Advanced Therapeutics, Vaccinations, and Precision Medicine in the Treatment and Management of Chronic Hepatitis B Viral Infections; Where Are We and Where Are We Going?

 ,
,  ,
, 
Abstract
:1. Unmet Need in Treatment
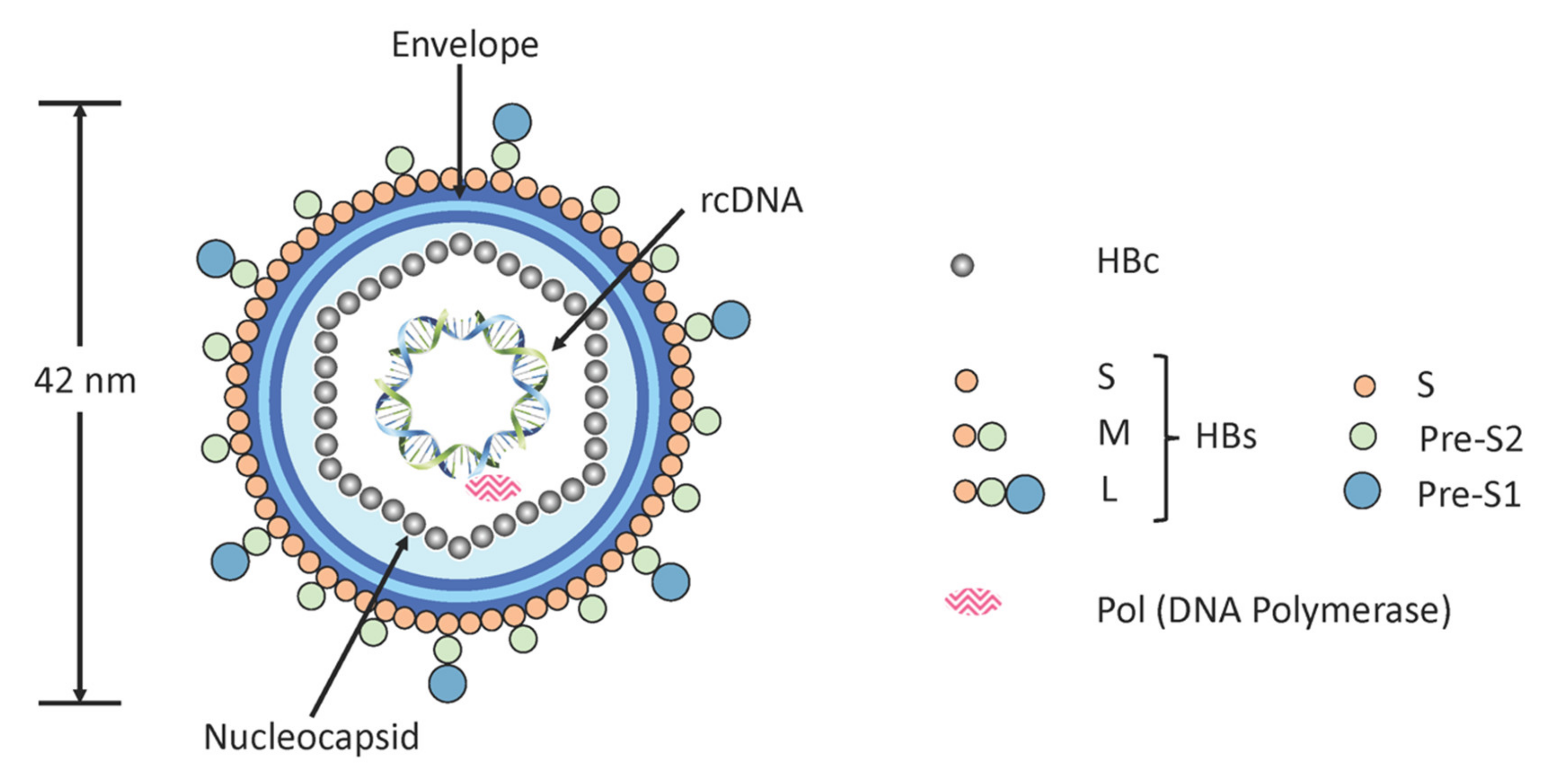
1.1. Virus Replication Cycle
1.2. Functional Cure
1.3. Genotypes of HBV
2. Currently Approved Treatments for CHB Infections
- interferon-alpha (IFNα) or PEGylated interferon-alpha (PEG-IFNα) and/or
2.1. Interferons
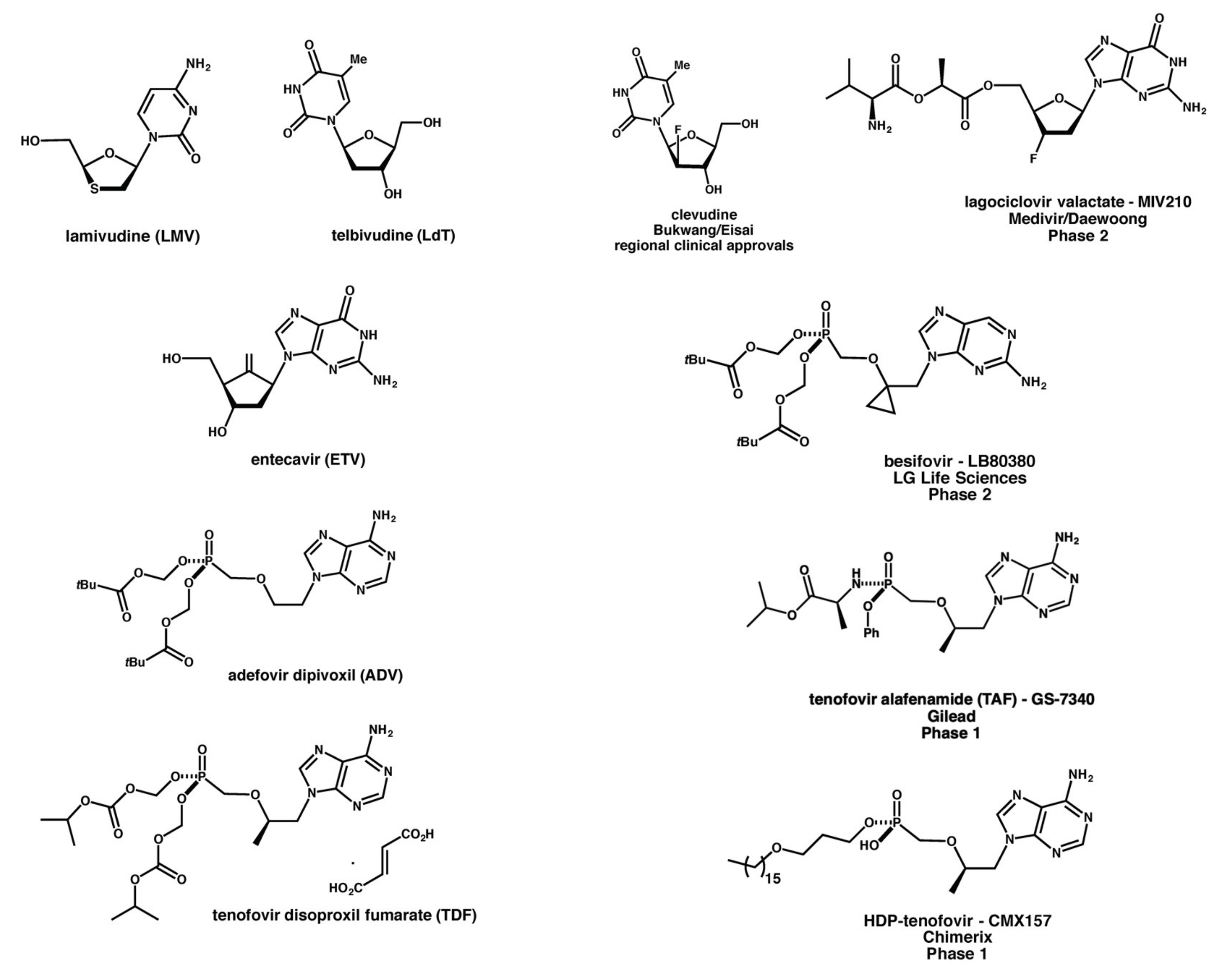
2.2. Nucleoside/Nucleotide Analogues
2.3. The Problem with Currently Approved Treatments
2.4. New Drugs in Pipeline
3. Advanced Therapeutic Approaches for CHB Treatment
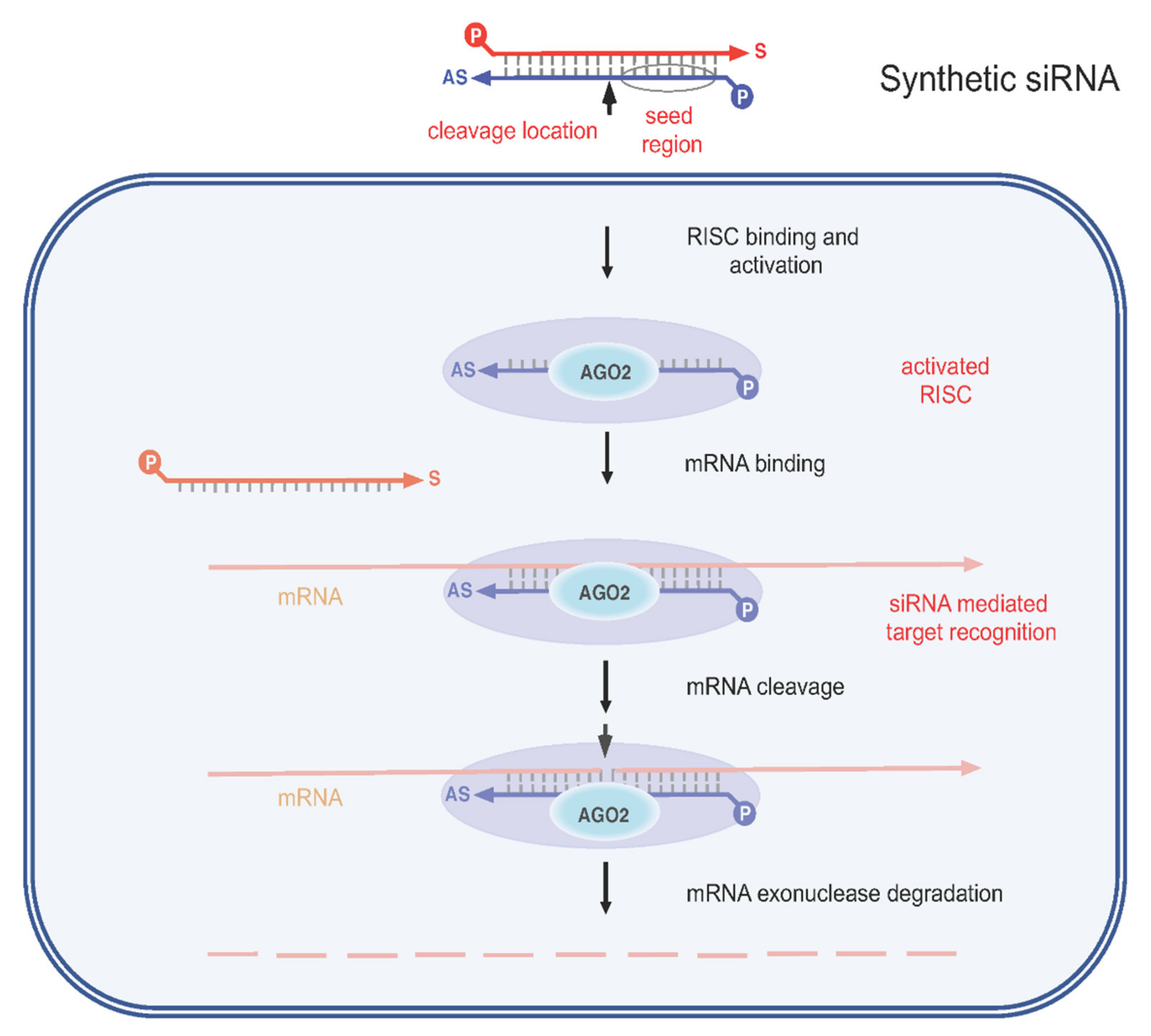
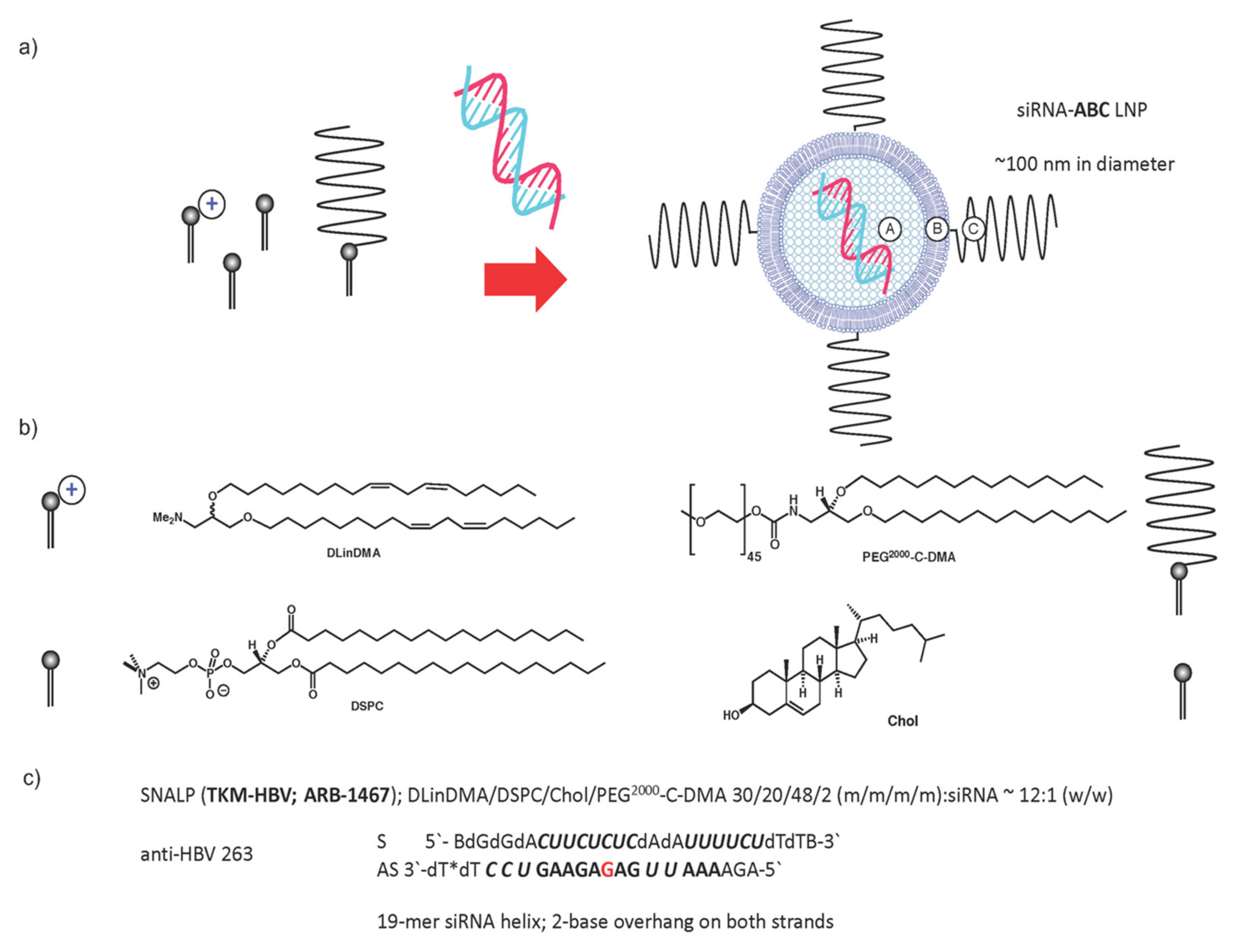
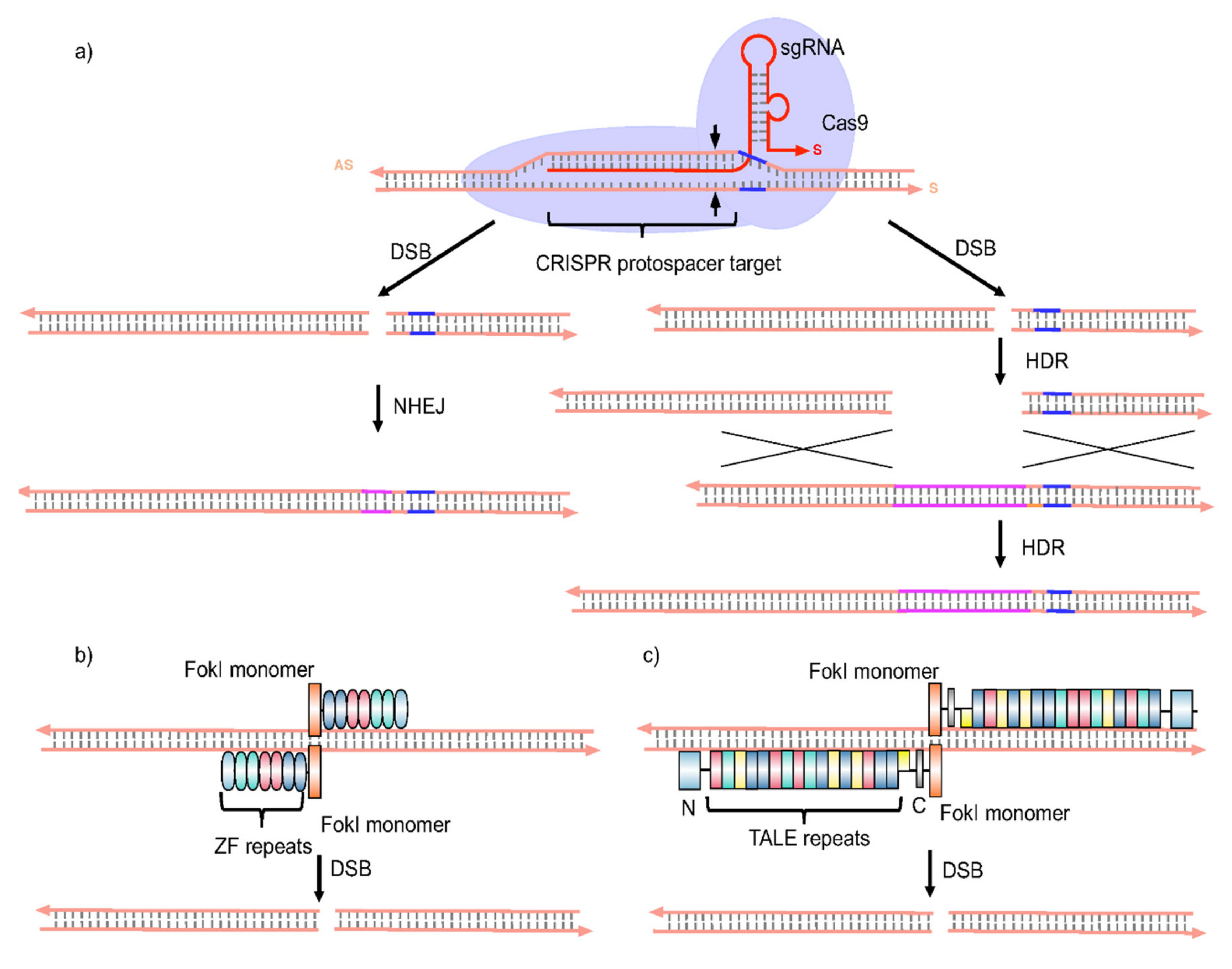
3.1. Gene Therapy Approaches
3.2. Therapies Targeting Innate and Adaptive Immune Responses
3.2.1. Innate Immunity
3.2.2. Adaptive Immunity
3.3. Therapeutic Vaccinations
4. Moving towards Precision Medicine Approaches for CHB Treatment
- Personalized medicine: This means understanding the genetic, immunological and/or metabolic individuality of patients in order to match individual patients with the most appropriate APIs for treatment of disease in these patients, i.e., “one size does not fit all.”
- Precision therapeutics: This means taking control of the delivery of APIs to target and/or selecting APIs for use with extreme target specificity.
4.1. Pharmacogenomics Studies
4.2. Next-Generation Gene Sequencing
4.3. Mass Spectrometric Studies
4.4. Diagnosis of HBV Infections
5. Where to Next and Why? The Future of HBV Treatment
- A commitment to improve fundamental understanding of the viral replication cycle, viral persistence, viral minichromosomal formation, viremia, HBeAg-positivity, identification of cellular receptors, other host –virus interactions, and viral genome heterogeneity (HBV genotype variation). All this to be made possible by using the very best possible cell and animal model systems. So too, studies on interactions with host immune responses must go hand in hand with basic research into underlying mechanisms, as mentioned above. Such an effort should result in more effective antiviral strategies that target different stages of HBV replication and restore host adaptive immune responses. Knowledge gained may then be used to devise combination therapeutic approaches that target CHB infection. In CHB infection, it is likely that combination therapy will be needed to obtain a functional cure. In recent years, various therapeutic approaches have been devised to suppress cccDNA formation and/or propagation as a primary and strategic way to overcome HBV persistence in vivo [142]. However, this remains a key unrealized goal that needs to be meet as a matter of high priority. Moreover, the growing consensus is that optimal API combinations, that would best achieve functional cure of CHB infections, that would best achieve functional cure of CHB infections, are combinations of API(s) that target the virus replication cycle and agents that target the immune system [10,22].
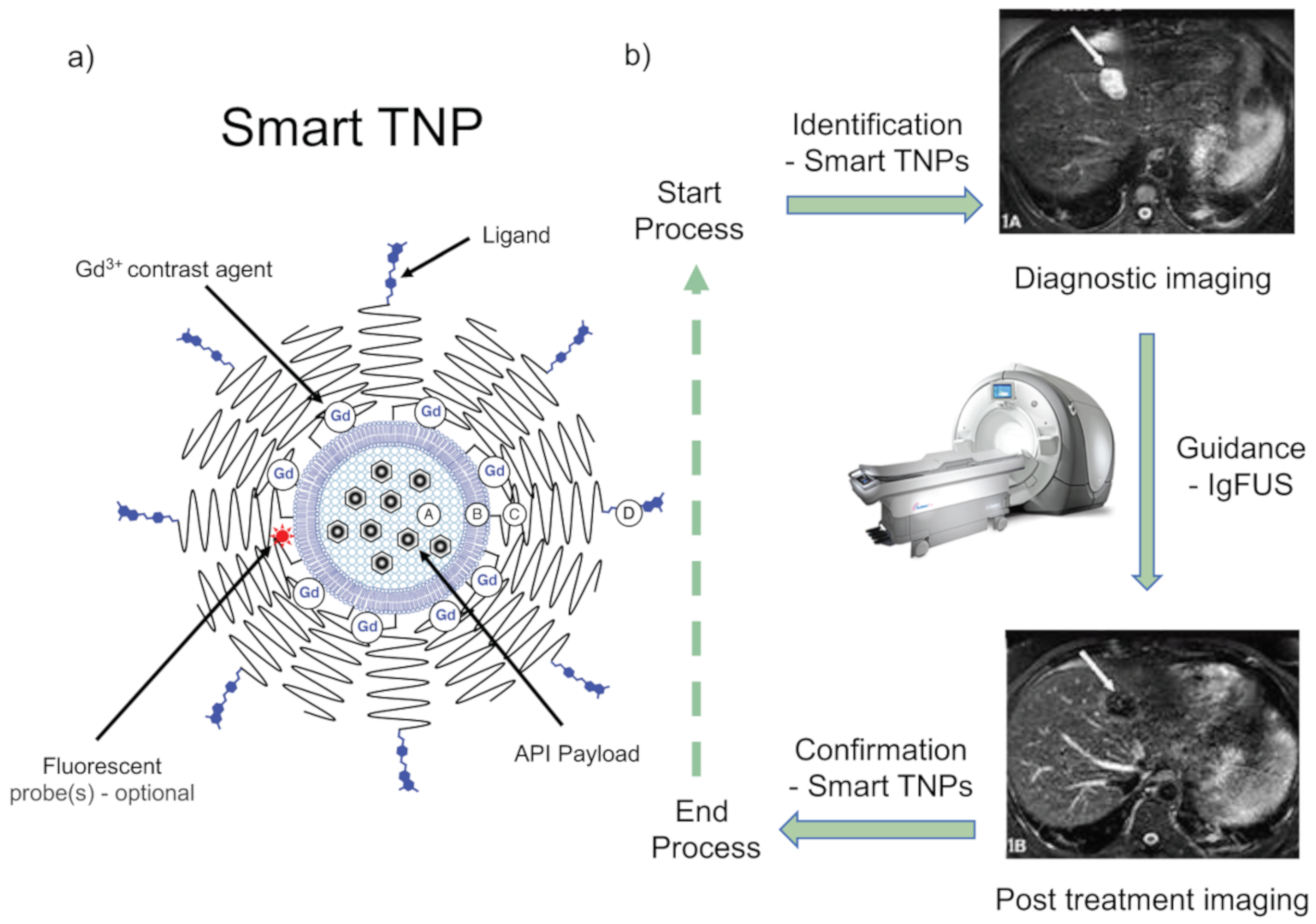
- A potential commitment towards PTAs for CHB treatment. As noted above, precision therapeutics means taking control of the delivery of APIs to target and/or selecting APIs for use with extreme target specificity. Accordingly, a PTA for CHB treatment would have the following basic elements (Figure 7) [56,143]:
- Identification of infected liver cells in situ, potentially achieved through the application of advanced diagnostic imaging techniques such as magnetic resonance imaging (MRI), near-infrared (NIFR) fluorescence, or resonance Raman, in appropriate combination with imaging nanoparticles (or theranostic nanoparticles (drug-delivery combined with imaging)).
- Guidance of theranostic nanoparticles to infected cells, potentially made possible by the exogenous application of a tissue irradiation technique, such as image-guided focused ultrasound (IgFUS), to direct theranostic nanoparticles (TNPs) to accumulate in infected cells [143], so as to clear infection there. Infected-cell receptor-specific targeting ligands may also be attached to TNPs in order to facilitate this process.
- Confirmation of therapeutic effects—potentially made possible by the accumulation of TNPs into infected cells. Infected cells should be visible for as long as infection continues.
Author Contributions
Funding
Conflicts of Interest
References
- Dandri, M.; Lutgehetmann, M.; Petersen, J. Experimental models and therapeutic approaches for HBV. Semin. Immunopathol. 2013, 35, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Heiberg, L.I.; Hogh, B. Horizontal transmission of hepatitis B virus-why discuss when we can vaccinate? J. Infect. Dis. 2012, 206, 464–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, E.M.; Tang, L.; Kottilil, S. Eradication strategies for chronic hepatitis B infection. Clin. Infect. Dis. 2016, 62, S318–S325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, M.; Watashi, K.; Tsukuda, S.; Aly, H.H.; Fukasawa, M.; Fujimoto, A.; Suzuki, R.; Aizaki, H.; Ito, T.; Koiwai, O.; et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun. 2014, 443, 808–813. [Google Scholar] [CrossRef] [Green Version]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [Green Version]
- Bock, C.T.; Schranz, P.; Schröder, C.H.; Zentgraf, H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef]
- Seeger, C.; Summers, J.; Mason, W.S. Viral DNA synthesis. Curr. Top. Microbiol. Immunol. 1991, 168, 41–60. [Google Scholar]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Norder, H.; Hammas, B.; Lee, S.D.; Bile, K.; Courouce, A.M.; Mushahwar, I.K.; Magnius, L.O. Genetic relatedness of hepatitis B viral strains of diverse geographical origin and natural variations in the primary structure of the surface antigen. J. Gen. Virol. 1993, 74, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A.; Restorp, K.; Norder, H.; Botha, J.F.; Magnius, L.O.; Kew, M.C. Full genome analysis of hepatitis B virus genotype E strains from South-Western Africa and Madagascar reveals low genetic variability. J. Med. Virol. 2005, 77, 47–52. [Google Scholar] [CrossRef]
- Singh, J.; Dickens, C.; Pahal, V.; Kumar, R.; Chaudhary, R.; Kramvis, A. First report of genotype E of hepatitis B virus in an Indian population. Intervirology 2009, 52, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Devesa, M.; Loureiro, C.L.; Rivas, Y.; Monsalve, F.; Cardona, N.; Duarte, M.C.; Poblete, F.; Gutierrez, M.F.; Botto, C.; Pujol, F.H. Subgenotype diversity of hepatitis B virus American genotype F in Amerindians from Venezuela and the general population of Colombia. J. Med. Virol. 2008, 80, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Trinh, T.N.; Abe, K. New complex recombinant genotype of hepatitis B virus identified in Vietnam. J. Virol. 2008, 82, 5657–5663. [Google Scholar]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 2009, 83, 10538–10547. [Google Scholar] [CrossRef] [Green Version]
- Papatheodoridis, G.; Buti, M.; Cornberg, M.; Janssen, H.L.A.; Mutimer, D.; Pol, S.; Raimondo, G.; Dusheiko, G.; Lok, A.; Marcellin, P. European Association for the Study of the Liver. EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. J. Hepatol. 2012, 57, 167–185. [Google Scholar]
- Buster, E.H.; Hansen, B.E.; Lau, G.K.; Piratvisuth, T.; Zeuzem, S.; Steyerberg, E.W.; Janssen, H.L.A. Factors that predict response of patients with hepatitis B e antigen-positive chronic hepatitis B to PEG interferon-α. Gastroenterology 2009, 137, 2002–2009. [Google Scholar] [CrossRef]
- Asselah, T.; Lada, O.; Moucari, R.; Martinot, M.; Boyer, N.; Marcellin, P. Interferon therapy for chronic hepatitis B. Clin. Liver 2007, 11, 839–849. [Google Scholar] [CrossRef]
- Janssen, H.L.; van Zonneveld, M.; Senturk, H.U.; Zeuzem, S.; Akarca, U.S.; Cakaloglu, Y.; Simon, C.; So, T.M.K.; Gerken, G.; de Man, R.A.; et al. HBV 99–01 Study Group; Rotterdam Foundation for Liver Research. PEGylated interferon α-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: A randomized trial. Lancet 2005, 365, 123–129. [Google Scholar] [CrossRef]
- Koumbi, L. Current and future antiviral drug therapies of hepatitis B chronic infection. World J. Hepatol. 2015, 7, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Tsai, N.; Petersen, J.; Flisiak, R.; Gurel, S.; Krastev, Z.; Schall, R.A.; Flaherty, J.F.; Martins, E.B.; Charuworn, P.; et al. Seven-year efficacy and safety of treatment with tenofovir disoproxil fumarate for chronic hepatitis B virus infection. Dig. Dis. Sci. 2015, 60, 1457–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampertico, P.; Liaw, Y.F. New perspectives in the therapy of chronic hepatitis B. Gut 2012, 61, 18–24. [Google Scholar] [CrossRef]
- Jung, T.Y.; Jun, D.W.; Lee, K.N.; Lee, H.L.; Lee, O.Y.; Yoon, B.C.; Choi, H.S. Fatal lactic acidosis in hepatitis B virus-associated decompensated cirrhosis treated with tenofovir: A case report. Medicine (Baltimore) 2017, 96, e7133. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, H.M.; Lim, J.K.; Pan, C.Q.; Nguyen, M.H.; Ray, K.W.; Mannalithara, A.; Trinh, H.; Chu, D.; Tran, T.; et al. Entecavir safety and effectiveness in a national cohort of chronic hepatitis B patients in the United States—The ENUMERATE study. Aliment. Pharmacol. Ther. 2014, 43, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourliere, M. Hepatitis B treatment: Could we extend the current treatment indication. Lancet Gastroenterol. Hepatol. 2017, 2, 543–544. [Google Scholar] [CrossRef]
- Verhelst, D.; Monge, M.; Meynard, J.-L.; Fouqueray, B.; Mougenot, B.; Girard, P.-M.; Ronco, P.; Rossert, J. Fanconi syndrome and renal failure induced by tenofovir: A first case report. Am. J. Kidney Dis. 2002, 40, 1331–1333. [Google Scholar] [CrossRef]
- Rodriguez-Nóvoa, S.; Alvarez, E.; Labarga, P.; Sorianom, V. Renal toxicity associated with tenofovir use. Expert. Opin. Drug Saf. 2010, 9, 545–559. [Google Scholar] [CrossRef]
- Viganò, M.; Brocchieri, A.; Spinetti, A.; Zaltron, S.; Mangia, G.; Facchetti, F.; Fugazza, A.; Castelli, F.; Colombo, M.; Lampertico, P. Tenofovir-induced Fanconi syndrome in chronic hepatitis B monoinfected patients that reverted after tenofovir withdrawal. J. Clin. Virol. 2014, 61, 600–603. [Google Scholar] [CrossRef]
- Lampertico, P.; Chan, H.L.; Janssen, H.L.; Strasser, S.I.; Schindler, R.; Berg, T. Review article: Long-term safety of nucleoside and nucleotide analogues in HBV-monoinfected patients. Aliment. Pharmacol. Ther. 2016, 44, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Lampertico, P.; Buti, M.; Fung, S.; Ahn, S.H.; Chuang, W.-L.; Tak, W.Y.; Ramji, A.; Chen, C.-Y.; Tam, E.; Bae, H.; et al. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide in virologically suppressed patients with chronic hepatitis B: A randomised, double-blind, phase 3, multicentre non-inferiority study. Lancet Gastroenterol. Hepatol. 2020, 5, 441–453. [Google Scholar] [CrossRef]
- Childs-Kean, L.M.; Egelund, E.F.; Jourjy, J. Tenofovir Alafenamide for the Treatment of Chronic Hepatitis B Monoinfection. Pharmacotherapy 2018, 38, 1051–1057. [Google Scholar] [CrossRef]
- Ogawa, E.; Furusyo, N.; Nguyen, M.H. Tenofovir alafenamide in the treatment of chronic hepatitis B: Design, development, and place in therapy. Drug Des. Devel. Ther. 2017, 11, 3197–3204. [Google Scholar] [CrossRef] [Green Version]
- Abdul Basit, S.; Dawood, A.; Ryan, J.; Gish, R. Tenofovir alafenamide for the treatment of chronic hepatitis B virus infection. Expert Rev. Clin. Pharmacol. 2017, 10, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.M.; Bojunga, J.; Hofmann, W.P.; Wunder, K.; Mihm, U.; Zeuzem, S.; Sarrazin, C. Severe lactic acidosis during treatment of chronic hepatitis B with entecavir in patients with impaired liver function. Hepatology 2009, 50, 2001–2006. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Alam, A.; Shukla, A.; Dashtseren, B.; Lesmana, C.R.A.; Duger, D.; Payawal, D.A.; Cuong, D.D.; Jargalsaikhan, G.; Cua, I.H.Y.; et al. An expert review on the use of tenofovir alafenamide for the treatment of chronic hepatitis B virus infection in Asia. J. Gastroenterol. 2020, 1–13. [Google Scholar] [CrossRef]
- Brouwer, W.P.; Xie, Q.; Sonneveld, M.J.; Zhang, N.; Zhang, Q.; Tabak, F.; Streinu-Cercel, A.; Wang, J.-Y.; Idilman, R.; Reesink, H.W.; et al. Adding pegylated interferon to entecavir for hepatitis B e antigen-positive chronic hepatitis B: A multicenter randomized trial (ARES study). Hepatology 2015, 61, 1512–1522. [Google Scholar] [CrossRef]
- Ward, H.; Tang, L.; Poonia, B.; Kottilil, S. Treatment of hepatitis B virus: An update. Future Microbiol. 2016, 11, 1581–1597. [Google Scholar] [CrossRef] [Green Version]
- Tajiri, K.; Shimizu, Y. Unsolved problems and future perspectives of hepatitis B virus vaccination. World J. Gastroenterol. 2015, 21, 7074–7083. [Google Scholar] [CrossRef]
- Papatheodoridis, G.V.; Vlachogiannakos, I.; Cholongitas, E.; Wursthorn, K.; Thomadakis, C.; Touloumi, G.; Petersen, J. Discontinuation of oral antivirals in chronic hepatitis B: A systematic review. Hepatology 2016, 63, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Song, I.H. Emerging therapeutics and relevant targets for chronic Hepatitis B. Turk. J. Gastroenterol. 2016, 27, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Seto, W.K.; Yuen, M.F. New pharmacological approaches to a functional cure of hepatitis B. Clin. Liver Dis. 2016, 8, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Rozema, D.B.; Hossbach, M.; John, M.; Hamilton, H.L.; Chu, Q.; Hegge, O.J.; Klein, J.J.; Wakefield, D.H.; Oropeza, C.E.; et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 2013, 21, 973–985. [Google Scholar] [CrossRef] [Green Version]
- McCaffrey, A.P. RNA interference inhibitors of hepatitis B virus. Ann. N. Y. Acad. Sci. 2009, 175, 15–23. [Google Scholar] [CrossRef]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef]
- Miller, A.D. Delivery of RNAi therapeutics: Work in progress. Expert Rev. Med. Devices 2013, 10, 781–811. [Google Scholar] [CrossRef]
- Miller, A.D. Delivering the promise of small non-coding RNA (ncRNA) therapeutics. Ther. Deliv. 2014, 5, 569–589. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Yuen, M.-F.; Chan, H.L.-Y.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef] [Green Version]
- Yuen, M.-F.; Locarnini, S.; Lim, T.H.; Strasser, S.; Sievert, W.; Cheng, W.; Thompson, A.; Given, B.; Schluep, T.; Hamilton, J.; et al. PS-080-Short term RNA interference therapy in chronic hepatitis B using JNJ-3989 brings majority of patients to HBsAg <100 IU/ml threshold. J. Hepatol. 2019, 70, E51–E52. [Google Scholar]
- Jackson, A.L.; Burchard, J.; Leake, D.; Reynolds, A.; Schelter, J.; Guo, J.; Johnson, J.M.; Lim, L.; Karpilow, J.; Nichols, K.; et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 2006, 12, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.K.; Choubdar, N.; Sadalapure, K.; Robert, F.; Wahba, A.S.; Pelletier, J.; Pinto, B.; Damha, M.J. 2′-Fluoro-4′-thioarabino-modified oligonucleotides: Conformational switches linked to siRNA activity. Nucleic Acids Res. 2007, 35, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Hean, J.; Crowther, C.; Ely, A.; Islam, R.U.; Barichievy, S.; Bloom, K.; Weinberg, M.S.; Van Otterlo, W.A.L.; De Koning, C.B.; Salazar, F.; et al. Inhibition of hepatitis B virus replication in vivo using lipoplexes containing altritol-modified antiviral siRNAs. Artif. DNA: PNA XNA 2010, 1, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona, S.; Jorgensen, M.R.; Kolli, S.; Crowther, C.; Salazar, F.H.; Marion, P.L.; Fujino, M.; Natori, Y.; Thanou, M.; Arbuthnot, P.; et al. Controlling HBV replication in vivo by intravenous administration of triggered PEGylated siRNA-nanoparticles. Mol. Pharm. 2009, 6, 706–717. [Google Scholar] [CrossRef]
- Kolli, S.; Wong, S.P.; Harbottle, R.; Johnston, B.; Thanou, M.; Miller, A.D. pH-Triggered nanoparticle mediated delivery of siRNA to liver cells in vitro and in vivo. Bioconjug. Chem. 2013, 24, 314–332. [Google Scholar] [CrossRef]
- Miller, A.D. Nanomedicine therapeutics and diagnostics are the goal. Ther. Deliv. 2016, 7, 431–456. [Google Scholar] [CrossRef]
- Miller, A.D. Synthetic nucleic acid delivery systems in gene therapy. In eLS; Wiley & Sons Ltd: Chichester, UK, 2017. [Google Scholar] [CrossRef]
- Starkey, J.L.; Chiari, E.F.; Isom, H.C. Hepatitis B virus (HBV)-specific short hairpin RNA is capable of reducing the formation of HBV covalently closed circular (CCC) DNA but has no effect on established CCC DNA in vitro. J. Gen. Virol. 2009, 9, 115–126. [Google Scholar] [CrossRef]
- Zhang, G.L.; Li, Y.X.; Zheng, S.Q.; Liu, M.; Li, X.; Tang, H. Suppression of hepatitis B virus replication by microRNA-199a-3p and microRNA-210. Antiviral. Res. 2010, 88, 169–175. [Google Scholar] [CrossRef]
- Chen, Y.; Shen, A.; Rider, P.J.; Yu, Y.; Wu, K.; Mu, Y. A liver-specific microRNA binds to a highly conserved RNA sequence of hepatitis B virus and negatively regulates viral gene expression and replication. FASEB J. 2011, 25, 4511–4521. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jiang, L.; Ji, X.; Yang, B.; Zhang, Y.; Fu, X.D. Hepatitis B Viral RNA Directly mediates down-regulation of the tumor suppressor microRNA miR-15a/miR-16–1 in hepatocytes. J. Biol. Chem. 2013, 288, 18484–18493. [Google Scholar] [CrossRef] [Green Version]
- Khee, S.G.; Yusof, Y.A.; Makpol, S. Expression of senescence associated microRNAs and target genes in cellular aging and modulation by to cotrienol-rich fraction. Oxid. Med. Cell. Longev. 2014, 2014, 725929. [Google Scholar] [PubMed] [Green Version]
- Huang, J.Y.; Chen, H.L.; Shih, C. MicroRNA miR-204 and miR-1236 inhibit hepatitis B virus replication via two different mechanisms. Sci. Rep. 2016, 6, 34740. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-Y.; Chou, S.-F.; Lee, J.-W.; Chen, H.-L.; Chen, C.-M.; Tao, M.-H.; Shih, C. MicroRNA-130a can inhibit hepatitis B virus replication via targeting PGC1α and PPARγ. RNA 2015, 21, 385–400. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, H.; Sun, H.; Fan, H.; Hu, Y.; Liu, M.; Li, X.; Tang, H. Hepatitis B virus-encoded microRNA controls viral replication. J. Virol. 2017, 91, e01919-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada-Tsutsumi, S.; Naito, Y.; Sato, S.; Takaoka, A.; Kawashima, K.; Isogawa, M.; Ochiya, T.; Tanaka, Y. The antiviral effects of human microRNA miR-302c-3p against hepatitis B virus infection. Aliment. Pharmacol. Ther. 2019, 49, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, J.; Zhang, S.; Sun, L.; Nan, Y.; Yao, H.; Fan, J.; Zhu, L.Y.; Yu, L. MicroRNA-802 induces hepatitis B virus replication and replication through regulating SMARCE1 expression in hepatocellular carcinoma. Cell Death Dis. 2019, 10, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Osakabe, Y.; Osakabe, K. Genome editing with engineered nucleases in plants. Plant Cell Physiol. 2015, 56, 389–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, G.; Zhang, X.; Naruse, K.; Nagahama, Y.; Hong, Y. Gene replacement by zinc finger nucleases in medaka embryos. Marine Biotechnol. 2014, 16, 739–747. [Google Scholar] [CrossRef]
- Menke, D.B. Engineering subtle targeted mutations into the mouse genome. Genesis 2013, 51, 605–618. [Google Scholar] [CrossRef]
- Zimmerman, K.A.; Fischer, K.P.; Joyce, M.A.; Tyrrell, D.L. Zinc finger proteins designed to specifically target duck hepatitis B virus covalently closed circular DNA inhibit viral transcription in tissue culture. J. Virol. 2008, 82, 8013–8021. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Kweon, J.; Kim, E.; Kim, S.; Kim, J.S. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res. 2012, 22, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cradick, T.J.; Keck, K.; Bradshaw, S.; Jamieson, A.C.; McCaffrey, A.P. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010, 18, 947–954. [Google Scholar] [CrossRef]
- Bloom, K.; Ely, A.; Mussolino, C.; Cathomen, T.; Arbuthnot, P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013, 21, 1889–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhang, W.; Lin, J.; Wang, F.; Wu, M.; Chen, C.; Zheng, Y.; Peng, X.; Li, J.; Yuan, Z. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol. Ther. 2013, 22, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Zhen, S.; Hua, L.; Liu, Y.H.; Gao, L.C.; Fu, J.; Wan, D.Y.; Dong, L.H.; Song, H.F.; Gao, X. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015, 22, 404–412. [Google Scholar] [CrossRef]
- Scott, T.; Moyo, B.; Nicholson, S.; Maepa, M.B.; Watashi, K.; Ely, A.; Weinberg, M.S.; Arbuthnot, P. ssAAVs containing cassettes encoding SaCas9 and guides targeting hepatitis B virus inactivate replication of the virus in cultured cells. Sci. Rep. 2017, 7, 7401. [Google Scholar] [CrossRef] [Green Version]
- Seeger, C.; Sohn, J.A. Complete spectrum of CRISPR/Cas9-induced mutations on HBV cccDNA. Mol. Ther. 2016, 24, 1258–1266. [Google Scholar] [CrossRef]
- Karimova, M.; Beschorner, N.; Dammermann, W.; Chemnitz, J.; Indenbirken, D.; Bockmann, J.-H.; Grundhoff, A.T.; Lüth, S.; Buchholz, F.; Wiesch, J.S.Z.; et al. CRISPR/Cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci. Rep. 2015, 5, 13734. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, T.; Fukuhara, T.; Ono, C.; Yamamoto, S.; Uemura, K.; Okamoto, T.; Sugiyama, M.; Motooka, D.; Nakamura, S.; Ikawa, M.; et al. Suppression of HBV replication by the expression of nickase- and nuclease dead-Cas9. Sci. Rep. 2017, 7, 6122. [Google Scholar] [CrossRef]
- Sakuma, T.; Masaki, K.; Abe-Chayama, H.; Mochida, K.; Yamamoto, T.; Chayama, K. Highly multiplexed CRISPR-Cas9-nuclease and Cas9-nickase vectors for inactivation of hepatitis B virus. Genes Cells 2016, 21, 1253–1262. [Google Scholar] [CrossRef]
- Wagner, D.L.; Amini, L.; Wendering, D.J.; Burkhardt, L.-M.; Akyüz, L.; Reinke, P.; Volk, H.-D.; Schmueck-Henneresse, M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2018, 25, 242–248. [Google Scholar] [CrossRef]
- Busca, A.; Kumar, A. Innate immune responses in hepatitis B virus (HBV) infection. Virol. J. 2014, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Broering, R.; Trippler, M.; Poggenpohl, L.; Fiedler, M.; Gerken, G.; Lu, M.; Schlaak, J. Toll-like receptor-mediated immune responses are attenuated in the presence of high levels of hepatitis B virus surface antigen. J. Viral Hepat. 2014, 21, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, M.; Mirzaei, V.; Bidaki, R.; Shabani, Z.; Daneshvar, H.; Karimi-Googheri, M.; Khaleghinia, M.; Afrooz, M.R.; Yousefpoor, Y.; Arababadi, M.K. Are RIG-1 and MDA5 expressions associated with chronic HBV infection? Viral Immunol. 2015, 28, 504–508. [Google Scholar] [CrossRef]
- Van Der Molen, R.G.; Sprengers, D.; Biesta, P.J.; Kusters, J.G.; Janssen, H.L.A. Favorable effect of adefovir on the number and functionality of myeloid dendritic cells of patients with chronic HBV. Hepatology 2006, 44, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-cell exhaustion in chronic hepatitis B infection: Current knowledge and clinical significance. Cell Death Dis. 2015, 6, e1694. [Google Scholar] [CrossRef]
- Zou, Z.Q.; Wang, L.; Wang, K.; Yu, J.G. Innate immune targets of hepatitis B virus infection. World J. Hepatol. 2016, 8, 716–725. [Google Scholar] [CrossRef] [Green Version]
- Pollicino, T.; Koumbi, L. Role natural killer group 2D-ligand interactions in hepatitis B infection. World J. Hepatol. 2015, 7, 819–824. [Google Scholar] [CrossRef]
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8+ T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 2003, 77, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology 2010, 138, 682–693.e4. [Google Scholar] [CrossRef]
- Schurich, A.; Khanna, P.; Lopes, A.R.; Han, K.J.; Peppa, D.; Micco, L.; Nebbia, G.; Kennedy, P.T.; Geretti, A.-M.; Dusheiko, G.; et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011, 53, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wu, H.; Tang, Z.; Zang, G. CTLA4 silencing with siRNA promotes deviation of Th1/Th2 in chronic hepatitis B patients. Cell Mol. Immunol. 2009, 6, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebbia, G.; Peppa, D.; Schurich, A.; Khanna, P.; Singh, H.D.; Cheng, Y.; Rosenberg, W.; Dusheiko, G.; Gilson, R.; ChinAleong, J.; et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS ONE 2012, 7, e47648. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Shi, Y.; Li, J.; Chen, F.; Chen, Z.; Zheng, M. Tim-3 expression on peripheral T cell subsets correlates with disease progression in hepatitis B infection. Virol. J. 2011, 8, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, K.; Böttinger, N.; Huang, L.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qasim, W.; Brunetto, M.R.; Gehring, A.J.; Xue, S.-A.; Schurich, A.; Khakpoor, A.; Zhan, H.; Ciccorossi, P.; Gilmour, K.; Cavallone, D.; et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J. Hepatol. 2015, 62, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Dubois, C.; Jacquier, E.; Dion, S.; Bourgine, M.; Godon, O.; Kratzer, R.; Lelu-Santolaria, K.; Evlachev, A.; Meritet, J.-F.; et al. TG1050, an immunotherapeutic to treat chronic hepatitis B, induces robust T cells and exerts an antiviral effect in HBV-persistent mice. Gut 2014, 64, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Kosinska, A.; Lu, M.; Roggendorf, M. New therapeutic vaccination strategies for the treatment of chronic hepatitis B. Virol. Sin. 2014, 29, 10–16. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, K.; Bai, W.; Jia, B.; Hu, H.; Zhou, N.; Zhang, X.; Xie, Y.; Bourgine, M.; Michel, M.-L.; et al. Adenoviral delivery of recombinant hepatitis B virus expressing foreign antigenic epitopes for immunotherapy of persistent viral infection. J. Virol. 2013, 88, 3004–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elvidge, S. Blockbuster expectations for hepatitis B therapeutic vaccine. Nat. Biotechnol. 2015, 33, 789. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Guerra, B.; Chavez, D.; Giavedoni, L.; Hodara, V.L.; Brasky, K.M.; Fosdick, A.; Frey, C.R.; Zheng, J.; Wolfgang, G.; et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013, 144, 1508–1517.e10. [Google Scholar] [CrossRef] [Green Version]
- Boni, C.; Janssen, H.L.; Rossi, M.; Yoon, S.K.; Vecchi, A.; Barili, V.; Yoshida, E.M.; Trinh, H.; Rodell, T.C.; Laccabue, D.; et al. Combined GS-4774 and tenofovir therapy can improve HBV-specific T-cell responses in patients with chronic hepatitis. Gastroenterology 2019, 157, 227–241.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcay, I.M.; Katrinli, S.; Ozdil, K.; Doganay, G.D.; Doganay, L. Host genetic factors affecting hepatitis B infection outcomes: Insights from genome-wide association studies. World J. Gastroenterol. 2018, 24, 3347–3360. [Google Scholar] [CrossRef]
- Kamatani, Y.; Wattanapokayakit, S.; Ochi, H.; Kawaguchi, T.; Takahashi, A.; Hosono, N.; Kubo, M.; Tsunoda, T.; Kamatani, N.; Kumada, H.; et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat. Genet. 2009, 41, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Mbarek, H.; Ochi, H.; Urabe, Y.; Kumar, V.; Kubo, M.; Hosono, N.; Takahashi, A.; Kamatani, Y.; Miki, D.; Abe, H.; et al. A genome-wide association study of chronic hepatitis B identified novel risk locus in a Japanese population. Hum. Mol. Genet. 2011, 20, 3884–3892. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Li, J.; Yao, J.; Yu, J.; Zhang, J.; Ning, Q.; Wen, Z.; Yang, D.; He, Y.; Kong, X.; et al. A genome-wide association study with DNA pooling identifies the variant rs11866328 in the GRIN2A gene that affects disease progression of chronic HBV infection. Viral Immunol. 2011, 24, 397–402. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, Y.; Zhai, X.; Dai, J.; Jin, G.; Wang, L.; Zhu, L.; Yang, Y.; Liu, J.; Chu, M.; et al. New loci associated with chronic hepatitis B virus infection in Han Chinese. Nat. Genet. 2013, 45, 1499–1503. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, J.-H.; Yu, S.J.; Yoon, J.-H.; Cheong, J.Y.; Cho, S.W.; Park, N.H.; Namgoong, S.; Shin, H.D. A genome-wide association study identified new variants associated with the risk of chronic hepatitis B. Hum. Mol. Genet. 2013, 22, 4233–4238. [Google Scholar] [CrossRef] [Green Version]
- Katrinli, S.; Niğdelioğlu, A.; Ozdil, K.; Dinler-Doganay, G.; Doganay, L. The association of variations in TLR genes and spontaneous immune control of hepatitis B virus. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-W.; Fann, C.S.-J.; Su, W.-H.; Wang, Y.C.; Weng, C.C.; Yu, C.-J.; Hsu, C.-L.; Hsieh, A.-R.; Chien, R.-N.; Chu, C.-M.; et al. A genome-wide association study on chronic HBV infection and its clinical progression in male Han-Taiwanese. PLoS ONE 2014, 9, e99724. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Nishida, N.; Khor, S.-S.; Honda, M.; Sugiyama, M.; Baba, N.; Yamada, K.; Sawada, N.; Tsugane, S.; Koike, K.; et al. Genome-wide association study identified new susceptible genetic variants in HLA class I region for hepatitis B virus-related hepatocellular carcinoma. Sci. Rep. 2018, 8, 7958. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Zhang, L.; Zhang, W.; Wu, X.; Li, Y.; Yan, B.; Zhu, X.; Liu, X.; Yang, C.; Xu, J.; et al. A genome-wide association study identifies polymorphisms in the HLA-DR region associated with non-response to hepatitis B vaccination in Chinese Han populations. Hum. Mol. Genet. 2013, 23, 2210–2219. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.-W.; Chen, C.-F.; Lai, S.-K.; Lin, H.H.; Chu, C.-C.; Wang, L.-Y. SNP rs7770370 in HLA-DPB1 loci as a major genetic determinant of response to booster hepatitis B vaccination: Results of a genome-wide association study. J. Gastroenterol. Hepatol. 2015, 30, 891–899. [Google Scholar] [CrossRef]
- Roh, E.Y.; Yoon, J.H.; In, J.W.; Lee, N.; Shin, S.; Song, E.Y. Association of HLA-DP variants with the responsiveness to Hepatitis B virus vaccination in Korean infants. Vaccine 2016, 34, 2602–2607. [Google Scholar] [CrossRef]
- Okada, Y.; Uno, N.; Sato, S.; Mori, S.; Sasaki, D.; Kaku, N.; Kosai, K.; Morinaga, Y.; Hasegawa, H.; Yanagihara, K. Strong influence of human leukocyte antigen-DP variants on response to hepatitis B vaccine in a Japanese population. Vaccine 2017, 35, 5662–5665. [Google Scholar] [CrossRef]
- Li, Y.; Si, L.; Zhai, Y.; Hu, Y.; Hu, Z.; Bei, J.-X.; Xie, B.; Ren, Q.; Cao, P.; Yang, F.; et al. Genome-wide association study identifies 8p21.3 associated with persistent hepatitis B virus infection among Chinese. Nat. Commun. 2016, 7, 11664. [Google Scholar] [CrossRef]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.-L.; Hui, J.; Marshall, J.; et al. A new arenavirus in a cluster of fatal transplant-associated diseases. New Engl. J. Med. 2008, 358, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Towner, J.S.; Sealy, T.K.; Khristova, M.L.; Albariño, C.G.; Conlan, S.; Reeder, S.A.; Quan, P.-L.; Lipkin, W.I.; Downing, R.; Tappero, J.W.; et al. Newly discovered Ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 2008, 4, e1000212. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Li, Y.; Yang, X.; Zhang, H.; Zhou, P.; Zhang, Y.; Shi, Z. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 2012, 86, 4620–4630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yozwiak, N.L.; Skewes-Cox, P.; Stenglein, M.D.; Balmaseda, A.; Harris, E.; DeRisi, J.L. Virus identification in unknown tropical febrile illness cases using deep sequencing. PLoS Neglected Trop. Dis. 2012, 6, e1485. [Google Scholar] [CrossRef] [Green Version]
- Wu, I.C.; Liu, W.C.; Chang, T.T. Applications of next-generation sequencing analysis for the detection of hepatocellular carcinoma-associated hepatitis B virus mutations. J. Biomed. Sci. 2018, 25, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Han, Y.; Chen, L.; Liu, F.; Hao, P.; Sheng, J.; Li, X.-H.; Yu, D.-M.; Gong, Q.-M.; Tian, F.; et al. Comparison of next-generation sequencing and clone-based sequencing in analysis of hepatitis B virus reverse transcriptase quasispecies heterogeneity. J. Clin. Microbiol. 2013, 51, 4087–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecuit, M.; Eloit, M. The human virome: New tools and concepts. Trends Microbiol. 2013, 21, 510–515. [Google Scholar] [CrossRef]
- Albalat, A.; Husi, H.; Stalmach, A.; Schanstra, J.P.; Mischak, H. Classical MALDI-MS versus CE-based ESI-MS proteomic profiling in urine for clinical applications. Bioanalysis 2014, 6, 247–266. [Google Scholar] [CrossRef]
- Luan, J.; Yuan, J.; Li, X.; Jin, S.; Yu, L.; Liao, M.; Zhang, H.; Xu, C.; He, Q.; Wen, B.; et al. Multiplex detection of 60 hepatitis B virus variants by MALDI-TOF mass spectrometry. Clin. Chem. 2009, 55, 1503–1509. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Xue, R.; Huang, X.; Zhang, D.; Dong, L.; Wu, H.; Shen, X. Proteomic profiling of hepatitis B virus-related hepatocellular carcinoma with magnetic bead-based matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Acta Biochim. Biophys. Sin. 2011, 43, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Ganova-Raeva, L.; Ramachandran, S.; Honisch, C.; Forbi, J.C.; Zhai, X.; Khudyakov, Y. Robust hepatitis B virus genotyping by mass spectrometry. J. Clin. Microbiol. 2010, 48, 4161–4168. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.P.; Kim, N.K.; Hwang, S.G.; Chung, H.J.; Kim, S.; Han, J.H.; Kim, H.T.; Rim, K.S.; Kang, M.S.; Yoo, W.; et al. Detection of hepatitis B virus YMDD variants using mass spectrometric analysis of oligonucleotide fragments. J. Hepatol. 2004, 40, 837–844. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guidelines for the Prevention, Care and Treatment of Persons with Chronic Hepatitis B Infection. 2015. Available online: http://www.who.int/hiv/pub/hepatitis/hepatitis-b-guidelines/en (accessed on 30 July 2020).
- Amini, A.; Varsaneux, O.; Kelly, H.; Tang, W.; Chen, W.; Boeras, D.; Falconer, J.; Tucker, J.D.; Chou, R.; Ishizaki, A.; et al. Diagnostic accuracy of tests to detect hepatitis B surface antigen: A systematic review of the literature and meta-analysis. BMC Infect. Dis. 2017, 17, 698. [Google Scholar] [CrossRef]
- Liu, Y.P.; Yao, C.Y. Rapid and quantitative detection of hepatitis B virus. World J. Gastroenterol. 2015, 21, 11954–11963. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, L.; Hu, M.; Wang, L.; Hu, J. Detection of hepatitis B virus by piezoelectric biosensor. J. Pharm. Biomed. Anal. 2002, 27, 341–345. [Google Scholar] [CrossRef]
- Huang, J.-T.; Yang, Y.; Hu, Y.-M.; Liu, X.; Liao, M.-Y.; Morgan, R.; Yuan, E.-F.; Li, X.; Liu, S.-M. A highly sensitive and robust method for hepatitis B virus covalently closed circular DNA detection in single cells and serum. J. Mol. Diagn. 2018, 20, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, M.; Bonnaud, B.; Arsac, M.; Lavocat, F.; Maisetti, J.; Kay, A.; Simon, F.; Zoulim, F.; Vernet, G. Microarray for hepatitis B virus genotyping and detection of 994 mutations along the genome. J. Clin. Microbiol. 2010, 48, 4207–4215. [Google Scholar] [CrossRef] [Green Version]
- Hua, W.; Zhang, G.; Guo, S.; Li, W.; Sun, L.; Xiang, G. Microarray-based genotyping and detection of drug-resistant HBV mutations from 620 Chinese patients with chronic HBV infection. Braz. J. Infect. Dis. 2015, 19, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Zhi, X.; Deng, M.; Yang, H.; Gao, G.; Qin, Q.; Fu, H.; Zhang, Y.; Chen, D.; Cui, D. A novel HBV genotypes detecting system combined with microfluidic chip, loop-mediated isothermal amplification and GMR sensors. Biosens. Bioelectron. 2014, 54, 372–377. [Google Scholar] [CrossRef]
- Brahmania, M.; Feld, J.; Arif, A.; Janssen, H.L. New therapeutic agents for chronic hepatitis B. Lancet Infect. Dis. 2016, 16, e10–e21. [Google Scholar] [CrossRef]
- Block, B.T.M.; Gish, R.G.; Guo, H.; Mehta, A.; Cuconati, A.; London, W.T.; Guo, J.-T. Chronic hepatitis B: What should be the goal for new therapies? Antivir. Res. 2013, 98, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Centelles, M.N.; Wright, M.; Tsolaki, M.; Amrahli, M.; Xu, X.Y.; Stebbing, J.; Miller, A.D.; Gedroyc, W.; Thanou, M. Image-guided thermosensitive liposomes for focused ultrasound drug delivery: Using NIRF-labelled lipids and topotecan to visualise the effects of hyperthermia in tumours. J. Control. Release 2018, 280, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, L.H.; Couvreur, P. Nanotechnology for therapy and imaging of liver diseases. J. Hepatol. 2011, 55, 1461–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, Y.; Wang, H.; Fu, Q.; Peng, J.; Wang, Y. Gold nanorod-based localized surface plasmon resonance biosensor for sensitive detection of hepatitis B virus in buffer, blood serum and plasma. Biosens. Bioelectron. 2010, 26, 404–410. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotypes | Sub-Genotypes | Serological Serotypes | Mode of Transmission | Geographical Distribution |
|---|---|---|---|---|
| A | A1, A2, A3 | adw | Patients most at risk of chronicity are those infected during early life (neonates and children). Adults are infected through sexual contact | Europe, North America, Sub-Saharan Africa and Western Africa |
| B | B1, B2–B5, B6 | adw, ayw | Perinatal (during childbirth associated trauma) —most common or vertical (via the placenta)—less common | Asia |
| C | C1–C3, C4, C5, C6–C11 | adw, ayr, adr | perinatal or vertical | Asia |
| D | D1–D6 | ayw | Patient infected through homosexual or or bisexual or heterosexual contact | Mediterranean area, Middle East and India |
| E | NA | ayw | Horizontal and homosexual | Sub-Saharan Africa and some other continents |
| F | F1–F4 | dw | Horizontal | Central America |
| G | dw | Horizontal | France, Germany, United States and Mexico | |
| H | dw | Horizontal | South America | |
| I | I1, I2 | dw | Pariental/horizontal | Vietnam and Laos |
| J | dw | Horizontal | Japan |
| Genotype | Response to Treatment | Reference | |
|---|---|---|---|
| IFNα or PEG-IFNα | NUCs | ||
| A | weak responder to IFNα; strong responder to PEG-IFNα | strong responder to NUCs; drug resistance noted | [21,22] |
| B | strong responder to IFNα; strong responder to PEG-IFNα | strong responder to NUCs; drug resistance noted | [21,22,23] |
| C | weak responder to IFNα; weak responder to PEG-IFNα | weak responder to NUCs | [21,23] |
| D | weak responder to IFNα; weak responder to PEG-IFNα | weak responder to NUCs | [21] |
| E | strong responder to IFNα; weak responder to PEG-IFNα | adequate responder to NUCs | [24,25] |
| F | weak responder to IFNα | adequate responder to NUCs | [24] |
| G | stronger response to IFNα | adequate responder to NUCs | [24] |
| H | weaker response to IFNα | adequate responder to NUCs | [24] |
| I | [25] | ||
| J | [25] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duraisamy, G.S.; Bhosale, D.; Lipenská, I.; Huvarova, I.; Růžek, D.; Windisch, M.P.; Miller, A.D. Advanced Therapeutics, Vaccinations, and Precision Medicine in the Treatment and Management of Chronic Hepatitis B Viral Infections; Where Are We and Where Are We Going? Viruses 2020, 12, 998. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090998
Duraisamy GS, Bhosale D, Lipenská I, Huvarova I, Růžek D, Windisch MP, Miller AD. Advanced Therapeutics, Vaccinations, and Precision Medicine in the Treatment and Management of Chronic Hepatitis B Viral Infections; Where Are We and Where Are We Going? Viruses. 2020; 12(9):998. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090998
Chicago/Turabian StyleDuraisamy, Ganesh Selvaraj, Dattatry Bhosale, Ivana Lipenská, Ivana Huvarova, Daniel Růžek, Marc P. Windisch, and Andrew D. Miller. 2020. "Advanced Therapeutics, Vaccinations, and Precision Medicine in the Treatment and Management of Chronic Hepatitis B Viral Infections; Where Are We and Where Are We Going?" Viruses 12, no. 9: 998. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090998