoviridae | 92 | Escherichia phage vB_EcoM_PHB05 | MF805809.1 |
| alia | MN850632 | 1 | 147,009 | 246 | 13 | 37.5 | Myoviridae | 93.1 | Enterobacteria phage ECGD1 | KU522583.1 |
| outra | MN850645 | 1 | 145,482 | 246 | 13 | 37.4 | Myoviridae | 93.8 | Enterobacteria phage ECGD1 | KU522583.1 |
| inny | MN850601 | 1 | 147,483 | 247 | 13 | 37.4 | Myoviridae | 92.4 | Enterobacteria phage ECGD1 | KU522583.1 |
| arall | MN850584 | 1 | 145,715 | 242 | 13 | 37.4 | Myoviridae | 94.6 | Escherichia phage vB_vPM_PD06 | MH816848.1 |
| kvi | MN850615 | 1 | 163,673 | 266 | - | 40.5 | Myoviridae; Krischvirus | 94.2 | Escherichia phage ECD7 | NC_041936.1 |
| kaaroe | MN850574 | 1 | 163,719 | 267 | - | 40.5 | Myoviridae; Krischvirus | 94.7 | Enterobacteria phage RB49 | NC_005066.1 |
| dhabil | MN850621 | 1 | 165,644 | 266 | 3 | 39.5 | Myoviridae; Dhakavirus | 87.5 | Enterobacteria phage JS10 | NC_012741.1 |
| dhaeg | MN850609 | 2 | 170,817 | 278 | 3 | 39.4 | Myoviridae; Dhakavirus | 87.4 | Enterobacteria phage JS10 | NC_012741.1 |
| mogra | MN850579 | 1 | 168,724 | 263 | 2 | 37.7 | Myoviridae; Mosigvirus | 91.1 | Escherichia phage vB_EcoM_PhAPEC2 | NC_024794.1 |
| mobillu | MN850622 | 1 | 163,063 | 255 | 2 | 37.7 | Myoviridae; Mosigvirus | 94.5 | Escherichia phage p000y | MK047718.1 |
| moha | MN850590 | 1 | 168,676 | 267 | 2 | 37.6 | Myoviridae; Mosigvirus | 94.8 | Escherichia phage APCEc01 | NC_029091.1 |
| moskry | MN850651 | 1 | 169,410 | 269 | 2 | 37.6 | Myoviridae; Mosigvirus | 93.2 | Escherichia virus vB_Eco_mar005P1 | LR027383.1 |
| teqskov | MN895437 | 1 | 165,017 | 257 | 6 | 35.4 | Myoviridae; Tequatrovirus | 91.7 | Yersinia phage phiD1 | NC_027353.1 |
| teqdroes | MN895438 | 1 | 166,833 | 269 | 10 | 35.4 | Myoviridae; Tequatrovirus | 88.6 | Escherichia phage T2 | LC348380.1 |
| teqhad | MN895434 | 1 | 167,892 | 270 | 10 | 35.3 | Myoviridae; Tequatrovirus | 90.1 | Escherichia phage T2 | LC348380.1 |
| teqhal | MN895435 | 2 | 168,070 | 266 | 11 | 35.4 | Myoviridae; Tequatrovirus | 93.9 | Escherichia phage slur13 | LN881737.1 |
| teqsoen | MN895436 | 1 | 166,468 | 268 | 10 | 35.5 | Myoviridae; Tequatrovirus | 91.7 | Yersinia phage phiD1 | NC_027353.1 |
| flopper | MN850594 | 1 | 52,092 | 78 | 1 | 44.2 | Chaseviridae; Carltongylesvirus | 87 | Escherichia phage ST32 | NC_047830.1 |
| damhaus | MN850602 | 1 | 51,154 | 89 | - | 44.1 | Drexlerviridae; Hanrivervirus | 85.8 | Shigella phage pSf-1 | NC_021331.1 |
| herni | MN850640 | 2 | 50,971 | 89 | - | 44.1 | Drexlerviridae; Hanrivervirus | 87.6 | Shigella phage pSf-1 | NC_021331.1 |
| grams | MN850567 | 1 | 49,530 | 83 | - | 44.1 | Drexlerviridae; Hanrivervirus | 87.1 | Shigella phage pSf-1 | NC_021331.1 |
| aaroes | MN850572 | 1 | 51,662 | 92 | - | 44.1 | Drexlerviridae; Hanrivervirus | 83 | Shigella phage pSf-1 | NC_021331.1 |
| aalborv | MN850591 | 1 | 46,660 | 79 | - | 43.9 | Drexlerviridae; Hanrivervirus | 86.9 | Shigella phage pSf-1 | NC_021331.1 |
| haarsle | MN850600 | 2 | 48,613 | 85 | - | 44 | Drexlerviridae; Hanrivervirus | 87.1 | Shigella phage pSf-1 | NC_021331.1 |
| egaa | MN850607 | 1 | 51,643 | 87 | - | 44.1 | Drexlerviridae; Hanrivervirus | 89.7 | Shigella phage pSf-1 | NC_021331.1 |
| vojen | MN850569 | 1 | 50,709 | 86 | - | 44.1 | Drexlerviridae; Hanrivervirus | 89.7 | Shigella phage pSf-1 | NC_021331.1 |
| tiwna | MN850643 | 1 | 51,014 | 85 | - | 44.6 | Drexlerviridae; Warwickvirus | 87.2 | Escherichia phage vB_Eco_Swan01 | NC_048202.1 |
| tonijn | MN850641 | 2 | 51,627 | 86 | - | 44.6 | Drexlerviridae; Warwickvirus | 88.4 | Escherichia phage vB_Eco_Swan01 | NC_048202.1 |
| tonnikala | MN850613 | 1 | 51,277 | 86 | - | 44.8 | Drexlerviridae; Warwickvirus | 86.4 | Escherichia phage vB_Eco_Swan01 | NC_048202.1 |
| atuna | MN850620 | 1 | 50,732 | 88 | - | 44.6 | Drexlerviridae; Warwickvirus | 84.9 | Escherichia virus vB_Eco_mar001J1 | NC_048204 |
| tunus | MN850638 | 1 | 51,111 | 87 | - | 44.8 | Drexlerviridae; Warwickvirus | 93.7 | Escherichia phage SECphi27 | NC_047938.1 |
| orkinos | MN850586 | 2 | 49,798 | 81 | - | 44.6 | Drexlerviridae; Warwickvirus | 91.3 | Escherichia phage SECphi27 | NC_047938.1 |
| ityhuna | MN850582 | 1 | 50,768 | 86 | - | 44.7 | Drexlerviridae; Warwickvirus | 93.3 | Escherichia phage SECphi27 | NC_047938.1 |
| tonn | MN850596 | 2 | 51,012 | 87 | - | 44.5 | Drexlerviridae; Warwickvirus | 94 | Escherichia phage vB_Eco_Swan01 | NC_048202.1 |
| tinuso | MN850634 | 1 | 50,856 | 86 | - | 44.8 | Drexlerviridae; Warwickvirus | 97.3 | Escherichia phage vB_Eco_Swan01 | NC_048202.1 |
| tunzivis | MN850604 | 1 | 50,596 | 84 | - | 44.6 | Drexlerviridae; Warwickvirus | 94.5 | Escherichia phage SECphi27 | NC_047938.1 |
| tuinn | MN850606 | 1 | 50,505 | 86 | - | 44.7 | Drexlerviridae; Warwickvirus | 94.8 | Escherichia phage vB_Eco_Swan01 | NC_048202.1 |
| Jahat | MK552105 | 1 | 51,101 | 87 | - | 45.7 | Drexlerviridae | 68.5 | Escherichia phage vB_Eco_Swan01 | NC_048202.1 |
| bob | MN850628 | 1 | 45,252 | 63 | - | 54.5 | Siphoviridae; Dhillonvirus | 88.6 | Escherichia phage SECphi18 | LT960609.1 |
| mckay | MN850629 | 1 | 44,443 | 63 | - | 54.5 | Siphoviridae; Dhillonvirus | 83.8 | Escherichia phage slur05 | NC_028901.1 |
| jat | MN850650 | 1 | 44,417 | 63 | - | 54.5 | Siphoviridae; Dhillonvirus | 89.4 | Escherichia phage Gluttony | NC_031113.1 |
| rolling | MN850575 | 1 | 46,017 | 64 | - | 54.2 | Siphoviridae; Dhillonvirus | 80.2 | Escherichia phage Sloth | KX534339.1 |
| welsh | MN850589 | 2 | 45,207 | 62 | - | 54.6 | Siphoviridae; Dhillonvirus | 83.8 | Escherichia phage B2 | KX534339.1 |
| buks | MN850616 | 1 | 40,308 | 62 | - | 49.7 | Siphoviridae; Jerseyvirus | 91.3 | Salmonella phage vB_SenS-Ent1 | NC_019539.1 |
| Skure | MK672798 | 1 | 59,474 | 92 | - | 44.6 | Siphoviridae; Seuratvirus | 90.4 | Escherichia phage vB_Eco_SLUR25 | LT907986.1 |
| Halfdan | MH362766 | 1 | 42,858 | 57 | - | 53.7 | Siphoviridae | 28.8 | Pseudomonas phage vB_PaeS_SCUT-S3 | MK165657.1 |
| Lilleen | MK629526 | 1 | 5342 | 6 | - | 46.9 | Microviridae; Gequatrovirus | 93.8 | Escherichia phage SECphi17 | LT960607.1 |
| Lilleput | MK629525 | 1 | 5490 | 6 | - | 47 | Microviridae; Gequatrovirus | 93.4 | Escherichia phage SECphi17 | LT960607.1 |
| Lilleto | MK629529 | 3 | 5492 | 6 | - | 46.8 | Microviridae; Gequatrovirus | 92.7 | Escherichia phage SECphi17 | LT960607.1 |
| Lilledu | MK791318 | 1 | 5483 | 6 | - | 47.2 | Microviridae; Gequatrovirus | 92.6 | Escherichia phage SECphi17 | LT960607.1 |
| lillemer | MN850599 | 1 | 5492 | 6 | - | 47.1 | Microviridae; Gequatrovirus | 94.5 | Escherichia phage SECphi17 | LT960607.1 |
| Lilleven | MK629527 | 1 | 6090 | 9 | - | 44.4 | Microviridae; Alphatrevirus | 93.9 | Enterobacteria phage St-1 | NC_012868.1 |
| sortsyn | MN850623 | 1 | 42,116 | 61 | - | 59 | Podoviridae; Murrayvirus | 92.3 | Enterobacteria phage IME_EC2 | KF591601.1 |
| sortregn | MN850588 | 1 | 38,200 | 53 | - | 59.3 | Podoviridae; Murrayvirus | 97.3 | Salmonella phage Lumpael | NC_048113.1 |
| Skarpretter | MK105855 | 1 | 42,042 | 63 | - | 55.8 | Podoviridae; Skarprettervirus | 37.9 | Escherichia phage C130_2 | MH363708.1 |
| sortkaff | MN850581 | 1 | 42,538 | 61 | - | 59.5 | Podoviridae; Sortsnevirus | 89.8 | Klebsiella phage vB_KpnS_IME279 | MF614100.1 |
| Sortsne | MK651787 | 1 | 41,912 | 62 | - | 60 | Podoviridae; Sortsnevirus | 67.6 | Klebsiella phage vB_KpnS_IME279 | MF614100.1 |
| aldrigsur | MN850592 | 1 | 42,379 | 55 | - | 55.7 | Autographviridae; Bonnellvirus | 71.9 | Enterobacteria phage J8-65 | NC_025445.1 |
| altidsur | MN850568 | 1 | 42,197 | 53 | - | 55.7 | Autographviridae; Bonnellvirus | 71.8 | Enterobacteria phage J8-65 | NC_025445.1 |
| forsur | MN850617 | 1 | 42,476 | 56 | - | 55.4 | Autographviridae; Bonnellvirus | 72 | Enterobacteria phage J8-65 | NC_025445.1 |
| glasur | MN850583 | 1 | 42,507 | 56 | - | 55.4 | Autographviridae; Bonnellvirus | 72.3 | Enterobacteria phage J8-65 | NC_025445.1 |
| Lidtsur | MK629528 | 1 | 42,291 | 56 | - | 54.6 | Autographviridae; Bonnellvirus | 69 | Enterobacteria phage J8-65 | NC_025445.1 |
| megetsur | MN850608 | 1 | 42,132 | 54 | - | 55.8 | Autographviridae; Bonnellvirus | 73.1 | Enterobacteria phage J8-65 | NC_025445.1 |
| mellemsur | MN850570 | 1 | 40,770 | 50 | - | 55.8 | Autographviridae; Bonnellvirus | 76.4 | Enterobacteria phage J8-65 | NC_025445.1 |
| smaasur | MN850625 | 1 | 41,110 | 50 | - | 55.4 | Autographviridae; Bonnellvirus | 93.3 | Enterobacteria phage J8-65 | NC_025445.1 |
| usur | MN850624 | 2 | 41,906 | 51 | - | 55.4 | Autographviridae; Bonnellvirus | 73.3 | Enterobacteria phage J8-65 | NC_025445.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Olsen, N.S.; Forero-Junco, L.; Kot, W.; Hansen, L.H. Exploring the Remarkable Diversity of Culturable Escherichia coli Phages in the Danish Wastewater Environment. Viruses 2020, 12, 986. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090986
AMA Style
Olsen NS, Forero-Junco L, Kot W, Hansen LH. Exploring the Remarkable Diversity of Culturable Escherichia coli Phages in the Danish Wastewater Environment. Viruses. 2020; 12(9):986. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090986
Chicago/Turabian StyleOlsen, Nikoline S., Laura Forero-Junco, Witold Kot, and Lars H. Hansen. 2020. "Exploring the Remarkable Diversity of Culturable Escherichia coli Phages in the Danish Wastewater Environment" Viruses 12, no. 9: 986. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090986
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.