
African Swine Fever Virus Structural Protein p17 Inhibits Cell Proliferation through ER Stress—ROS Mediated Cell Cycle Arrest

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents and Antibodies
2.2. D117L/p17 Expression Plasmids
2.3. Cell Culture and Transfection
2.4. Cell Proliferation and LDH Release Analysis
2.5. Cell Apoptosis and Cell Cycle Distribution Analysis
2.6. Western Blot Analysis
2.7. Confocal Microscopy
2.8. Measurement of Cellular Reactive Oxygen Species (ROS) and Ca2+
2.9. Statistical Analysis
3. Results
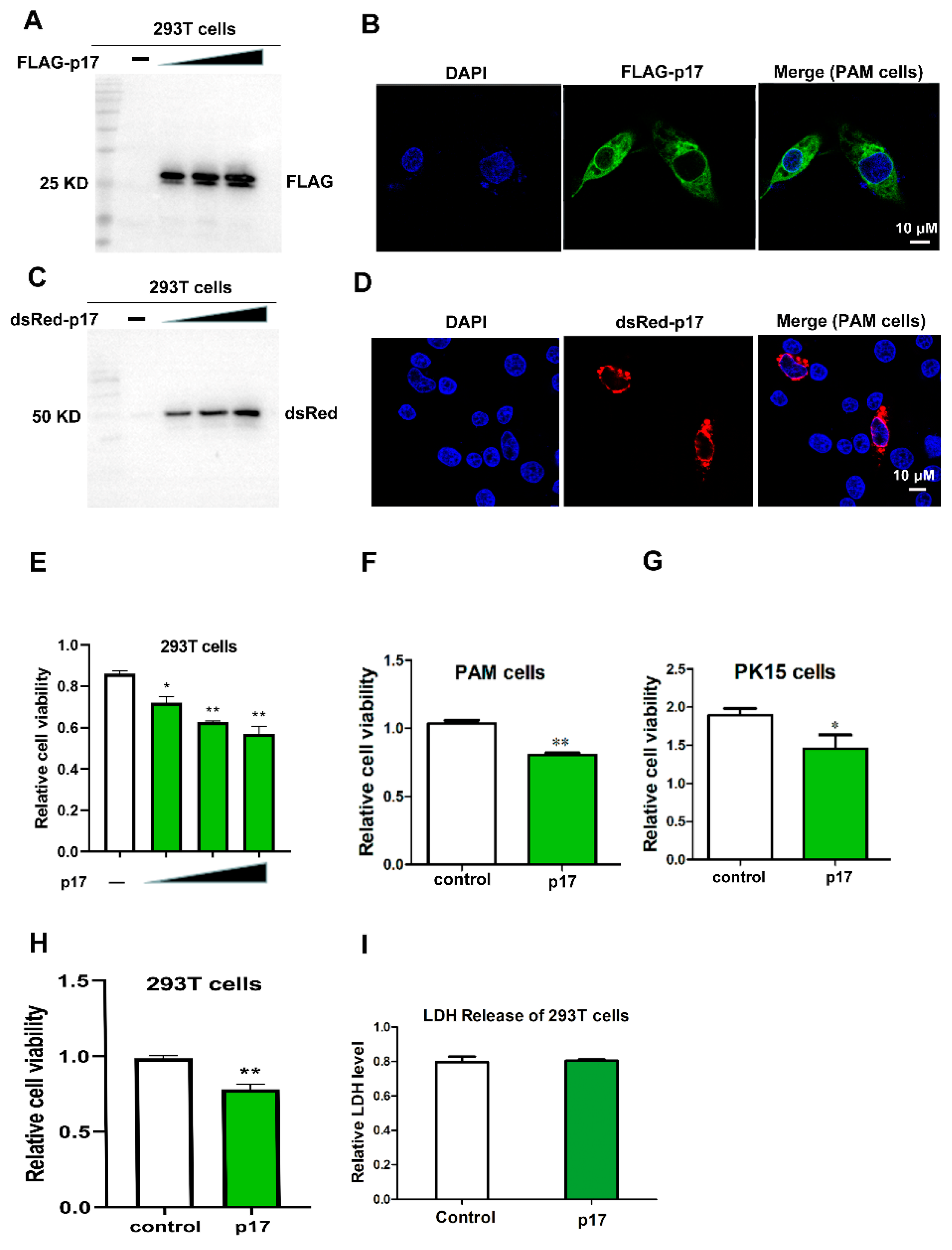
3.1. The Expression of p17 Protein in 293T and PAM Cells
3.2. Effects of p17 Expression on Cell Proliferation and LDH Release
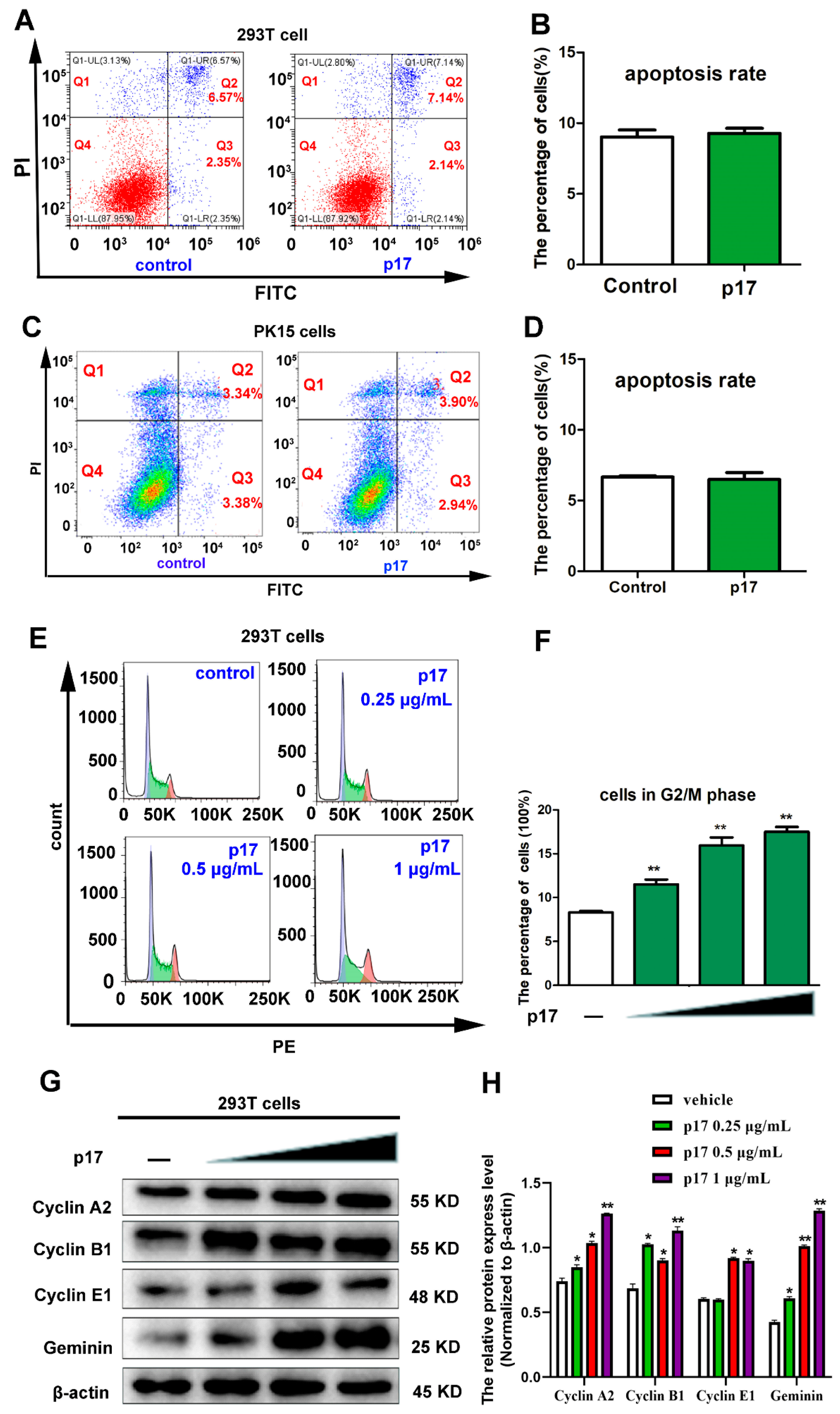
3.3. Effects of p17 on Cell Apoptosis and Cell Cycle Progression
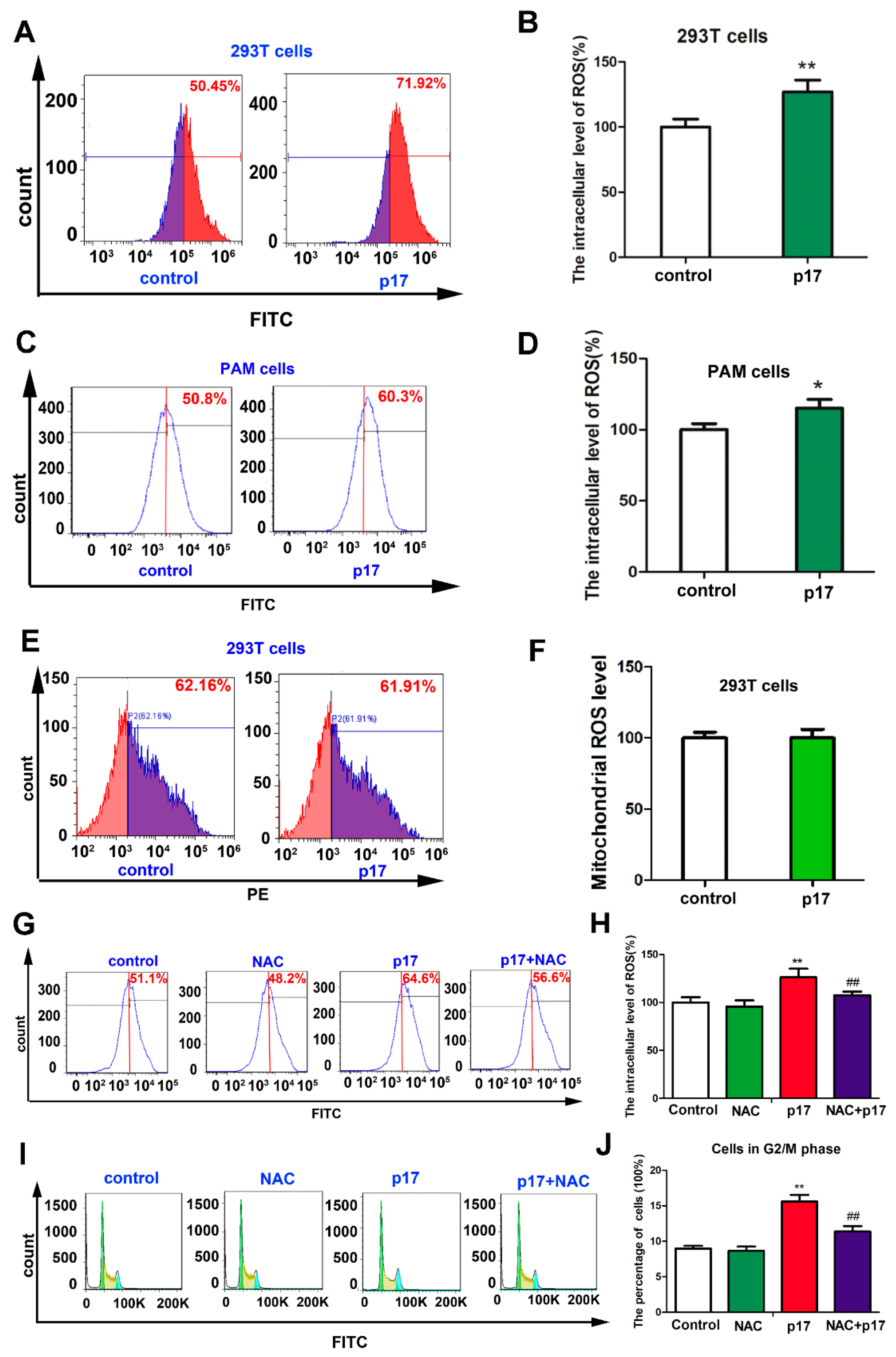
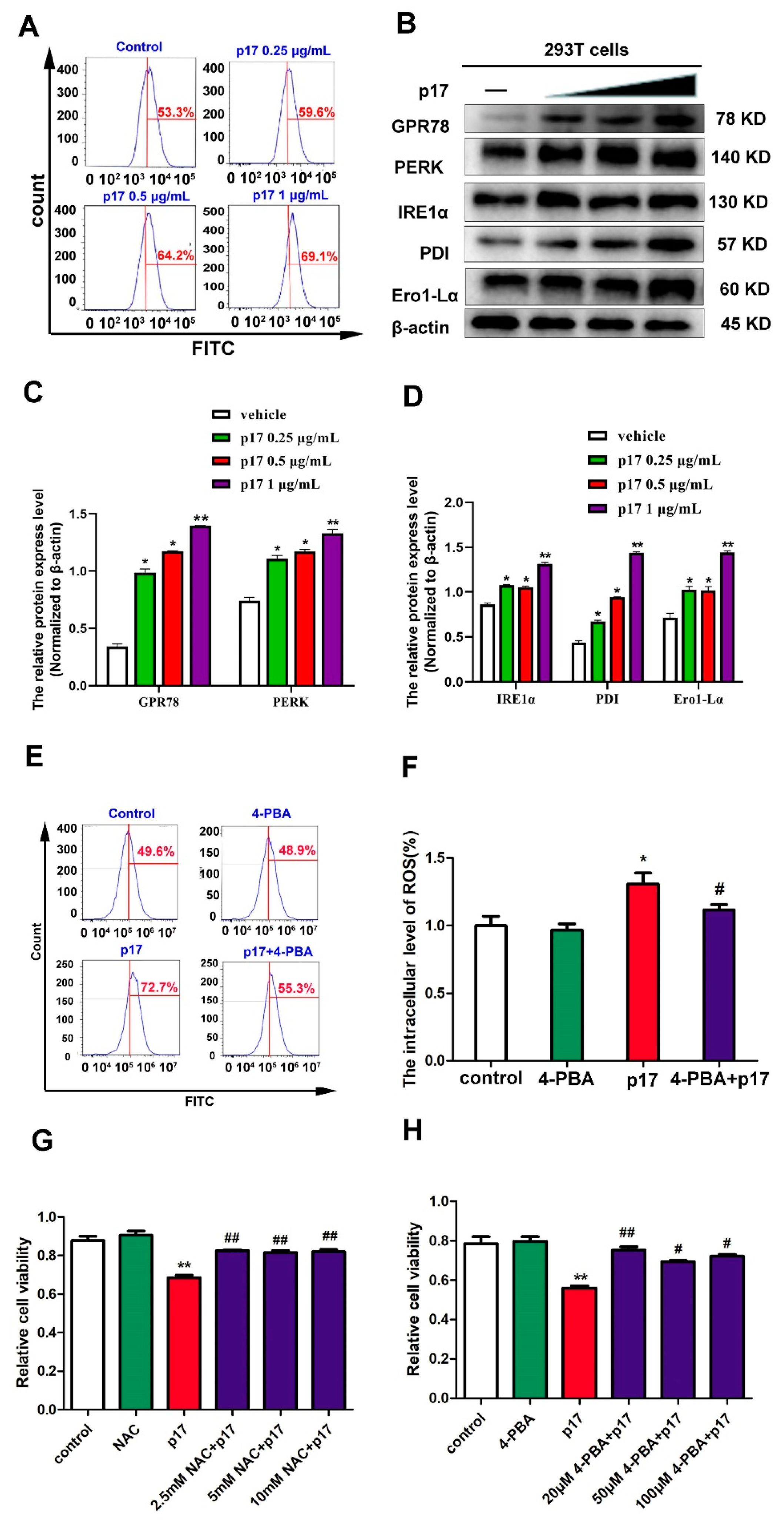
3.4. ASFV p17-Induced Cell Cycle Arrest Through the Production of Reactive Oxygen Species (ROS)
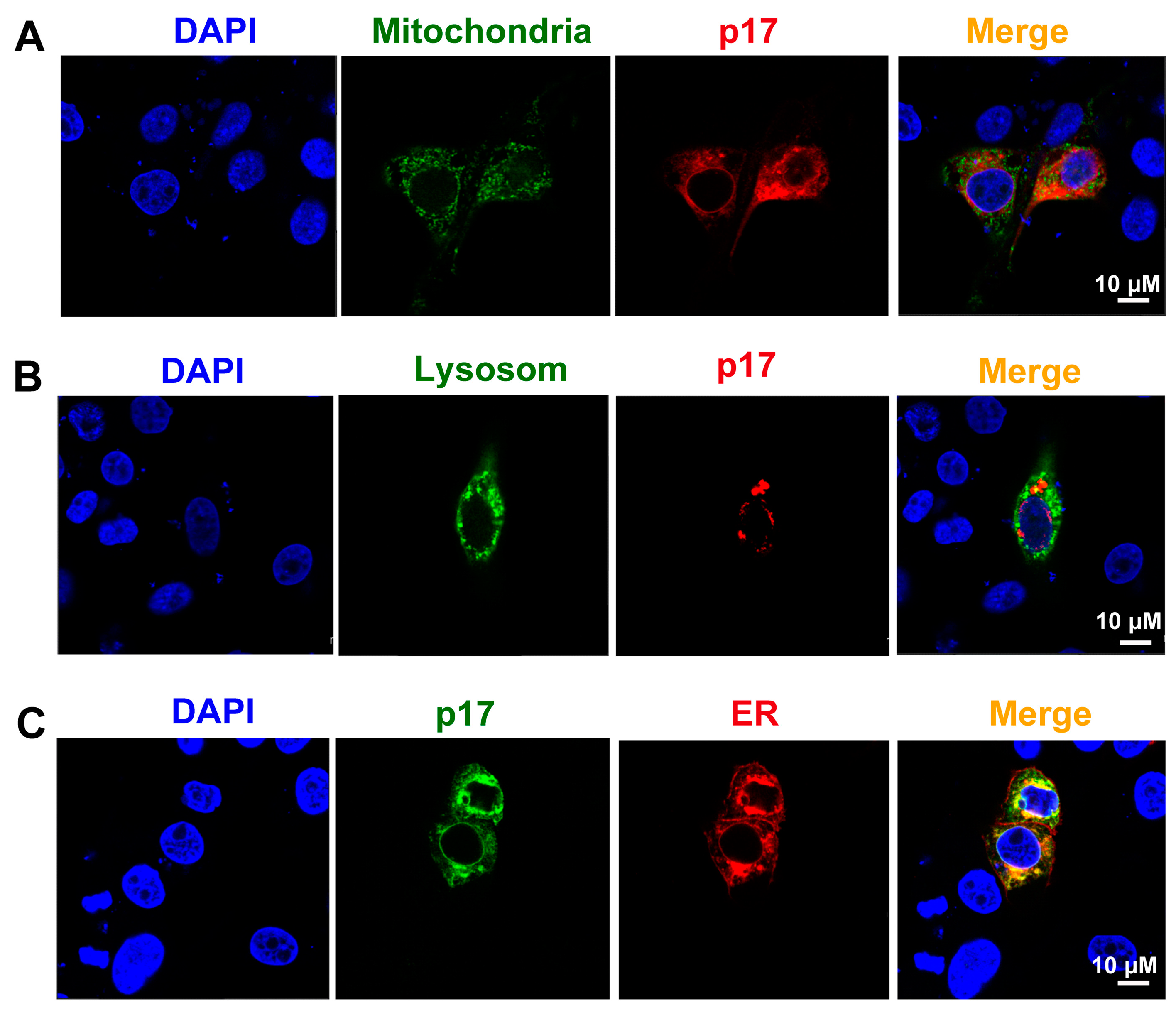
3.5. ASFV p17 Was Localized in the Endoplasmic Reticulum (ER) and Induced ER Stress
3.6. ASFV p17 Inhibited Cell Proliferation through ER Stress and ROS-Mediated Cell Cycle Arrest
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rai, A.; Pruitt, S.; Ramirez-Medina, E.; Vuono, E.A.; Silva, E.; Velazquez-Salinas, L.; Carrillo, C.; Borca, M.V.; Gladue, D.P. Identification of a Continuously Stable and Commercially Available Cell Line for the Identification of Infectious African Swine Fever Virus in Clinical Samples. Viruses 2020, 12, 820. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, J.; Wu, Y.; Chen, H.; Zhang, S.; Li, J.; Xin, T.; Jia, H.; Hou, S.; Jiang, Y.; et al. Inhibition of cGAS-STING-TBK1 signaling pathway by DP96R of ASFV China 2018/1. Biochem. Biophys. Res. Commun. 2018, 506, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Borca, M.V.; O’Donnell, V.; Holinka, L.G.; Risatti, G.R.; Ramirez-Medina, E.; Vuono, E.A.; Shi, J.; Pruitt, S.; Rai, A.; Silva, E.; et al. Deletion of CD2-like gene from the genome of African swine fever virus strain Georgia does not attenuate virulence in swine. Sci. Rep. 2020, 10, 494. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Huhr, J.; Blome, S.; Conraths, F.J.; Probst, C. Stability of African Swine Fever Virus in Carcasses of Domestic Pigs and Wild Boar Experimentally Infected with the ASFV “Estonia 2014” Isolate. Viruses 2020, 12, 1118. [Google Scholar] [CrossRef]
- O’Donnell, V.; Holinka, L.G.; Gladue, D.P.; Sanford, B.; Krug, P.W.; Lu, X.; Arzt, J.; Reese, B.; Carrillo, C.; Risatti, G.R.; et al. African Swine Fever Virus Georgia Isolate Harboring Deletions of MGF360 and MGF505 Genes Is Attenuated in Swine and Confers Protection against Challenge with Virulent Parental Virus. J. Virol. 2015, 89, 6048–6056. [Google Scholar] [CrossRef] [Green Version]
- Franzoni, G.; Graham, S.P.; Giudici, S.D.; Bonelli, P.; Pilo, G.; Anfossi, A.G.; Pittau, M.; Nicolussi, P.S.; Laddomada, A.; Oggiano, A. Characterization of the interaction of African swine fever virus with monocytes and derived macrophage subsets. Vet. MicroBiol. 2017, 198, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Franzoni, G.; Graham, S.P.; Sanna, G.; Angioi, P.; Fiori, M.S.; Anfossi, A.; Amadori, M.; Dei Giudici, S.; Oggiano, A. Interaction of porcine monocyte-derived dendritic cells with African swine fever viruses of diverse virulence. Vet. Microbiol. 2018, 216, 190–197. [Google Scholar] [CrossRef]
- Oura, C.A.; Powell, P.P.; Anderson, E.; Parkhouse, R.M. The pathogenesis of African swine fever in the resistant bushpig. J. Gen. Virol. 1998, 79 Pt 6, 1439–1443. [Google Scholar] [CrossRef] [Green Version]
- Salguero, F.J.; Sanchez-Cordon, P.J.; Nunez, A.; Fernandez de Marco, M.; Gomez-Villamandos, J.C. Proinflammatory cytokines induce lymphocyte apoptosis in acute African swine fever infection. J. Comp. Pathol. 2005, 132, 289–302. [Google Scholar] [CrossRef]
- Oura, C.A.; Powell, P.P.; Parkhouse, R.M. Detection of African swine fever virus in infected pig tissues by immunocytochemistry and in sity hybridisation. J. Virol. Methods 1998, 72, 205–217. [Google Scholar] [CrossRef]
- Ramiro-Ibanez, F.; Ortega, A.; Brun, A.; Escribano, J.M.; Alonso, C. Apoptosis: A mechanism of cell killing and lymphoid organ impairment during acute African swine fever virus infection. J. Gen. Virol. 1996, 77 Pt 9, 2209–2219. [Google Scholar] [CrossRef]
- Sierra, M.A.; Quezada, M.; Fernandez, A.; Carrasco, L.; Gomez-Villamandos, J.C.; Martin de las Mulas, J.; Sanchez-Vizcaino, J.M. Experimental African swine fever: Evidence of the virus in interstitial tissues of the kidney. Vet. Pathol. 1989, 26, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Cabezas, M.; Munoz-Moreno, R.; Galindo, I.; Cuesta-Geijo, M.A.; Alonso, C. A179L, a new viral Bcl2 homolog targeting Beclin 1 autophagy related protein. Curr. Mol. Med. 2013, 13, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.L.; Connell, S.; Benfield, C.T.O.; Dixon, L.K. The Genetics of Life and Death: Virus-Host Interactions Underpinning Resistance to African Swine Fever, a Viral Hemorrhagic Disease. Front. Genet. 2019, 10, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Villamandos, J.C.; Hervas, J.; Mendez, A.; Carrasco, L.; Martin de las Mulas, J.; Villeda, C.J.; Wilkinson, P.J.; Sierra, M.A. Experimental African swine fever: Apoptosis of lymphocytes and virus replication in other cells. J. Gen. Virol. 1995, 76 Pt 9, 2399–2405. [Google Scholar] [CrossRef]
- Dixon, L.K.; Sanchez-Cordon, P.J.; Galindo, I.; Alonso, C. Investigations of Pro- and Anti-Apoptotic Factors Affecting African Swine Fever Virus Replication and Pathogenesis. Viruses 2017, 9, 241. [Google Scholar] [CrossRef] [Green Version]
- Alonso, C.; Galindo, I.; Cuesta-Geijo, M.A.; Cabezas, M.; Hernaez, B.; Munoz-Moreno, R. African swine fever virus-cell interactions: From virus entry to cell survival. Virus Res. 2013, 173, 42–57. [Google Scholar] [CrossRef]
- Banjara, S.; Shimmon, G.L.; Dixon, L.K.; Netherton, C.L.; Hinds, M.G.; Kvansakul, M. Crystal Structure of African Swine Fever Virus A179L with the Autophagy Regulator Beclin. Viruses 2019, 11, 789. [Google Scholar] [CrossRef] [Green Version]
- Andres, G.; Charro, D.; Matamoros, T.; Dillard, R.S.; Abrescia, N.G.A. The cryo-EM structure of African swine fever virus unravels a unique architecture comprising two icosahedral protein capsids and two lipoprotein membranes. J. Biol. Chem. 2020, 295, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Luo, Y.; Wang, Y.; Li, S.; Zhao, Z.; Bi, Y.; Sun, J.; Peng, R.; Song, H.; Zhu, D.; et al. Cryo-EM Structure of the African Swine Fever Virus. Cell Host Microbe 2019, 26, 836–843. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; et al. Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Epifano, C.; Krijnse-Locker, J.; Salas, M.L.; Rodriguez, J.M.; Salas, J. The African swine fever virus nonstructural protein pB602L is required for formation of the icosahedral capsid of the virus particle. J. Virol. 2006, 80, 12260–12270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Sanyal, S.; Bruzzone, R. Breaking Bad: How Viruses Subvert the Cell Cycle. Front. Cell Infect. Microbiol. 2018, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Simoes, M.; Martins, C.; Ferreira, F. Host DNA damage response facilitates African swine fever virus infection. Vet. Microbiol. 2013, 165, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Quintas, A.; Sanchez, E.G.; Sabina, P.; Nogal, M.; Carrasco, L.; Revilla, Y. Regulation of host translational machinery by African swine fever virus. PLoS Pathog. 2009, 5, e1000562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arooz, T.; Yam, C.H.; Siu, W.Y.; Lau, A.; Li, K.K.; Poon, R.Y. On the concentrations of cyclins and cyclin-dependent kinases in extracts of cultured human cells. Biochemistry 2000, 39, 9494–9501. [Google Scholar] [CrossRef]
- Zhou, F.; Yuan, Q.; Zhang, W.; Niu, M.; Fu, H.; Qiu, Q.; Mao, G.; Wang, H.; Wen, L.; Wang, H.; et al. MiR-663a Stimulates Proliferation and Suppresses Early Apoptosis of Human Spermatogonial Stem Cells by Targeting NFIX and Regulating Cell Cycle. Mol. Ther. Nucleic Acids 2018, 12, 319–336. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Wang, C.; Wu, J. Expression of geminin, p16, and Ki67 in cervical intraepithelial neoplasm and normal tissues. Medicine 2017, 96, e7302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Z.; Hao, Q.; Tan, W.; Sun, J.; Li, J.; Chen, C.W.; Li, Z.; Meng, Y.; Zhou, Y.; et al. The Cdk2-c-Myc-miR-571 Axis Regulates DNA Replication and Genomic Stability by Targeting Geminin. Cancer Res. 2019, 79, 4896–4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.F.; Dong, Q.; Bai, Y.; Yuan, J.; Xu, Q.; Cao, C.; Liu, X. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic. Biol. Med. 2017, 103, 177–187. [Google Scholar] [CrossRef] [PubMed]
- D’Angiolella, V.; Santarpia, C.; Grieco, D. Oxidative stress overrides the spindle checkpoint. Cell Cycle 2007, 6, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Fan, R.F.; Yang, D.B.; Zhang, D.; Wang, L. Puerarin reverses cadmium-induced lysosomal dysfunction in primary rat proximal tubular cells via inhibiting Nrf2 pathway. Biochem. Pharmacol. 2019, 162, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Feng, N.; Wang, Y.; Noll, L.; Xu, S.; Liu, X.; Lu, N.; Zou, H.; Gu, J.; Yuan, Y.; et al. Effects of zearalenone and its derivatives on the synthesis and secretion of mammalian sex steroid hormones: A review. Food Chem. Toxicol. 2019, 126, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Singh-Mallah, G.; Nair, S.; Sandberg, M.; Mallard, C.; Hagberg, H. The Role of Mitochondrial and Endoplasmic Reticulum Reactive Oxygen Species Production in Models of Perinatal Brain Injury. Antioxid. Redox Signal. 2019, 31, 643–663. [Google Scholar] [CrossRef]
- Zheng, W.L.; Wang, B.J.; Wang, L.; Shan, Y.P.; Zou, H.; Song, R.L.; Wang, T.; Gu, J.H.; Yuan, Y.; Liu, X.Z.; et al. ROS-Mediated Cell Cycle Arrest and Apoptosis Induced by Zearalenone in Mouse Sertoli Cells via ER Stress and the ATP/AMPK Pathway. Toxins 2018, 10, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Netherton, C.L.; Parsley, J.C.; Wileman, T. African swine fever virus inhibits induction of the stress-induced proapoptotic transcription factor CHOP/GADD153. J. Virol. 2004, 78, 10825–10828. [Google Scholar] [CrossRef] [Green Version]
- Araki, K.; Iemura, S.; Kamiya, Y.; Ron, D.; Kato, K.; Natsume, T.; Nagata, K. Ero1-alpha and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol. 2013, 202, 861–874. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, N.; Wang, H.; Liu, X.; Shao, Q.; Ao, D.; Xu, Y.; Jiang, S.; Luo, J.; Zhang, J.; Chen, N.; et al. African Swine Fever Virus Structural Protein p17 Inhibits Cell Proliferation through ER Stress—ROS Mediated Cell Cycle Arrest. Viruses 2021, 13, 21. https://0-doi-org.brum.beds.ac.uk/10.3390/v13010021
Xia N, Wang H, Liu X, Shao Q, Ao D, Xu Y, Jiang S, Luo J, Zhang J, Chen N, et al. African Swine Fever Virus Structural Protein p17 Inhibits Cell Proliferation through ER Stress—ROS Mediated Cell Cycle Arrest. Viruses. 2021; 13(1):21. https://0-doi-org.brum.beds.ac.uk/10.3390/v13010021
Chicago/Turabian StyleXia, Nengwen, Hui Wang, Xueliang Liu, Qi Shao, Da Ao, Yulin Xu, Sen Jiang, Jia Luo, Jiajia Zhang, Nanhua Chen, and et al. 2021. "African Swine Fever Virus Structural Protein p17 Inhibits Cell Proliferation through ER Stress—ROS Mediated Cell Cycle Arrest" Viruses 13, no. 1: 21. https://0-doi-org.brum.beds.ac.uk/10.3390/v13010021