
Combinatorial F-G Immunogens as Nipah and Respiratory Syncytial Virus Vaccine Candidates

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antigen Design and Protein Expression
2.2. In Vitro Protein Characterisation by SDS-PAGE and SEC
2.3. ELISAs
2.4. Negative Stain Transmission Electron Microscopy (TEM)
2.5. Animal Immunisation
2.6. Micro-Fusion Inhibition Test (mFIT)
2.7. Generating Lentiviral-Based Pseudoparticles
2.8. Pseudovirus Neutralisation Assay
2.9. Plaque Reduction Neutralisation Tests (PRNTs)
3. Results
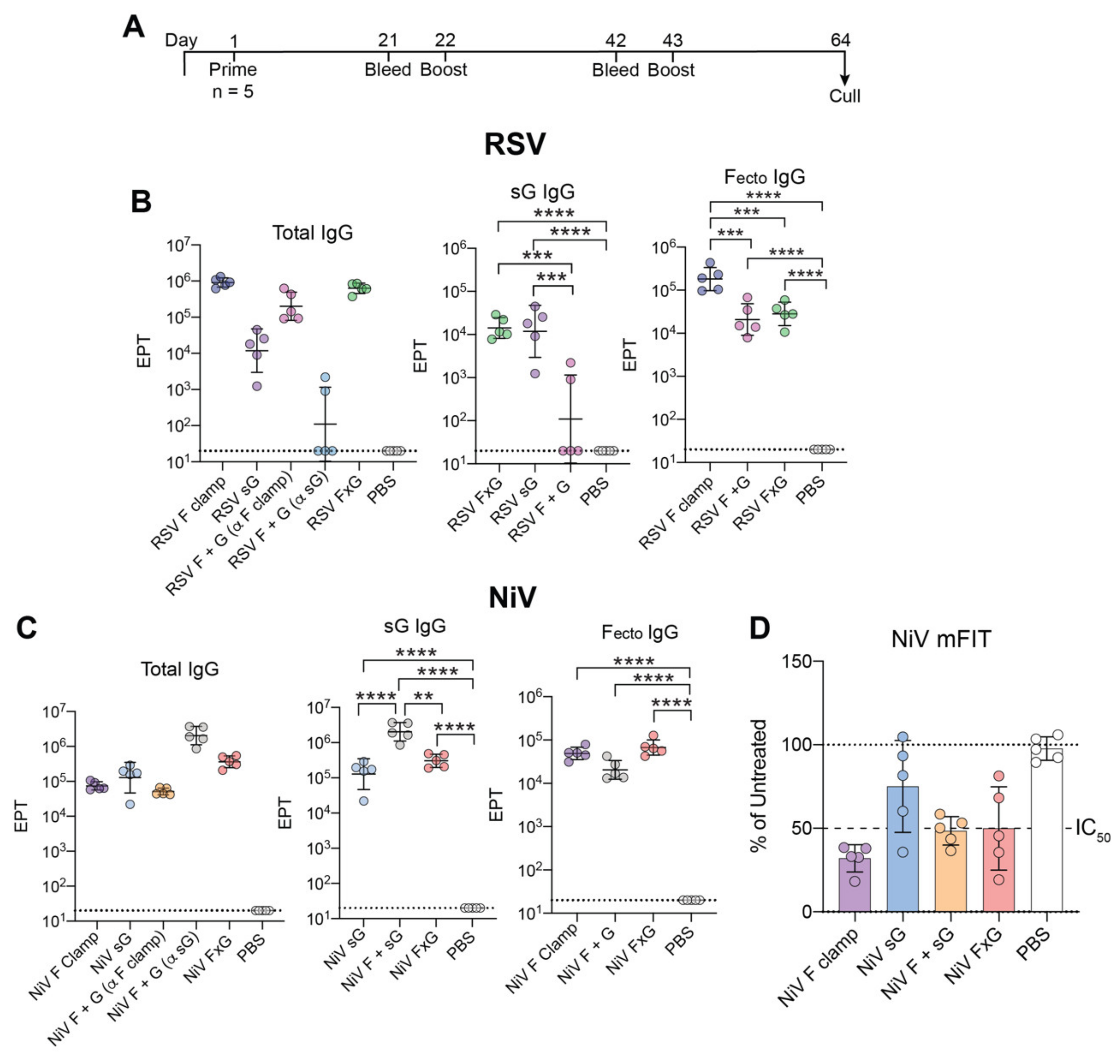
3.1. Rational Design of FxG Results in Expression of Immunogens Containing Antigenically-Sound F and G Glycoproteins
3.2. FxG Antigens Elicit an Immune Response Directed against Both Protein Targets
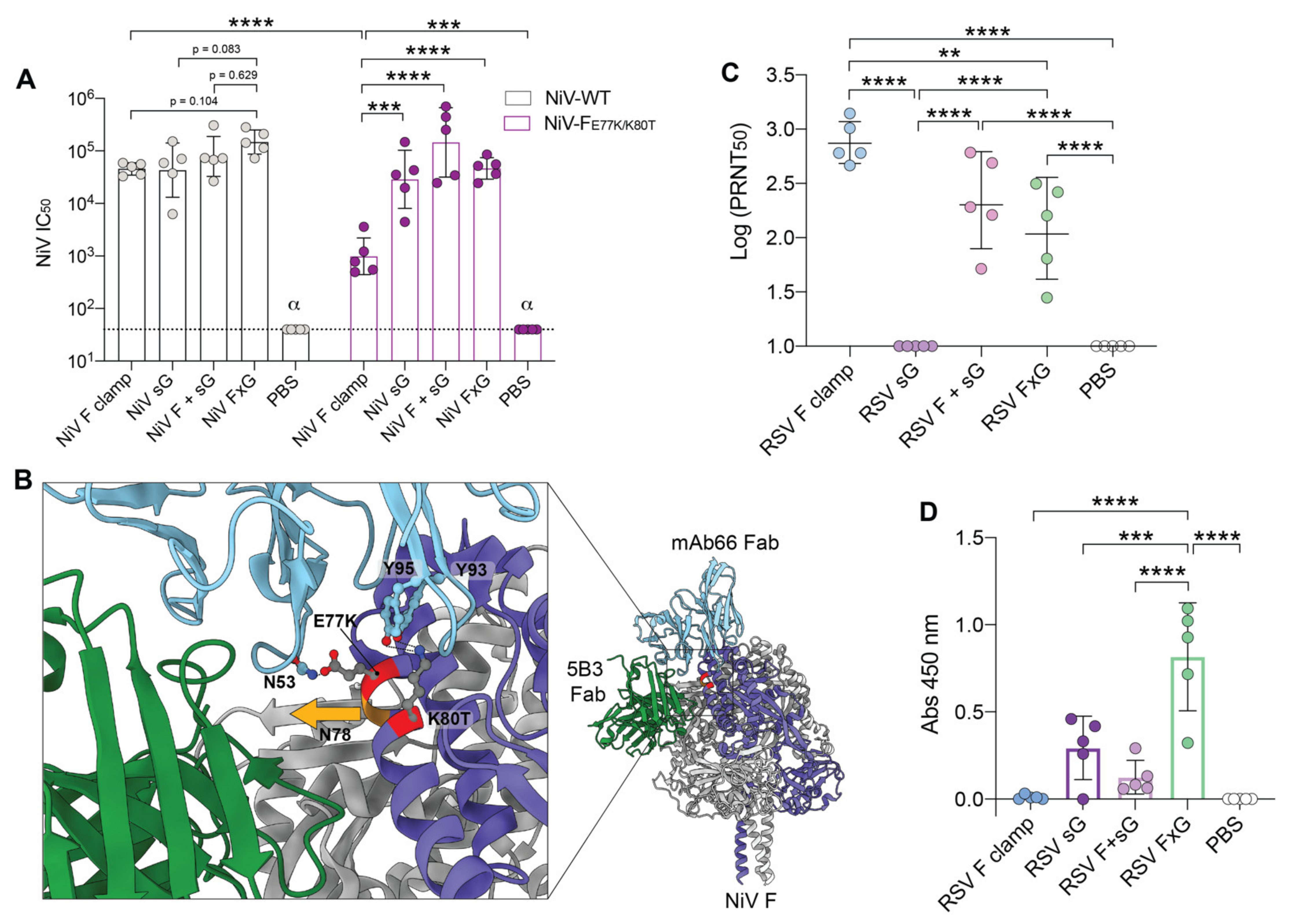
3.3. Two Vaccine Targets Allow for More Efficient Neutralisation of a NiV Pseudovirus Mutant
3.4. RSV FxG Elicits Immune Responses against the Cross-Protective Central Cysteine Domain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xuan, C.; Shi, Y.; Qi, J.; Zhang, W.; Xiao, H.; Gao, G.F. Structural vaccinology: Structure-based design of influenza A virus hemagglutinin subtype-specific subunit vaccines. Protein Cell 2011, 2, 997–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, G.-Y.; Geng, H.; Pancera, M.; Xu, K.; Cheng, C.; Acharya, P.; Chambers, M.; Druz, A.; Tsybovsky, Y.; Wanninger, T.G.; et al. Structure-Based Design of a Soluble Prefusion-Closed HIV-1 Env Trimer with Reduced CD4 Affinity and Improved Immunogenicity. J. Virol. 2017, 91, e02268-16. [Google Scholar] [CrossRef] [Green Version]
- Kulp, D.W.; Steichen, J.M.; Pauthner, M.; Hu, X.; Schiffner, T.; Liguori, A.; Cottrell, C.A.; Havenar-Daughton, C.; Ozorowski, G.; Georgeson, E.; et al. Structure-based design of native-like HIV-1 envelope trimers to silence non-neutralizing epitopes and eliminate CD4 binding. Nat. Commun. 2017, 8, 1655. [Google Scholar] [CrossRef] [PubMed]
- Loomis, R.J.; Stewart-Jones, G.B.E.; Tsybovsky, Y.; Caringal, R.T.; Morabito, K.M.; McLellan, J.S.; Chamberlain, A.L.; Nugent, S.T.; Hutchinson, G.B.; Kueltzo, L.A.; et al. Structure-Based Design of Nipah Virus Vaccines: A Generalizable Approach to Paramyxovirus Immunogen Development. Front. Immunol. 2020, 11, 842. [Google Scholar] [CrossRef]
- Rutten, L.; Gilman, M.S.A.; Blokland, S.; Juraszek, J.; McLellan, J.S.; Langedijk, J.P.M. Structure-Based Design of Prefusion-Stabilized Filovirus Glycoprotein Trimers. Cell Rep. 2020, 30, 4540–4550.e4543. [Google Scholar] [CrossRef]
- Hsieh, C.-L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.-C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501. [Google Scholar] [CrossRef]
- Sesterhenn, F.; Bonet, J.; Correia, B.E. Structure-based immunogen design—leading the way to the new age of precision vaccines. Curr. Opin. Struct. Biol. 2018, 51, 163–169. [Google Scholar] [CrossRef]
- Jardetzky, T.S.; Lamb, R.A. Activation of Paramyxovirus Membrane Fusion and Virus Entry. Curr. Opin. Virol. 2014, 5, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.A.; Paterson, R.G.; Jardetzky, T.S. Paramyxovirus membrane fusion: Lessons from the F and HN atomic structures. Virology 2006, 344, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Bonaparte, M.I.; Dimitrov, A.S.; Bossart, K.N.; Crameri, G.; Mungall, B.A.; Bishop, K.A.; Choudhry, V.; Dimitrov, D.S.; Wang, L.F.; Eaton, B.T.; et al. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10652–10657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossart, K.N.; Wang, L.F.; Flora, M.N.; Chua, K.B.; Lam, S.K.; Eaton, B.T.; Broder, C.C. Membrane fusion tropism and heterotypic functional activities of the Nipah virus and Hendra virus envelope glycoproteins. J. Virol. 2002, 76, 11186–11198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrete, O.A.; Levroney, E.L.; Aguilar, H.C.; Bertolotti-Ciarlet, A.; Nazarian, R.; Tajyar, S.; Lee, B. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 2005, 436, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Rajashankar, K.R.; Chan, Y.P.; Himanen, J.P.; Broder, C.C.; Nikolov, D.B. Host cell recognition by the henipaviruses: Crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. USA 2008, 105, 9953–9958. [Google Scholar] [CrossRef] [Green Version]
- Bossart, K.N.; Geisbert, T.W.; Feldmann, H.; Zhu, Z.; Feldmann, F.; Geisbert, J.B.; Yan, L.; Feng, Y.-R.; Brining, D.; Scott, D.; et al. A neutralizing human monoclonal antibody protects African Green monkeys from Hendra virus challenge. Sci. Transl. Med. 2011, 3, 105ra103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossart, K.N.; Zhu, Z.; Middleton, D.; Klippel, J.; Crameri, G.; Bingham, J.; McEachern, J.A.; Green, D.; Hancock, T.J.; Chan, Y.P.; et al. A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute nipah virus infection. PLoS Pathog. 2009, 5, e1000642. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cross, R.W.; Doyle, M.P.; Kose, N.; Mousa, J.J.; Annand, E.J.; Borisevich, V.; Agans, K.N.; Sutton, R.; Nargi, R.; et al. Potent Henipavirus Neutralization by Antibodies Recognizing Diverse Sites on Hendra and Nipah Virus Receptor Binding Protein. Cell 2020, 183, 1536–1550.e1517. [Google Scholar] [CrossRef]
- Zhu, Z.; Bossart, K.N.; Bishop, K.A.; Crameri, G.; Dimitrov, A.S.; McEachern, J.A.; Feng, Y.; Middleton, D.; Wang, L.-F.; Broder, C.C.; et al. Exceptionally Potent Cross-Reactive Neutralization of Nipah and Hendra Viruses by a Human Monoclonal Antibody. J. Infect. Dis. 2008, 197, 846–853. [Google Scholar] [CrossRef]
- Zhu, Z.; Dimitrov, A.S.; Bossart, K.N.; Crameri, G.; Bishop, K.A.; Choudhry, V.; Mungall, B.A.; Feng, Y.-R.; Choudhary, A.; Zhang, M.-Y.; et al. Potent neutralization of Hendra and Nipah viruses by human monoclonal antibodies. J. Virol. 2006, 80, 891–899. [Google Scholar] [CrossRef] [Green Version]
- Bossart, K.N.; Crameri, G.; Dimitrov, A.S.; Mungall, B.A.; Feng, Y.-R.; Patch, J.R.; Choudhary, A.; Wang, L.-F.; Eaton, B.T.; Broder, C.C. Receptor Binding, Fusion Inhibition, and Induction of Cross-Reactive Neutralizing Antibodies by a Soluble G Glycoprotein of Hendra Virus. J. Virol. 2005, 79, 6690. [Google Scholar] [CrossRef] [Green Version]
- Bossart, K.N.; Rockx, B.; Feldmann, F.; Brining, D.; Scott, D.; LaCasse, R.; Geisbert, J.B.; Feng, Y.R.; Chan, Y.P.; Hickey, A.C.; et al. A Hendra virus G glycoprotein subunit vaccine protects African green monkeys from Nipah virus challenge. Sci. Transl. Med. 2012, 4, 146ra107. [Google Scholar] [CrossRef] [Green Version]
- Mire, C.E.; Geisbert, J.B.; Agans, K.N.; Feng, Y.-R.; Fenton, K.A.; Bossart, K.N.; Yan, L.; Chan, Y.-P.; Broder, C.C.; Geisbert, T.W. A Recombinant Hendra Virus G Glycoprotein Subunit Vaccine Protects Nonhuman Primates against Hendra Virus Challenge. J. Virol. 2014, 88, 4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallister, J.; Middleton, D.; Wang, L.-F.; Klein, R.; Haining, J.; Robinson, R.; Yamada, M.; White, J.; Payne, J.; Feng, Y.-R.; et al. A recombinant Hendra virus G glycoprotein-based subunit vaccine protects ferrets from lethal Hendra virus challenge. Vaccine 2011, 29, 5623–5630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallister, J.A.; Klein, R.; Arkinstall, R.; Haining, J.; Long, F.; White, J.R.; Payne, J.; Feng, Y.-R.; Wang, L.-F.; Broder, C.C.; et al. Vaccination of ferrets with a recombinant G glycoprotein subunit vaccine provides protection against Nipah virus disease for over 12 months. Virol. J. 2013, 10, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, D.; Pallister, J.; Klein, R.; Feng, Y.-R.; Haining, J.; Arkinstall, R.; Frazer, L.; Huang, J.-A.; Edwards, N.; Wareing, M.; et al. Hendra virus vaccine, a one health approach to protecting horse, human, and environmental health. Emerg. Infect. Dis. 2014, 20, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Battles, M.B.; McLellan, J.S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef]
- Satake, M.; Coligan, J.E.; Elango, N.; Norrby, E.; Venkatesan, S. Respiratory syncytial virus envelope glycoprotein (G) has a novel structure. Nucleic Acids Res. 1985, 13, 7795–7812. [Google Scholar] [CrossRef] [Green Version]
- Wertz, G.W.; Collins, P.L.; Huang, Y.; Gruber, C.; Levine, S.; Ball, L.A. Nucleotide sequence of the G protein gene of human respiratory syncytial virus reveals an unusual type of viral membrane protein. Proc. Natl. Acad. Sci. USA 1985, 82, 4075–4079. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.G.; Ritschel, T.; Pascual, G.; Brakenhoff, J.P.J.; Keogh, E.; Furmanova-Hollenstein, P.; Lanckacker, E.; Wadia, J.S.; Gilman, M.S.A.; Williamson, R.A.; et al. Structural basis for recognition of the central conserved region of RSV G by neutralizing human antibodies. PLoS Pathog. 2018, 14, e1006935. [Google Scholar] [CrossRef] [Green Version]
- Haynes, L.M.; Caidi, H.; Radu, G.U.; Miao, C.; Harcourt, J.L.; Tripp, R.A.; Anderson, L.J. Therapeutic Monoclonal Antibody Treatment Targeting Respiratory Syncytial Virus (RSV) G Protein Mediates Viral Clearance and Reduces the Pathogenesis of RSV Infection in BALB/c Mice. J. Infect. Dis. 2009, 200, 439–447. [Google Scholar] [CrossRef]
- Fedechkin, S.O.; George, N.L.; Wolff, J.T.; Kauvar, L.M.; DuBois, R.M. Structures of respiratory syncytial virus G antigen bound to broadly neutralizing antibodies. Sci. Immunol. 2018, 3, eaar3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortjens, B.; Yasuda, E.; Yu, X.; Wagner, K.; Claassen, Y.B.; Bakker, A.Q.; van Woensel, J.B.M.; Beaumont, T. Broadly Reactive Anti-Respiratory Syncytial Virus G Antibodies from Exposed Individuals Effectively Inhibit Infection of Primary Airway Epithelial Cells. J. Virol. 2017, 91, e02357-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirkova, T.; Lin, S.; Oomens, A.G.P.; Gaston, K.A.; Boyoglu-Barnum, S.; Meng, J.; Stobart, C.C.; Cotton, C.U.; Hartert, T.V.; Moore, M.L.; et al. CX3CR1 is an important surface molecule for respiratory syncytial virus infection in human airway epithelial cells. J. Gen. Virol. 2015, 96, 2543–2556. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.I.; Piepenhagen, P.A.; Kishko, M.; DiNapoli, J.M.; Groppo, R.P.; Zhang, L.; Almond, J.; Kleanthous, H.; Delagrave, S.; Parrington, M. CX3CR1 Is Expressed in Differentiated Human Ciliated Airway Cells and Co-Localizes with Respiratory Syncytial Virus on Cilia in a G Protein-Dependent Manner. PLoS ONE 2015, 10, e0130517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.M.; McNally, B.A.; Ioannidis, I.; Flano, E.; Teng, M.N.; Oomens, A.G.; Walsh, E.E.; Peeples, M.E. Respiratory Syncytial Virus Uses CX3CR1 as a Receptor on Primary Human Airway Epithelial Cultures. PLoS Pathog. 2015, 11, e1005318. [Google Scholar] [CrossRef] [Green Version]
- Tripp, R.A.; Jones, L.P.; Haynes, L.M.; Zheng, H.; Murphy, P.M.; Anderson, L.J. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat. Immunol. 2001, 2, 732–738. [Google Scholar] [CrossRef]
- Chan, Y.-P.; Lu, M.; Dutta, S.; Yan, L.; Barr, J.; Flora, M.; Feng, Y.-R.; Xu, K.; Nikolov, D.B.; Wang, L.-F.; et al. Biochemical, conformational, and immunogenic analysis of soluble trimeric forms of henipavirus fusion glycoproteins. J. Virol. 2012, 86, 11457–11471. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.C.; Popa, A.; Chang, A.; Masante, C.; Dutch, R.E. Viral entry mechanisms: The increasing diversity of paramyxovirus entry. FEBS J. 2009, 276, 7217–7227. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-F.; Harcourt, B.H.; Yu, M.; Tamin, A.; Rota, P.A.; Bellini, W.J.; Eaton, B.T. Molecular biology of Hendra and Nipah viruses. Microbes Infect. 2001, 3, 279–287. [Google Scholar] [CrossRef]
- Avanzato, V.A.; Oguntuyo, K.Y.; Escalera-Zamudio, M.; Gutierrez, B.; Golden, M.; Kosakovsky Pond, S.L.; Pryce, R.; Walter, T.S.; Seow, J.; Doores, K.J.; et al. A structural basis for antibody-mediated neutralization of Nipah virus reveals a site of vulnerability at the fusion glycoprotein apex. Proc. Natl. Acad. Sci. USA 2019, 116, 25057. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.V.; Chan, Y.-P.; Park, Y.-J.; Snijder, J.; Da Silva, S.C.; Vu, B.; Yan, L.; Feng, Y.-R.; Rockx, B.; Geisbert, T.W.; et al. An antibody against the F glycoprotein inhibits Nipah and Hendra virus infections. Nat. Struct. Mol. Biol. 2019, 26, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Pedrera, M.; Macchi, F.; McLean, R.K.; Franceschi, V.; Thakur, N.; Russo, L.; Medfai, L.; Todd, S.; Tchilian, E.Z.; Audonnet, J.-C.; et al. Bovine Herpesvirus-4-Vectored Delivery of Nipah Virus Glycoproteins Enhances T Cell Immunogenicity in Pigs. Vaccines 2020, 8, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingartl, H.M.; Berhane, Y.; Caswell, J.L.; Loosmore, S.; Audonnet, J.-C.; Roth, J.A.; Czub, M. Recombinant Nipah Virus Vaccines Protect Pigs against Challenge. J. Virol. 2006, 80, 7929–7938. [Google Scholar] [CrossRef] [Green Version]
- Battles, M.B.; Más, V.; Olmedillas, E.; Cano, O.; Vázquez, M.; Rodríguez, L.; Melero, J.A.; McLellan, J.S. Structure and immunogenicity of pre-fusion-stabilized human metapneumovirus F glycoprotein. Nat. Commun. 2017, 8, 1528. [Google Scholar] [CrossRef] [PubMed]
- Gilman, M.S.; Castellanos, C.A.; Chen, M.; Ngwuta, J.O.; Goodwin, E.; Moin, S.M.; Mas, V.; Melero, J.A.; Wright, P.F.; Graham, B.S.; et al. Rapid profiling of RSV antibody repertoires from the memory B cells of naturally infected adult donors. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magro, M.; Mas, V.; Chappell, K.; Vázquez, M.; Cano, O.; Luque, D.; Terrón, M.C.; Melero, J.A.; Palomo, C. Neutralizing antibodies against the preactive form of respiratory syncytial virus fusion protein offer unique possibilities for clinical intervention. Proc. Natl. Acad. Sci. USA 2012, 109, 3089–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngwuta, J.O.; Chen, M.; Modjarrad, K.; Joyce, M.G.; Kanekiyo, M.; Kumar, A.; Yassine, H.M.; Moin, S.M.; Killikelly, A.M.; Chuang, G.Y.; et al. Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl. Med. 2015, 7, 309ra162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, B.R.; Walsh, E.E. Formalin-inactivated respiratory syncytial virus vaccine induces antibodies to the fusion glycoprotein that are deficient in fusion-inhibiting activity. J. Clin. Microbiol. 1988, 26, 1595–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US National Library of Medicine. Dose, Safety, Tolerability and Immunogenicity of a Stabilized Prefusion RSV F Subunit Protein Vaccine, VRC-RSVRGP084-00-VP (DS-Cav1), Alone or With Alum Adjuvant, in Healthy Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT03049488 (accessed on 15 December 2020).
- US National Library of Medicine. Study of Safety, Reactogenicity and Immunogenicity of GlaxoSmithKline’s (GSK)Respiratory Syncytial Virus (RSV)Maternal Unadjuvanted Vaccine in Healthy Pregnant Women (Aged 18 to 40 Years) and Their Infants. Available online: https://clinicaltrials.gov/ct2/show/NCT04126213 (accessed on 15 December 2020).
- Samy, N.; Reichhardt, D.; Schmidt, D.; Chen, L.M.; Silbernagl, G.; Vidojkovic, S.; Meyer, T.P.; Jordan, E.; Adams, T.; Weidenthaler, H.; et al. Safety and immunogenicity of novel modified vaccinia Ankara-vectored RSV vaccine: A randomized phase I clinical trial. Vaccine 2020, 38, 2608–2619. [Google Scholar] [CrossRef] [PubMed]
- Karron, R.A.; Luongo, C.; Thumar, B.; Loehr, K.M.; Englund, J.A.; Collins, P.L.; Buchholz, U.J. A gene deletion that up-regulates viral gene expression yields an attenuated RSV vaccine with improved antibody responses in children. Sci. Transl. Med. 2015, 7, 312ra175. [Google Scholar] [CrossRef] [Green Version]
- US National Library of Medicine. Safety and Efficacy of BARS13 in the Elderly. Available online: https://clinicaltrials.gov/ct2/show/NCT04681833 (accessed on 12 October 2020).
- Isaacs, A.; Li, Z.; Cheung, S.T.M.; Wijesundara, D.K.; McMillan, C.L.D.; Modhiran, N.; Young, P.R.; Ranasinghe, C.; Watterson, D.; Chappell, K.J. Adjuvant Selection for Influenza and RSV Prefusion Subunit Vaccines. Vaccines 2021, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Watterson, D.; Wijesundara, D.; Modhiran, N.; Mordant, F.; Li, Z.; Avumegah, M.; McMillan, C.; Lackenby, J.; Guilfoyle, K.; van Amerongen, G.; et al. Molecular clamp stabilised Spike protein for protection against SARS-CoV-2. Res. Sq. Prepr. 2020, v1. [Google Scholar] [CrossRef]
- Wijesundara, D.K.; Avumegah, M.S.; Lackenby, J.; Modhiran, N.; Isaacs, A.; Young, P.R.; Watterson, D.; Chappell, K.J. Rapid Response Subunit Vaccine Design in the Absence of Structural Information. Front. Immunol. 2020, 11, 592370. [Google Scholar] [CrossRef] [PubMed]
- Frey, G.; Chen, J.; Rits-Volloch, S.; Freeman, M.M.; Zolla-Pazner, S.; Chen, B. Distinct conformational states of HIV-1 gp41 are recognized by neutralizing and non-neutralizing antibodies. Nat. Struct. Mol. Biol. 2010, 17, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Watterson, D.; Chang, C.-W.; Che, X.-Y.; Li, X.-Q.; Ericsson, D.J.; Qiu, L.-W.; Cai, J.-P.; Chen, J.; Fry, S.R.; et al. Structural and Functional Characterization of a Cross-Reactive Dengue Virus Neutralizing Antibody that Recognizes a Cryptic Epitope. Structure 2018, 26, 51–59.e54. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.L.; Seldon, T.; Smede, M.; Linville, A.; Chin, D.Y.; Barnard, R.; Mahler, S.M.; Munster, D.; Hart, D.; Gray, P.P.; et al. A method for rapid, ligation-independent reformatting of recombinant monoclonal antibodies. J. Immunol. Methods 2010, 354, 85–90. [Google Scholar] [CrossRef]
- Corti, D.; Bianchi, S.; Vanzetta, F.; Minola, A.; Perez, L.; Agatic, G.; Guarino, B.; Silacci, C.; Marcandalli, J.; Marsland, B.J.; et al. Cross-neutralization of four paramyxoviruses by a human monoclonal antibody. Nature 2013, 501, 439–443. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV Fusion Glycoprotein Trimer Bound to a Prefusion-Specific Neutralizing Antibody. Science 2013, 340, 1113. [Google Scholar] [CrossRef] [Green Version]
- McLellan, J.S.; Chen, M.; Chang, J.-S.; Yang, Y.; Kim, A.; Graham, B.S.; Kwong, P.D. Structure of a Major Antigenic Site on the Respiratory Syncytial Virus Fusion Glycoprotein in Complex with Neutralizing Antibody 101F. J. Virol. 2010, 84, 12236–12244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, J.S.; Chen, M.; Kim, A.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structural basis of respiratory syncytial virus neutralization by motavizumab. Nat. Struct. Mol. Biol. 2010, 17, 248–250. [Google Scholar] [CrossRef]
- Wu, H.; Pfarr, D.S.; Johnson, S.; Brewah, Y.A.; Woods, R.M.; Patel, N.K.; White, W.I.; Young, J.F.; Kiener, P.A. Development of Motavizumab, an Ultra-potent Antibody for the Prevention of Respiratory Syncytial Virus Infection in the Upper and Lower Respiratory Tract. J. Mol. Biol. 2007, 368, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Bingham, P.; Hierholzer, J.C. Neutralization of respiratory syncytial virus by individual and mixtures of F and G protein monoclonal antibodies. J. Virol. 1988, 62, 4232–4238. [Google Scholar] [CrossRef] [Green Version]
- Collarini, E.J.; Lee, F.E.; Foord, O.; Park, M.; Sperinde, G.; Wu, H.; Harriman, W.D.; Carroll, S.F.; Ellsworth, S.L.; Anderson, L.J.; et al. Potent high-affinity antibodies for treatment and prophylaxis of respiratory syncytial virus derived from B cells of infected patients. J. Immunol. 2009, 183, 6338–6345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, N.; Conceicao, C.; Isaacs, A.; Human, S.; Modhiran, N.; McLean, R.K.; Pedrera, M.; Tan, T.K.; Rijal, P.; Townsend, A.; et al. Micro-fusion inhibition tests: Quantifying antibody neutralization of virus-mediated cell–cell fusion. J. Gen. Virol. 2020, 102, jgv001506. [Google Scholar] [CrossRef]
- Chappell, K.J.; Watterson, D.; Young, P.R. Chimeric Molecules and Uses Thereof. 2018. Available online: https://patents.google.com/patent/WO2018176103A1/en (accessed on 12 October 2020).
- Leroux-Roels, G.; De Boever, F.; Maes, C.; Nguyen, T.L.-A.; Baker, S.; Gonzalez Lopez, A. Safety and immunogenicity of a respiratory syncytial virus fusion glycoprotein F subunit vaccine in healthy adults: Results of a phase 1, randomized, observer-blind, controlled, dosage-escalation study. Vaccine 2019, 37, 2694–2703. [Google Scholar] [CrossRef] [PubMed]
- Karron, R.A.; Buonagurio, D.A.; Georgiu, A.F.; Whitehead, S.S.; Adamus, J.E.; Clements-Mann, M.L.; Harris, D.O.; Randolph, V.B.; Udem, S.A.; Murphy, B.R.; et al. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: Clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. USA 1997, 94, 13961–13966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Techaarpornkul, S.; Barretto, N.; Peeples, M.E. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 2001, 75, 6825–6834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, H.C.; Murray, J.; Nuñez Castrejon, A.M.; DuBois, R.M.; Tripp, R.A. Respiratory Syncytial Virus (RSV) G Protein Vaccines With Central Conserved Domain Mutations Induce CX3C-CX3CR1 Blocking Antibodies. Viruses 2021, 13, 352. [Google Scholar] [CrossRef]
- Crank, M.C.; Ruckwardt, T.J.; Chen, M.; Morabito, K.M.; Phung, E.; Costner, P.J.; Holman, L.A.; Hickman, S.P.; Berkowitz, N.M.; Gordon, I.J.; et al. A proof of concept for structure-based vaccine design targeting RSV in humans. Science 2019, 365, 505. [Google Scholar] [CrossRef]
- Steff, A.-M.; Monroe, J.; Friedrich, K.; Chandramouli, S.; Nguyen, T.L.-A.; Tian, S.; Vandepaer, S.; Toussaint, J.-F.; Carfi, A. Pre-fusion RSV F strongly boosts pre-fusion specific neutralizing responses in cattle pre-exposed to bovine RSV. Nat. Commun. 2017, 8, 1085. [Google Scholar] [CrossRef] [Green Version]
- Caidi, H.; Harcourt, J.L.; Tripp, R.A.; Anderson, L.J.; Haynes, L.M. Combination Therapy Using Monoclonal Antibodies against Respiratory Syncytial Virus (RSV) G Glycoprotein Protects from RSV Disease in BALB/c Mice. PLoS ONE 2012, 7, e51485. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Mason, C.S.; Jones, L.P.; Crabtree, J.; Jorquera, P.A.; Tripp, R.A. Antibodies to the central conserved region of respiratory syncytial virus (RSV) G protein block RSV G protein CX3C-CX3CR1 binding and cross-neutralize RSV A and B strains. Viral Immunol. 2012, 25, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.N.; Power, U.F.; Robert, A.; Haeuw, J.F.; Helffer, K.; Perez, A.; Asin, M.A.; Corvaia, N.; Libon, C. The respiratory syncytial virus G protein conserved domain induces a persistent and protective antibody response in rodents. PLoS ONE 2012, 7, e34331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Choi, Y.; Haynes, L.M.; Harcourt, J.L.; Anderson, L.J.; Jones, L.P.; Tripp, R.A. Vaccination To Induce Antibodies Blocking the CX3C-CX3CR1 Interaction of Respiratory Syncytial Virus G Protein Reduces Pulmonary Inflammation and Virus Replication in Mice. J. Virol. 2010, 84, 1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]





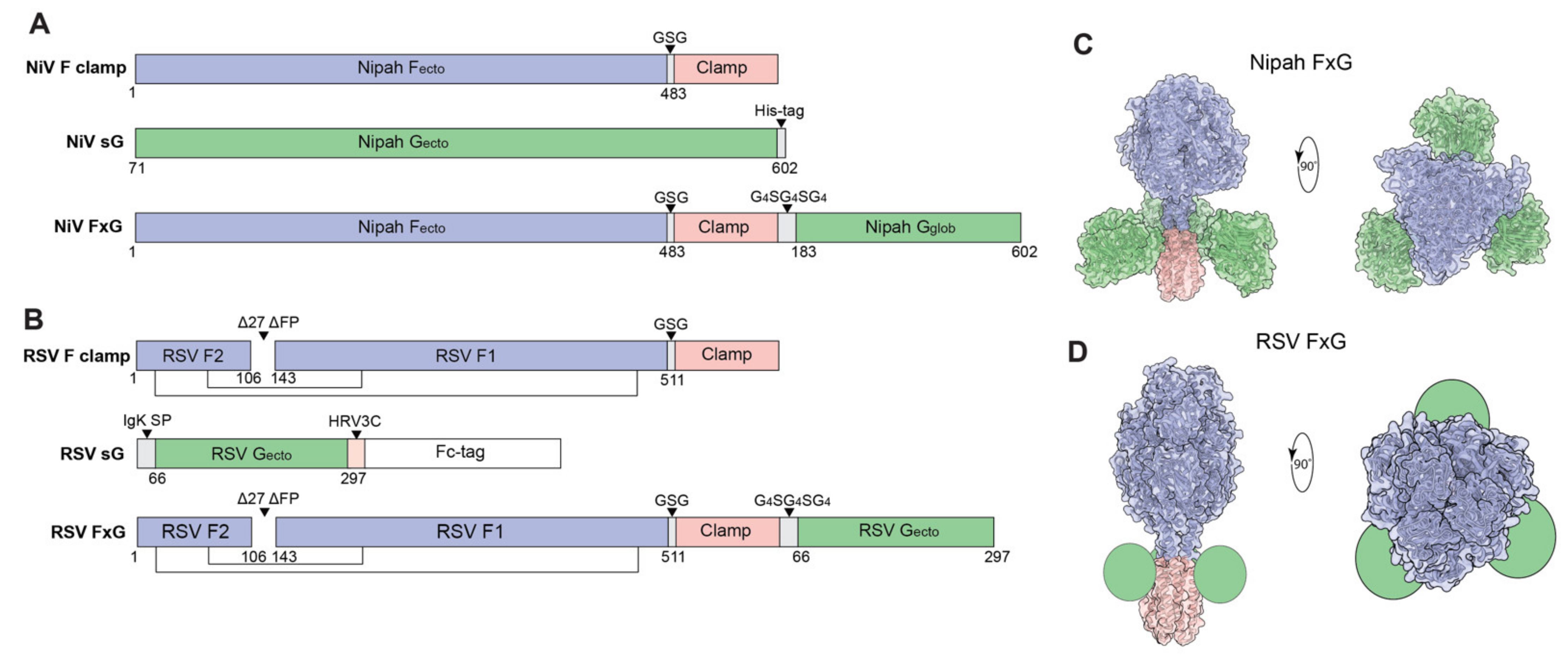{kind=link}
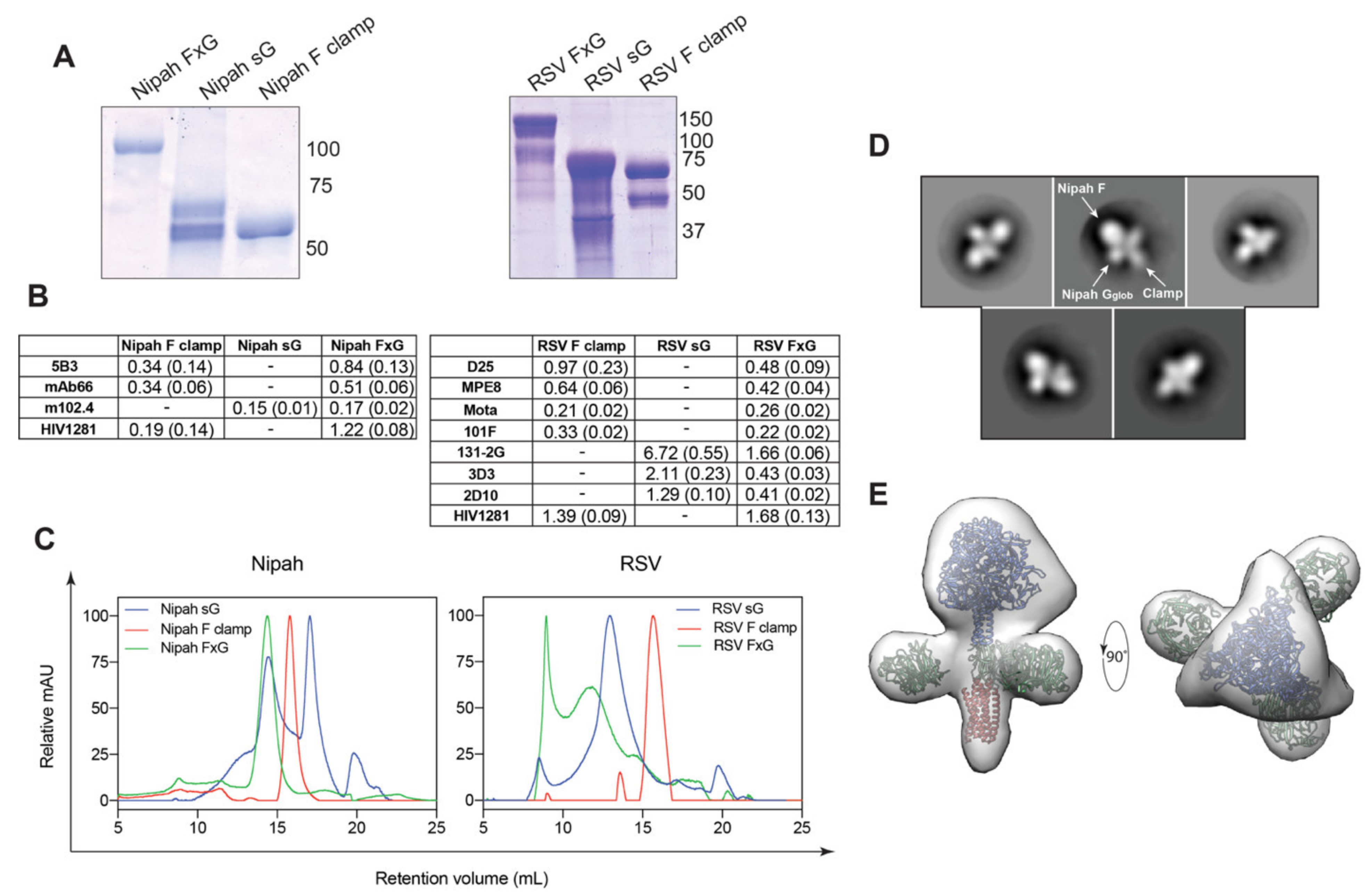{kind=link}
{kind=link}
{kind=link}
| Target | mAb | Specificity | Reference |
|---|---|---|---|
| NiV | 5B3 | F | [41] |
| mAb66 | F | [40] | |
| m102.4 | G | [19] | |
| RSV | MPE8 | F (Site III) | [60] |
| D25 | F (Site Ø) | [61] | |
| 101F | F (Site IV) | [62] | |
| Motavizumab | F (Site II) | [63,64] | |
| 131-2G | G (CCD) | [65] | |
| 3D3 | G (CCD) | [66] | |
| 2D10 | G (CCD) | [31] | |
| Clamp | HIV1281 | HIV gp41 postfusion core | [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isaacs, A.; Cheung, S.T.M.; Thakur, N.; Jaberolansar, N.; Young, A.; Modhiran, N.; Bailey, D.; Graham, S.P.; Young, P.R.; Chappell, K.J.; et al. Combinatorial F-G Immunogens as Nipah and Respiratory Syncytial Virus Vaccine Candidates. Viruses 2021, 13, 1942. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101942
Isaacs A, Cheung STM, Thakur N, Jaberolansar N, Young A, Modhiran N, Bailey D, Graham SP, Young PR, Chappell KJ, et al. Combinatorial F-G Immunogens as Nipah and Respiratory Syncytial Virus Vaccine Candidates. Viruses. 2021; 13(10):1942. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101942
Chicago/Turabian StyleIsaacs, Ariel, Stacey T. M. Cheung, Nazia Thakur, Noushin Jaberolansar, Andrew Young, Naphak Modhiran, Dalan Bailey, Simon P. Graham, Paul R. Young, Keith J. Chappell, and et al. 2021. "Combinatorial F-G Immunogens as Nipah and Respiratory Syncytial Virus Vaccine Candidates" Viruses 13, no. 10: 1942. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101942