
Characterization of a Novel Mitovirus of the Sand Fly Lutzomyia longipalpis Using Genomic and Virus–Host Interaction Signatures

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Acquisition and Processing of RNA Libraries
2.2. Profile HMM Screening and Progressive Assembly
2.3. Phylogenetic Analysis
2.4. Analysis of Small RNA Libraries
2.5. Dinucleotide and Codon Usage Analyses
2.6. Amplification and Sanger Sequencing
2.7. Analyses of Public Libraries
3. Results
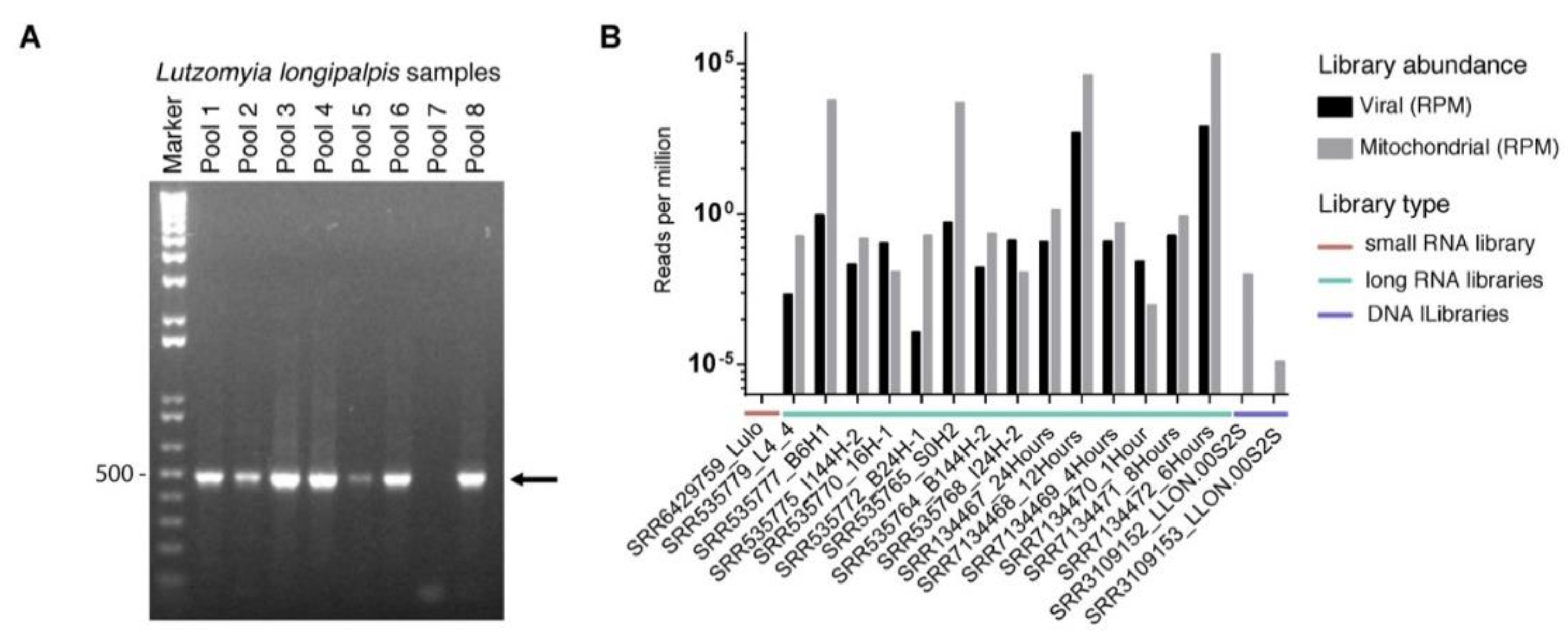
3.1. Identification of Viral Sequences in Lu. longipalpis Datasets
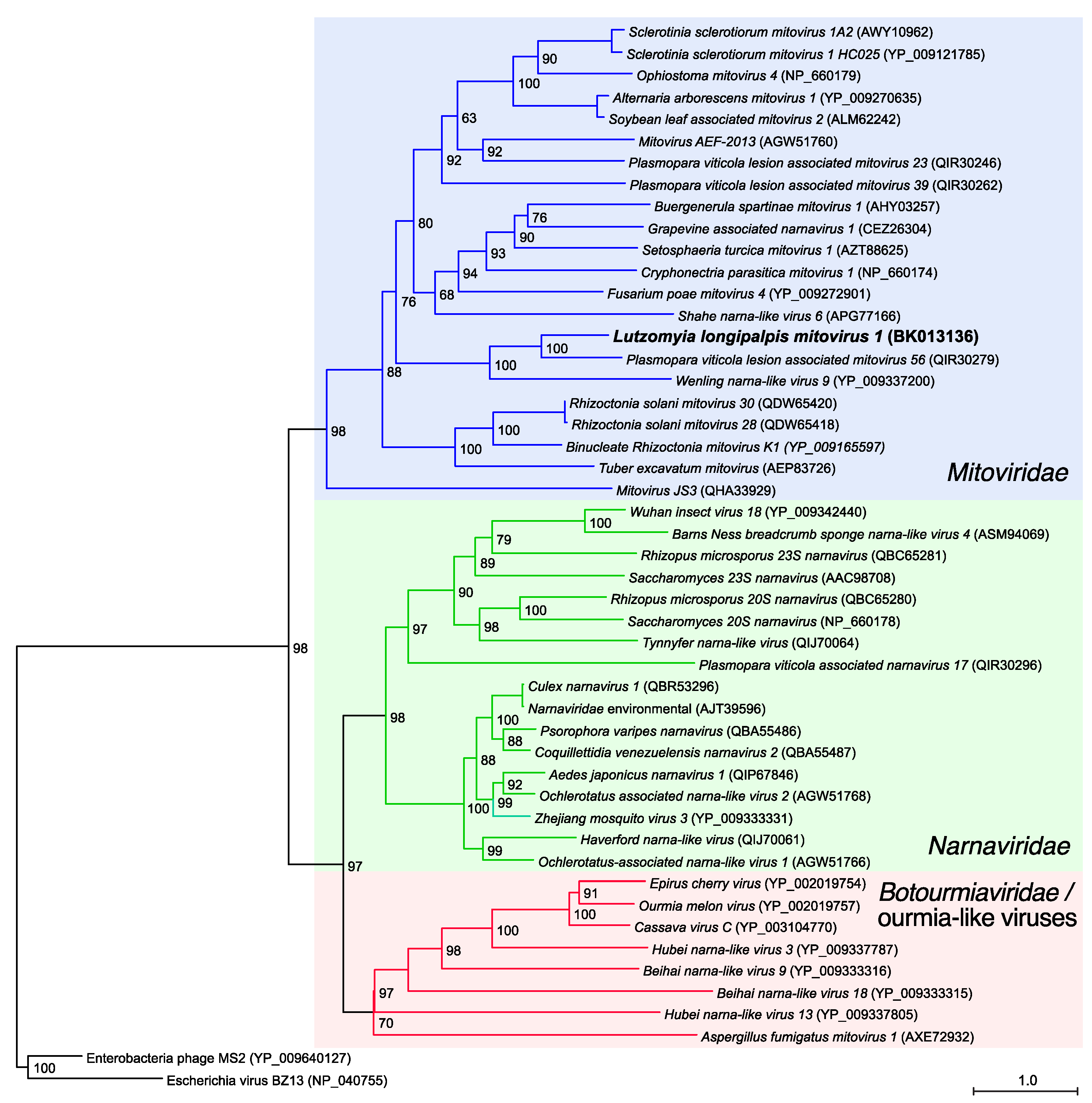
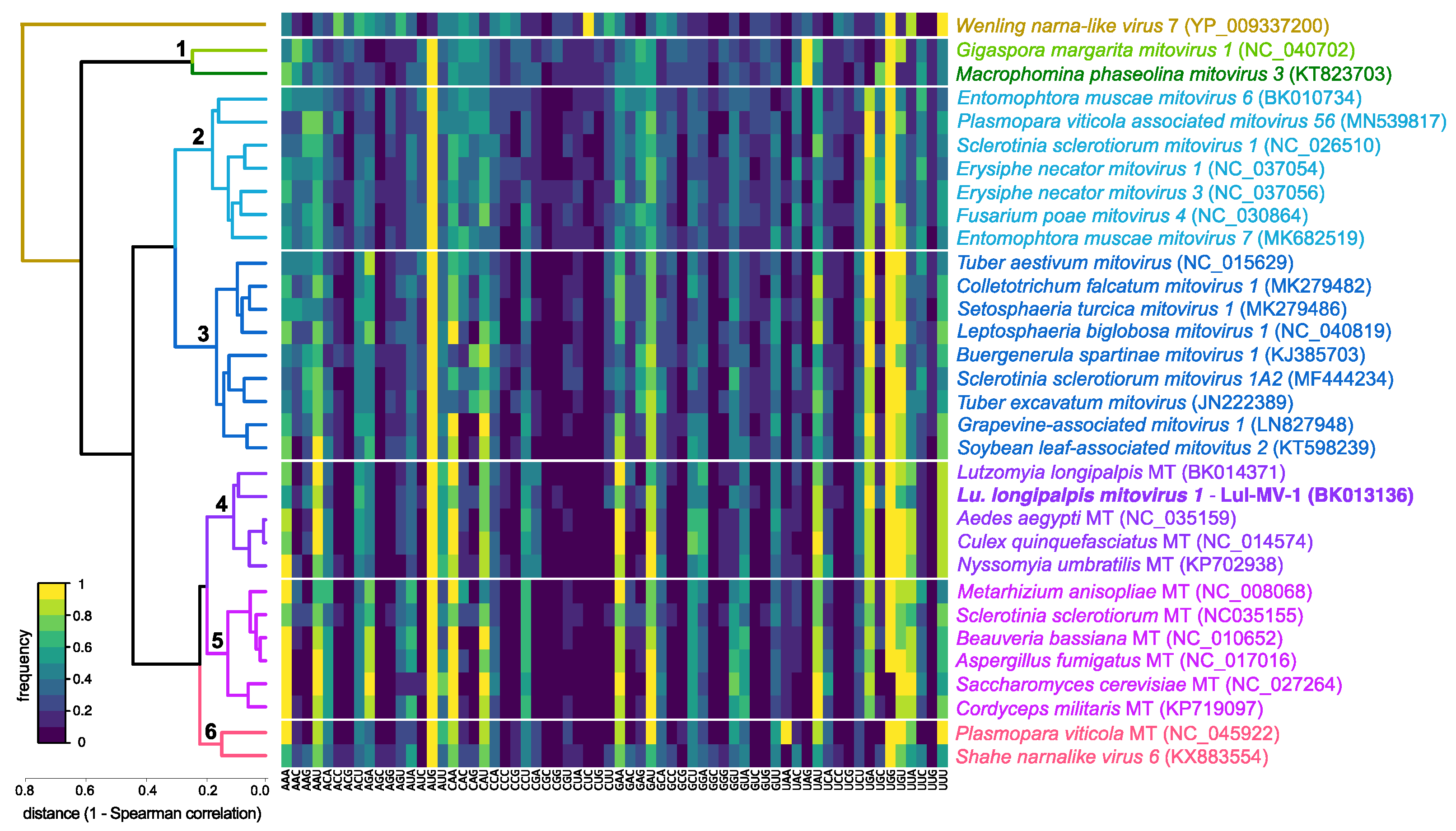
3.2. Phylogenetic and Genome-Based Characterization of Lu. longipalpis Mitovirus 1
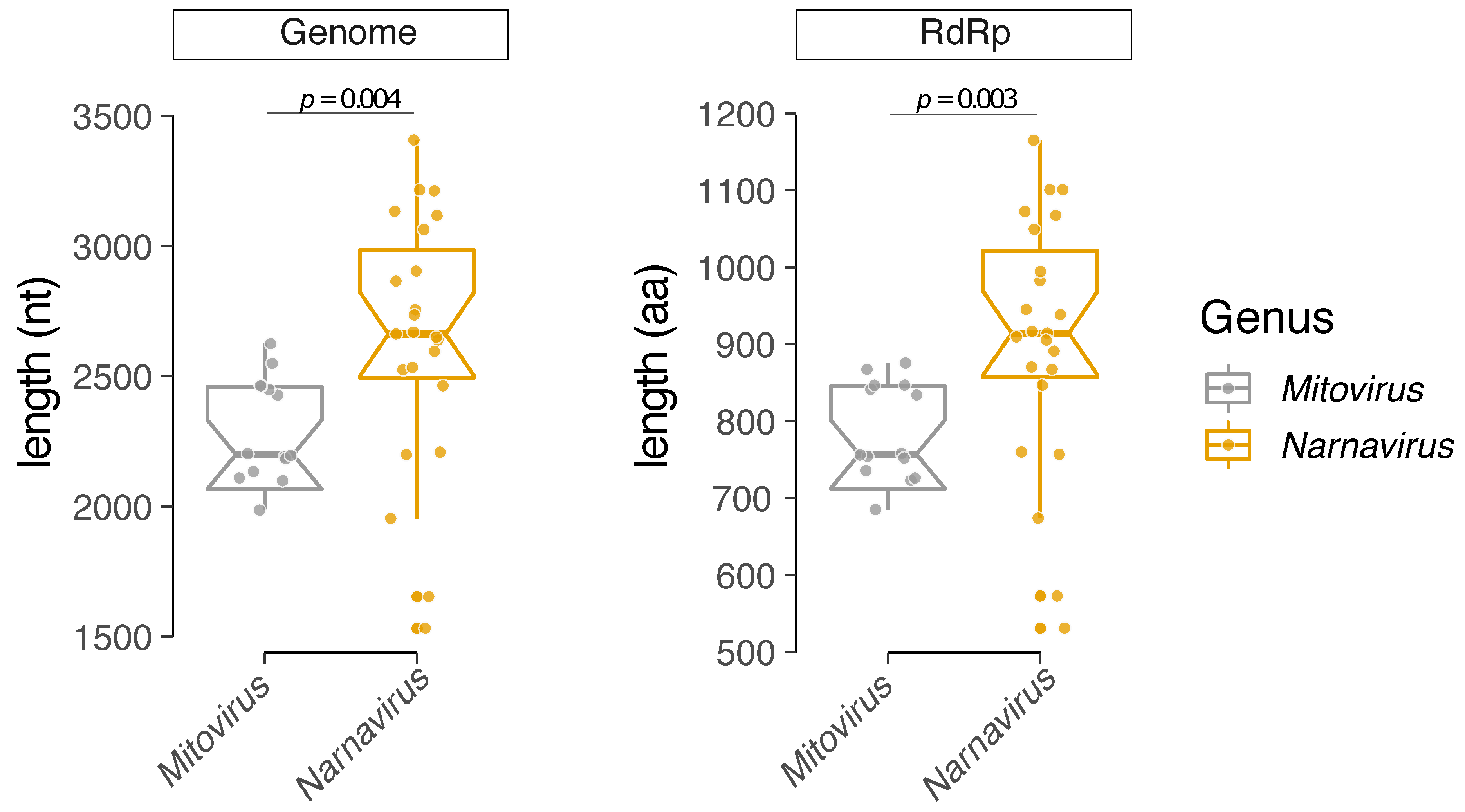
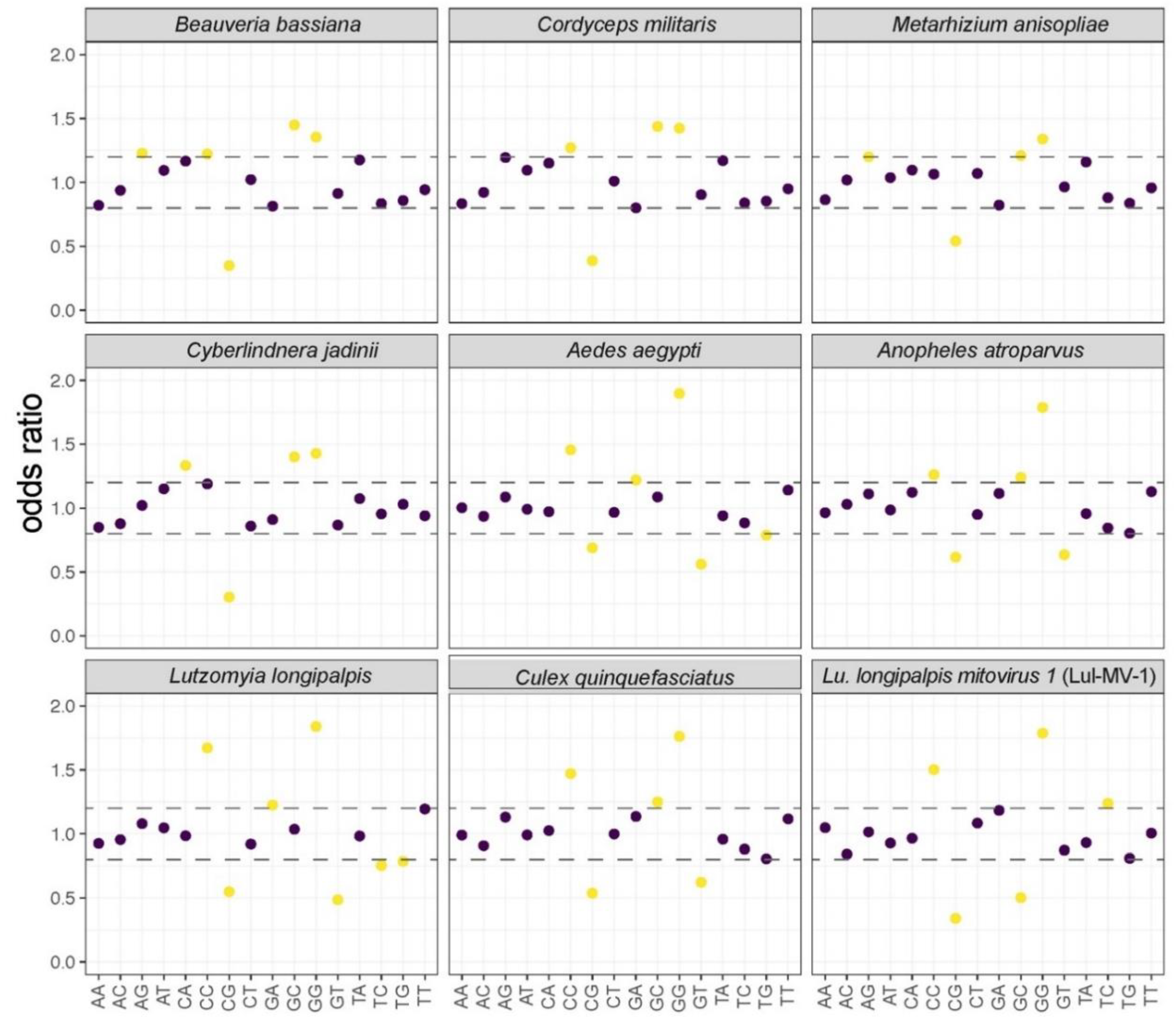
3.3. Comparative Analysis of Structural and Compositional Features
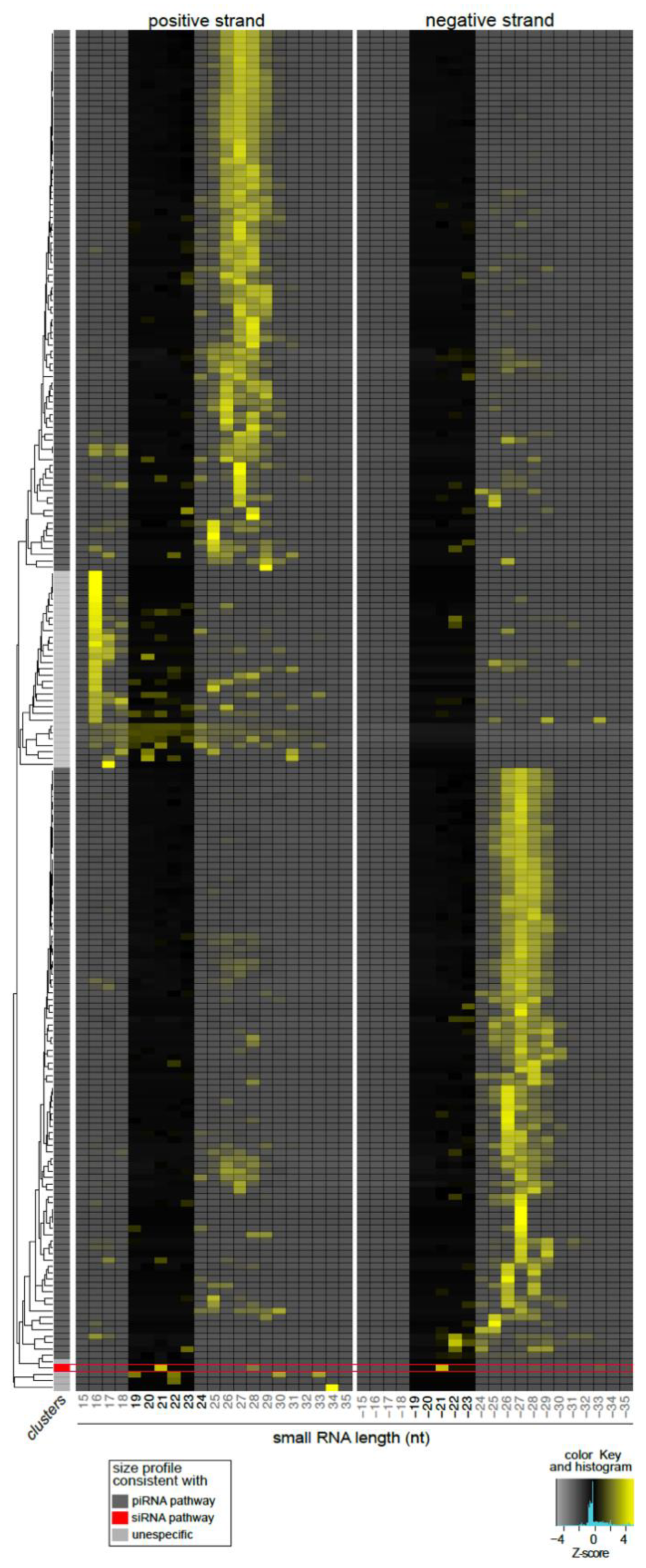
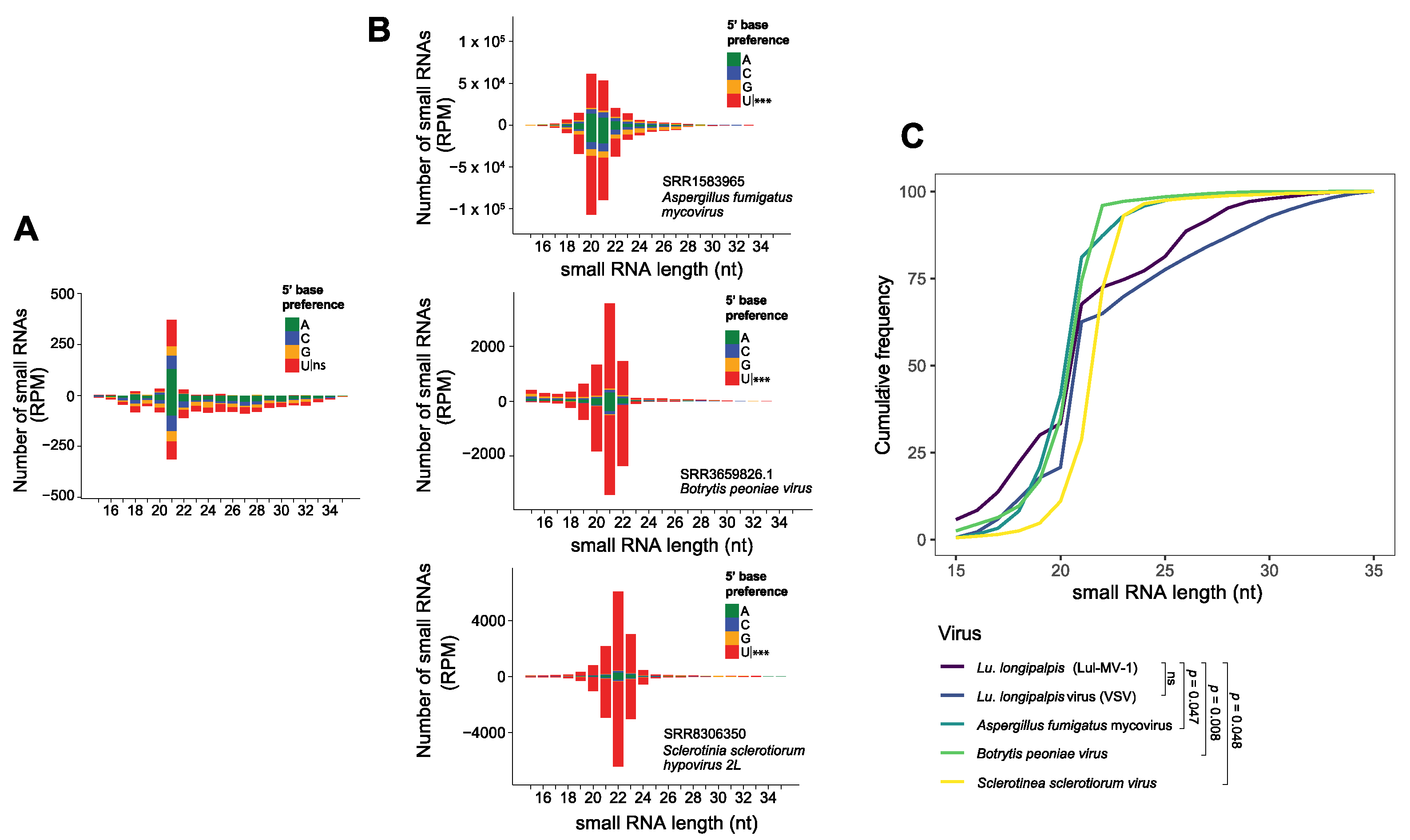
3.4. Lul-MV-1 Is Targeted by the Lu. longipalpis siRNA Pathway
3.5. Prevalence of Lul-MV-1 in Lu. longipalpis Colony and Public Datasets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hyman, P.; Abedon, S.T. Smaller Fleas: Viruses of Microorganisms. Scientifica 2012, 2012, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-Z.; Shi, M.; Holmes, E.C. Using Metagenomics to Characterize an Expanding Virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef]
- Graham, E.B.; Paez-Espino, D.; Brislawn, C.; Hofmockel, K.S.; Wu, R.; Kyrpides, N.C.; Jansson, J.K.; McDermott, J.E. Untapped viral diversity in global soil metagenomes. bioRxiv 2019. [CrossRef]
- Ng, T.F.F.; Willner, D.L.; Lim, Y.W.; Schmieder, R.; Chau, B.; Nilsson, C.; Anthony, S.; Ruan, Y.; Rohwer, F.; Breitbart, M. Broad Surveys of DNA Viral Diversity Obtained through Viral Metagenomics of Mosquitoes. PLoS ONE 2011, 6, e20579. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef]
- Reyes, A.P.; Alves, J.M.; Durham, A.M.; Gruber, A. Use of profile hidden Markov models in viral discovery: Current insights. Adv. Genom Genet. 2017, 7, 29–45. [Google Scholar] [CrossRef] [Green Version]
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol 2018, 92, e01031-17. [Google Scholar] [CrossRef] [Green Version]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. JVI 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [Green Version]
- Fancello, L.; Raoult, D.; Desnues, C. Computational tools for viral metagenomics and their application in clinical research. Virology 2012, 434, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.Y. Viral pathogen discovery. Curr. Opin. Microbiol. 2013, 16, 468–478. [Google Scholar] [CrossRef] [Green Version]
- Pallen, M.J. Diagnostic metagenomics: Potential applications to bacterial, viral and parasitic infections. Parasitology 2014, 141, 1856–1862. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Roux, S.; Chan, L.-K.; Egan, R.; Malmstrom, R.R.; McMahon, K.D.; Sullivan, M.B. Ecogenomics of virophages and their giant virus hosts assessed through time series metagenomics. Nat. Commun. 2017, 8, 858. [Google Scholar] [CrossRef]
- Novella, I.S.; Clarke, D.K.; Quer, J.; Duarte, E.A.; Lee, C.H.; Weaver, S.C.; Elena, S.F.; Moya, A.; Domingo, E.; Holland, J.J. Extreme fitness differences in mammalian and insect hosts after continuous replication of vesicular stomatitis virus in sandfly cells. J. Virol. 1995, 69, 6805–6809. [Google Scholar] [CrossRef] [Green Version]
- Belalov, I.S.; Lukashev, A.N. Causes and Implications of Codon Usage Bias in RNA Viruses. PLoS ONE 2013, 8, e56642. [Google Scholar] [CrossRef]
- Lobo, F.P.; Mota, B.E.F.; Pena, S.D.J.; Azevedo, V.; Macedo, A.M.; Tauch, A.; Machado, C.R.; Franco, G.R. Virus–host Coevolution: Common Patterns of Nucleotide Motif Usage in Flaviviridae and Their Hosts. PLoS ONE 2009, 4, e6282. [Google Scholar] [CrossRef] [Green Version]
- Biswas, K.; Palchoudhury, S.; Chakraborty, P.; Bhattacharyya, U.; Ghosh, D.; Debnath, P.; Ramadugu, C.; Keremane, M.; Khetarpal, R.; Lee, R. Codon Usage Bias Analysis of Citrus tristeza Virus: Higher Codon Adaptation to Citrus reticulata Host. Viruses 2019, 11, 331. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Tan, Z.; Zeng, G.; Peng, J. Comprehensive Analysis of Simple Sequence Repeats in Pre-miRNAs. Mol. Biol. Evol. 2010, 27, 2227–2232. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Tan, Z.; Zeng, G. Microsatellite is an important component of complete Hepatitis C virus genomes. Infect. Genet. Evol. 2011, 11, 1646–1654. [Google Scholar] [CrossRef]
- Di Giallonardo, F.; Schlub, T.E.; Shi, M.; Holmes, E.C. Dinucleotide Composition in Animal RNA Viruses Is Shaped More by Virus Family than by Host Species. J. Virol 2017, 91, e02381-16. [Google Scholar] [CrossRef] [Green Version]
- Obbard, D.J.; Gordon, K.H.J.; Buck, A.H.; Jiggins, F.M. The evolution of RNAi as a defence against viruses and transposable elements. Phil. Trans. R. Soc. B 2009, 364, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, E.R.G.R.; Olmo, R.P.; Paro, S.; Ferreira, F.V.; de Faria, I.J.D.S.; Todjro, Y.M.H.; Lobo, F.P.; Kroon, E.G.; Meignin, C.; Gatherer, D.; et al. Sequence-independent characterization of viruses based on the pattern of viral small RNAs produced by the host. Nucleic Acids Res. 2015, 43, 6191–6206. [Google Scholar] [CrossRef]
- Aguiar, E.R.G.R.; de Almeida, J.P.P.; Queiroz, L.R.; Oliveira, L.S.; Olmo, R.P.; de Faria, I.J.; da S Imler, J.-L.; Gruber, A.; Matthews, B.J.; Marques, J.T. A single unidirectional piRNA cluster similar to the flamenco locus is the major source of EVE-derived transcription and small RNAs in Aedes aegypti mosquitoes. RNA 2020, 26, 581–594. [Google Scholar] [CrossRef]
- Aguiar, E.R.G.R.; Olmo, R.P.; Marques, J.T. Virus-derived small RNAs: Molecular footprints of host-pathogen interactions: Virus-derived small RNAs. WIREs RNA 2016, 7, 824–837. [Google Scholar] [CrossRef]
- Webster, C.L.; Waldron, F.M.; Robertson, S.; Crowson, D.; Ferrari, G.; Quintana, J.F.; Brouqui, J.-M.; Bayne, E.H.; Longdon, B.; Buck, A.H.; et al. The Discovery, Distribution, and Evolution of Viruses Associated with Drosophila melanogaster. PLoS Biol 2015, 13, e1002210. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, M.M.; Klein, T.A.; Kochel, T.; Quandelacy, T.M.; Smith, B.L.; Villinski, J.; Bethell, D.; Tyner, S.; Se, Y.; Lon, C.; et al. Malaria and other vector-borne infection surveillance in the U.S. Department of Defense Armed Forces Health Surveillance Center-Global Emerging Infections Surveillance program: Review of 2009 accomplishments. BMC Public Health 2011, 11, S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, E.; Pettersson, J.; Higgs, S.; Charrel, R.; de Lamballerie, X. Emerging arboviruses: Why today? One Health 2017, 4, 1–13. [Google Scholar] [CrossRef]
- Ayhan, N.; Prudhomme, J.; Laroche, L.; Bañuls, A.-L.; Charrel, R.N. Broader Geographical Distribution of Toscana Virus in the Mediterranean Region Suggests the Existence of Larger Varieties of Sand Fly Vectors. Microorganisms 2020, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- Minard, G.; Mavingui, P.; Moro, C. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit Vectors 2013, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Calisher, C.H.; Higgs, S. The Discovery of Arthropod-Specific Viruses in Hematophagous Arthropods: An Open Door to Understanding the Mechanisms of Arbovirus and Arthropod Evolution? Annu. Rev. Entomol. 2018, 63, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.V.; Aguiar, E.R.G.R.; Olmo, R.P.; de Oliveira, K.P.V.; Silva, E.G.; Sant′Anna, M.R.V.; Gontijo, N.D.F.; Kroon, E.G.; Imler, J.L.; Marques, J.T. The small non-coding RNA response to virus infection in the Leishmania vector Lutzomyia longipalpis. PLoS Negl. Trop. Dis. 2018, 12, e0006569. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.; Chung, B.Y.-W.; Bass, D.; Moureau, G.; Tang, S.; McAlister, E.; Culverwell, C.L.; Glücksman, E.; Wang, H.; Brown, T.D.K.; et al. Novel Virus Discovery and Genome Reconstruction from Field RNA Samples Reveals Highly Divergent Viruses in Dipteran Hosts. PLoS ONE 2013, 8, e80720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. In International Committee on Taxonomy of Viruses; King, A.M.Q. (Ed.) Academic Press: Waltham, MA, USA, 2012; ISBN 978-0-12-384684-6. [Google Scholar]
- Hillman, B.I.; Cai, G. The Family Narnaviridae. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2013; Volume 86, pp. 149–176. ISBN 978-0-12-394315-6. [Google Scholar]
- ICTV Master Species List 2019.v1 (MSL #35). Available online: https://talk.ictvonline.org/files/master-species-lists/m/msl/9601/download (accessed on 27 August 2020).
- Koonin, E.V.; Dolja, V.V. Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements. Microbiol. Mol. Biol. Rev. 2014, 78, 278–303. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, Y.; Abraham, A.; Uesaka, K.; Kondo, H.; Suga, H.; Suzuki, N.; Chiba, S. Novel Mitoviruses and a Unique Tymo-Like Virus in Hypovirulent and Virulent Strains of the Fusarium Head Blight Fungus, Fusarium boothii. Viruses 2018, 10, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nibert, M.; Debat, H.; Manny, A.; Grigoriev, I.; De Fine Licht, H. Mitovirus and Mitochondrial Coding Sequences from Basal Fungus Entomophthora muscae. Viruses 2019, 11, 351. [Google Scholar] [CrossRef] [Green Version]
- Espino-Vázquez, A.N.; Bermúdez-Barrientos, J.R.; Cabrera-Rangel, J.F.; Córdova-López, G.; Cardoso-Martínez, F.; Martínez-Vázquez, A.; Camarena-Pozos, D.A.; Mondo, S.J.; Pawlowska, T.E.; Abreu-Goodger, C.; et al. Narnaviruses: Novel players in fungal–bacterial symbioses. ISME J. 2020, 14, 1743–1754. [Google Scholar] [CrossRef]
- Lin, Y.; Zhou, J.; Zhou, X.; Shuai, S.; Zhou, R.; An, H.; Fang, S.; Zhang, S.; Deng, Q. A novel narnavirus from the plant-pathogenic fungus Magnaporthe oryzae. Arch. Virol. 2020, 165, 1235–1240. [Google Scholar] [CrossRef]
- Pearson, M.N.; Beever, R.E.; Boine, B.; Arthur, K. Mycoviruses of filamentous fungi and their relevance to plant pathology. Mol. Plant. Pathol. 2009, 10, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Dolja, V.V.; Koonin, E.V. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2018, 244, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Ohkita, S.; Lee, Y.; Nguyen, Q.; Ikeda, K.; Suzuki, N.; Nakayashiki, H. Three ourmia-like viruses and their associated RNAs in Pyricularia oryzae. Virology 2019, 534, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef]
- Brenner, S.E.; Chothia, C.; Hubbard, T.J.P. Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc. Natl. Acad. Sci. USA 1998, 95, 6073–6078. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Karplus, K.; Barrett, C.; Hughey, R.; Haussler, D.; Hubbard, T.; Chothia, C. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 1998, 284, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Skewes-Cox, P.; Sharpton, T.J.; Pollard, K.S.; DeRisi, J.L. Profile Hidden Markov Models for the Detection of Viruses within Metagenomic Sequence Data. PLoS ONE 2014, 9, e105067. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, S.; Bulla, I.; Ziller, M.; Pohlmann, A.; Harder, T.; Stanke, M. ClassyFlu: Classification of Influenza A Viruses with Discriminatively Trained Profile-HMMs. PLoS ONE 2014, 9, e84558. [Google Scholar] [CrossRef]
- Masembe, C.; Phan, M.V.T.; Robertson, D.L.; Cotten, M. Increased resolution of African swine fever virus genome patterns based on profile HMMs of protein domains. Virus Evol. 2020, 6, veaa044. [Google Scholar] [CrossRef]
- Bramley, J.C.; Yenkin, A.L.; Zaydman, M.A.; DiAntonio, A.; Milbrandt, J.D.; Buchser, W.J. Domain-centric database to uncover structure of minimally characterized viral genomes. Sci. Data 2020, 7, 202. [Google Scholar] [CrossRef]
- Alves, J.M.P.; de Oliveira, A.L.; Sandberg, T.O.M.; Moreno-Gallego, J.L.; de Toledo, M.A.F.; de Moura, E.M.M.; Oliveira, L.S.; Durham, A.M.; Mehnert, D.U.; de A. Zanotto, P.M.; et al. GenSeed-HMM: A Tool for Progressive Assembly Using Profile HMMs as Seeds and its Application in Alpavirinae Viral Discovery from Metagenomic Data. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladner, J.T.; Beitzel, B.; Chain, P.S.G.; Davenport, M.G.; Donaldson, E.; Frieman, M.; Kugelman, J.; Kuhn, J.H.; O′Rear, J.; Sabeti, P.C.; et al. Standards for Sequencing Viral Genomes in the Era of High-Throughput Sequencing. mBio 2014, 5, e01360-14. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An Interactive Tool for Rooted Phylogenetic Trees and Networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, J.T.; Wang, J.-P.; Wang, X.; de Oliveira, K.P.V.; Gao, C.; Aguiar, E.R.G.R.; Jafari, N.; Carthew, R.W. Functional Specialization of the Small Interfering RNA Pathway in Response to Virus Infection. PLoS Pathog. 2013, 9, e1003579. [Google Scholar] [CrossRef]
- Wei, T.; Simko, V. Package ‘corrplot’. Available online: https://cran.r-project.org/web/packages/corrplot/corrplot.pdf (accessed on 15 August 2020).
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Use R! Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Maumus, F.; Fiston-Lavier, A.-S.; Quesneville, H. Impact of transposable elements on insect genomes and biology. Curr. Opin. Insect Sci. 2015, 7, 30–36. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Katzourakis, A. Endogenous viruses: Connecting recent and ancient viral evolution. Virology 2015, 479–480, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, M.J.; Bruenn, J.A.; Taylor, D.J. Phylogeny, integration and expression of sigma virus-like genes in Drosophila. Mol. Phylogenet. Evol. 2012, 65, 251–258. [Google Scholar] [CrossRef]
- Virus Taxonomy: The Classification and Nomenclature of Viruses The 9th Report of the ICTV (2011). Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/ (accessed on 27 August 2020).
- Shackelton, L.A.; Holmes, E.C. The role of alternative genetic codes in viral evolution and emergence. J. Theor. Biol. 2008, 254, 128–134. [Google Scholar] [CrossRef]
- Bruenn, J.A.; Warner, B.E.; Yerramsetty, P. Widespread mitovirus sequences in plant genomes. PeerJ 2015, 3, e876. [Google Scholar] [CrossRef] [Green Version]
- Chiapello, M.; Rodríguez-Romero, J.; Ayllón, M.A.; Turina, M. Analysis of the virome associated to grapevine downy mildew lesions reveals new mycovirus lineages. Virus Evol. 2020, veaa058. [Google Scholar] [CrossRef]
- Zoll, J.; Verweij, P.E.; Melchers, W.J.G. Discovery and characterization of novel Aspergillus fumigatus mycoviruses. PLoS ONE 2018, 13, e0200511. [Google Scholar] [CrossRef] [PubMed]
- DeRisi, J.L.; Huber, G.; Kistler, A.; Retallack, H.; Wilkinson, M.; Yllanes, D. An exploration of ambigrammatic sequences in narnaviruses. Sci. Rep. 2019, 9, 17982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinan, A.M.; Lukhovitskaya, N.I.; Olendraite, I.; Firth, A.E. A case for a negative-strand coding sequence in a group of positive-sense RNA viruses. Virus Evol. 2020. [CrossRef]
- Velazquez-Salinas, L.; Zarate, S.; Eschbaumer, M.; Pereira Lobo, F.; Gladue, D.P.; Arzt, J.; Novella, I.S.; Rodriguez, L.L. Selective Factors Associated with the Evolution of Codon Usage in Natural Populations of Arboviruses. PLoS ONE 2016, 11, e0159943. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Miesen, P.; van Rij, R.P. Antiviral RNAi in Insects and Mammals: Parallels and Differences. Viruses 2019, 11, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayhan, N.; Charrel, R.N. Of phlebotomines (sandflies) and viruses: A comprehensive perspective on a complex situation. Curr. Opin. Insect Sci. 2017, 22, 117–124. [Google Scholar] [CrossRef]
- Alkan, C.; Bichaud, L.; de Lamballerie, X.; Alten, B.; Gould, E.A.; Charrel, R.N. Sandfly-borne phleboviruses of Eurasia and Africa: Epidemiology, genetic diversity, geographic range, control measures. Antivir. Res. 2013, 100, 54–74. [Google Scholar] [CrossRef] [Green Version]
- Nibert, M.L.; Vong, M.; Fugate, K.K.; Debat, H.J. Evidence for contemporary plant mitoviruses. Virology 2018, 518, 14–24. [Google Scholar] [CrossRef]
- Stough, J.M.A.; Beaudoin, A.J.; Schloss, P.D. Coding-Complete RNA Virus Genomes Assembled from Murine Cecal Metatranscriptomes. Microbiol. Resour. Announc. 2020, 9, e00018-20. [Google Scholar] [CrossRef] [Green Version]
- Richaud, A.; Frézal, L.; Tahan, S.; Jiang, H.; Blatter, J.A.; Zhao, G.; Kaur, T.; Wang, D.; Félix, M.-A. Vertical transmission in Caenorhabditis nematodes of RNA molecules encoding a viral RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2019, 116, 24738–24747. [Google Scholar] [CrossRef] [Green Version]
- Mahar, J.E.; Shi, M.; Hall, R.N.; Strive, T.; Holmes, E.C. Comparative Analysis of RNA Virome Composition in Rabbits and Associated Ectoparasites. J. Virol. 2020, 94, e02119-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, J.A.; Liu, R.M.; Bennett, S.N. RNA shotgun metagenomic sequencing of northern California (USA) mosquitoes uncovers viruses, bacteria, and fungi. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göertz, G.; Miesen, P.; Overheul, G.; van Rij, R.; van Oers, M.; Pijlman, G. Mosquito Small RNA Responses to West Nile and Insect-Specific Virus Infections in Aedes and Culex Mosquito Cells. Viruses 2019, 11, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nibert, M.L. Mitovirus UGA (Trp) codon usage parallels that of host mitochondria. Virology 2017, 507, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Je, M.; Kim, H.; Son, H.S. Analysis of the codon usage pattern of the RdRP gene of mycovirus infecting Aspergillus spp. Virol. J. 2019, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.-R. Dissimilation of synonymous codon usage bias in virus–host coevolution due to translational selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Wang, L.; Cai, Y.; Tao, W.; Zhang, Y.-J.; Huang, B. Mitochondrial genome of the entomophthoroid fungus Conidiobolus heterosporus provides insights into evolution of basal fungi. Appl. Microbiol. Biotechnol. 2019, 103, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Ramakodi, M.P.; Singh, B.; Wells, J.D.; Guerrero, F.; Ray, D.A. A 454 sequencing approach to dipteran mitochondrial genome research. Genomics 2015, 105, 53–60. [Google Scholar] [CrossRef]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.-S.; Imrie, A.; Holmes, E.C. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca, P.; Ferreira, F.; da Silva, F.; Oliveira, L.S.; Marques, J.T.; Goes-Neto, A.; Aguiar, E.; Gruber, A. Characterization of a Novel Mitovirus of the Sand Fly Lutzomyia longipalpis Using Genomic and Virus–Host Interaction Signatures. Viruses 2021, 13, 9. https://0-doi-org.brum.beds.ac.uk/10.3390/v13010009
Fonseca P, Ferreira F, da Silva F, Oliveira LS, Marques JT, Goes-Neto A, Aguiar E, Gruber A. Characterization of a Novel Mitovirus of the Sand Fly Lutzomyia longipalpis Using Genomic and Virus–Host Interaction Signatures. Viruses. 2021; 13(1):9. https://0-doi-org.brum.beds.ac.uk/10.3390/v13010009
Chicago/Turabian StyleFonseca, Paula, Flavia Ferreira, Felipe da Silva, Liliane Santana Oliveira, João Trindade Marques, Aristóteles Goes-Neto, Eric Aguiar, and Arthur Gruber. 2021. "Characterization of a Novel Mitovirus of the Sand Fly Lutzomyia longipalpis Using Genomic and Virus–Host Interaction Signatures" Viruses 13, no. 1: 9. https://0-doi-org.brum.beds.ac.uk/10.3390/v13010009