
Effects of the Newly Isolated T4-like Phage on Transmission of Plasmid-Borne Antibiotic Resistance Genes via Generalized Transduction

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains and Plasmids
2.2. Acquisition of Phage
2.3. Identification of Phage
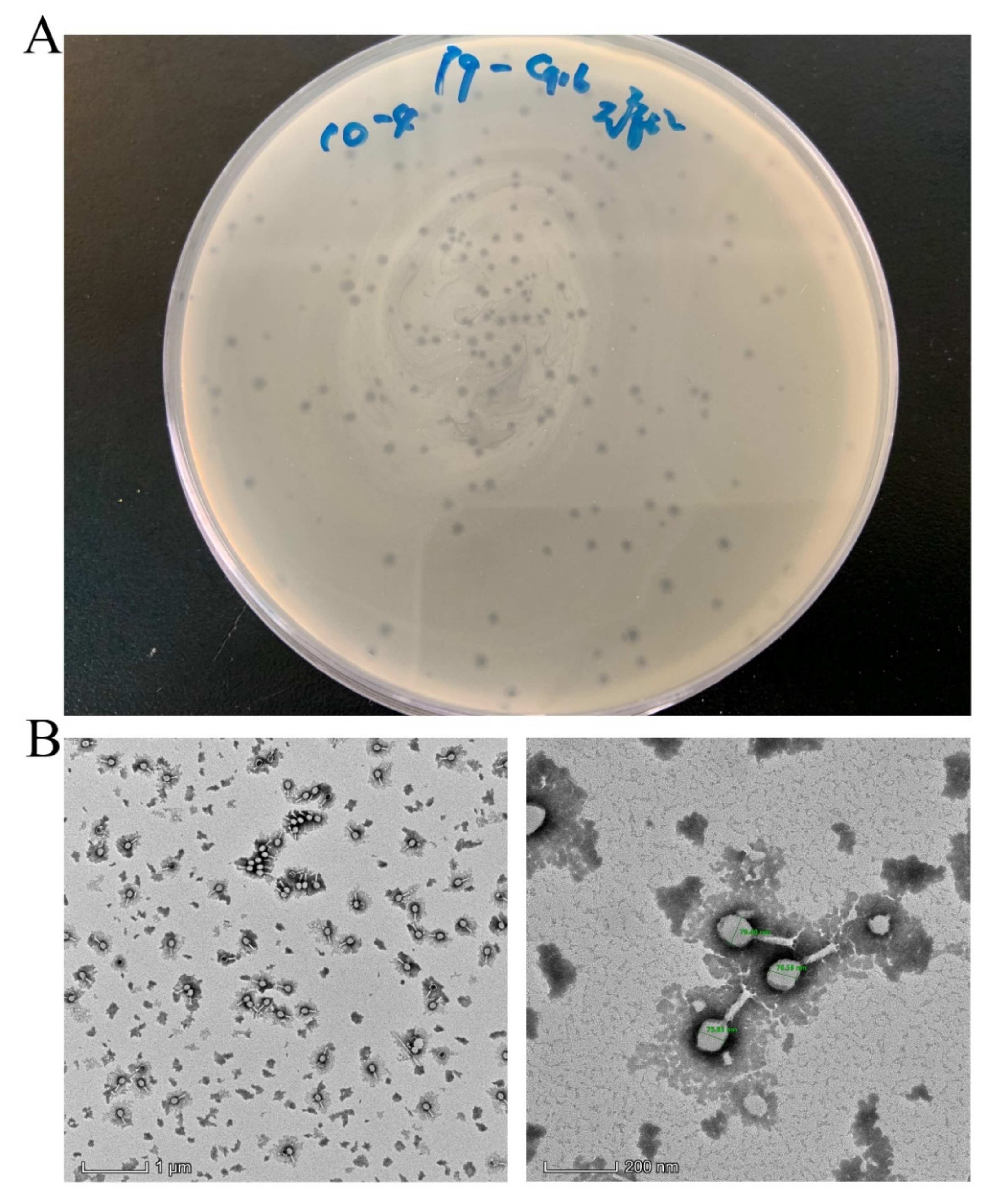
2.3.1. Transmission Electron Microscopy
2.3.2. Optimal Multiplicity of Infection
2.3.3. One Step Growth Curve
2.3.4. Physical Stability of the Phage
2.4. Bacterial and Bacteriophage DNA Extraction
2.5. Lytic Phage Genome Sequencing and Assembly
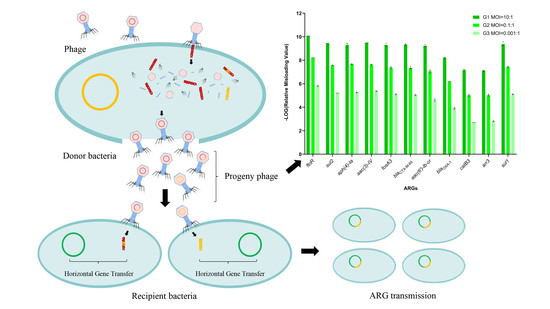
2.6. Effect of Lytic Phage Generalized Transduction on Plasmid-Borne ARGs
3. Results
3.1. Isolation and Characterization of Lytic Phage
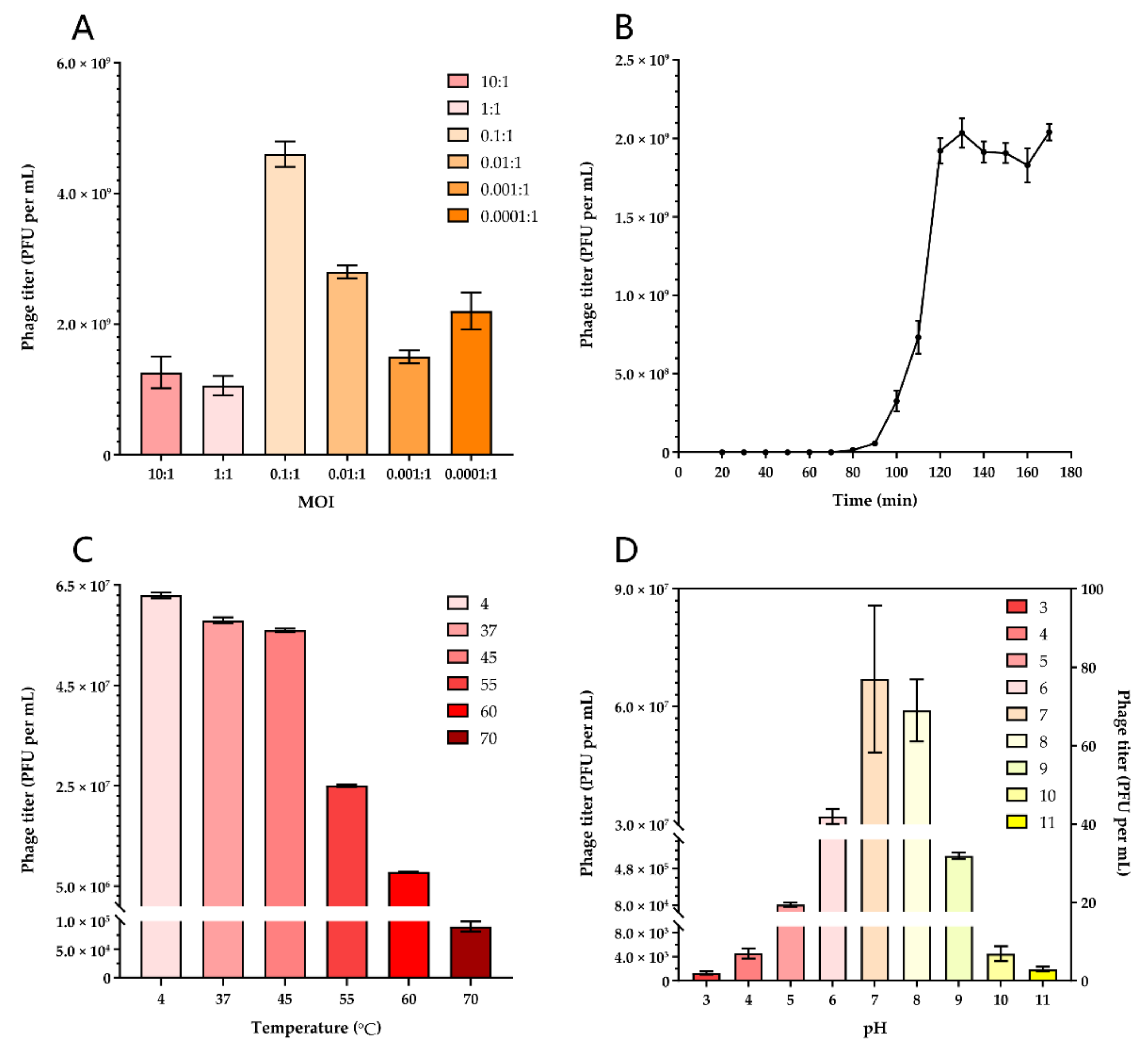
3.2. Optimal Multiplicity of Infection of Lytic Phage
3.3. One-Step Growth Curve
3.4. Phage Stability Tests
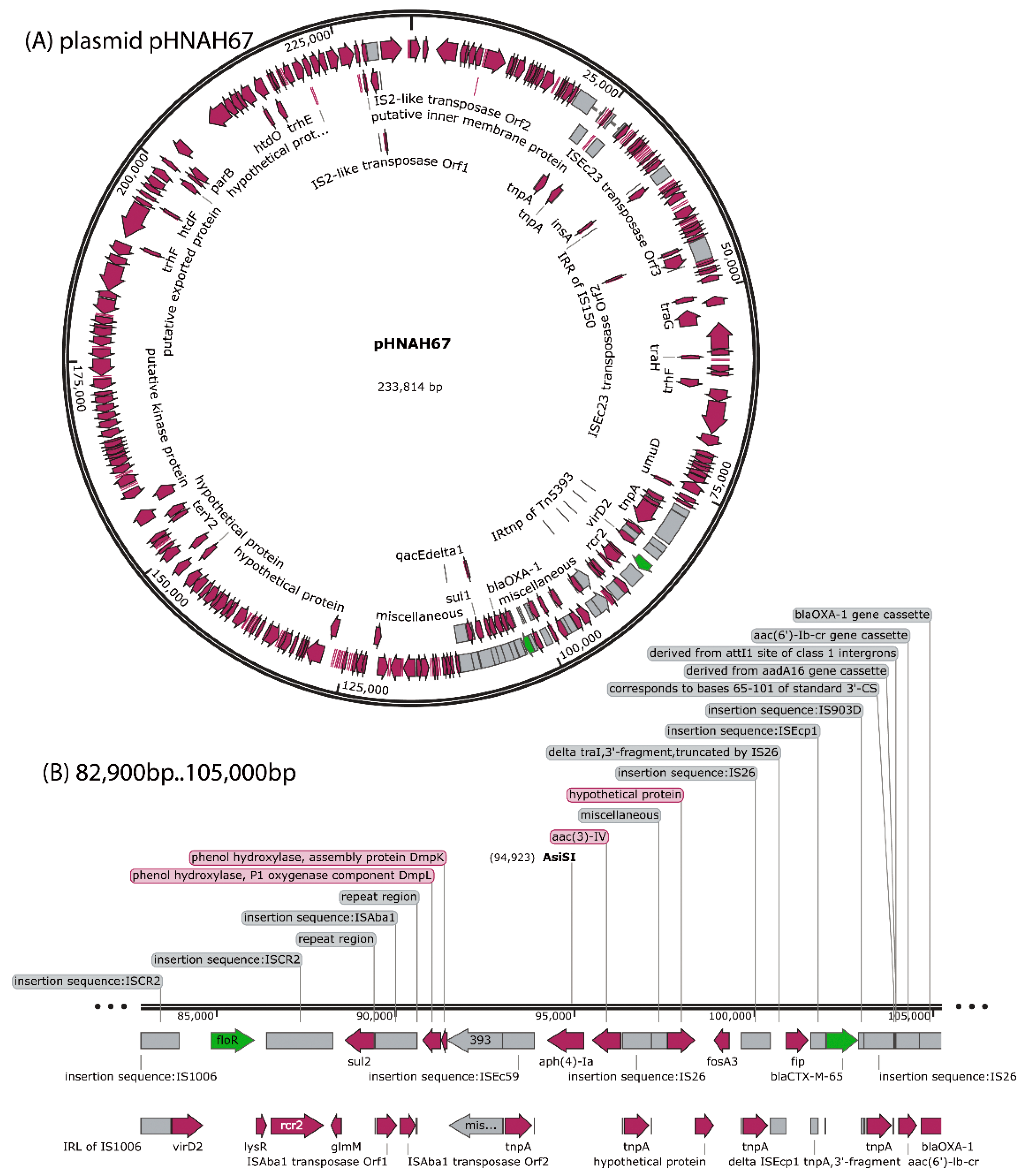
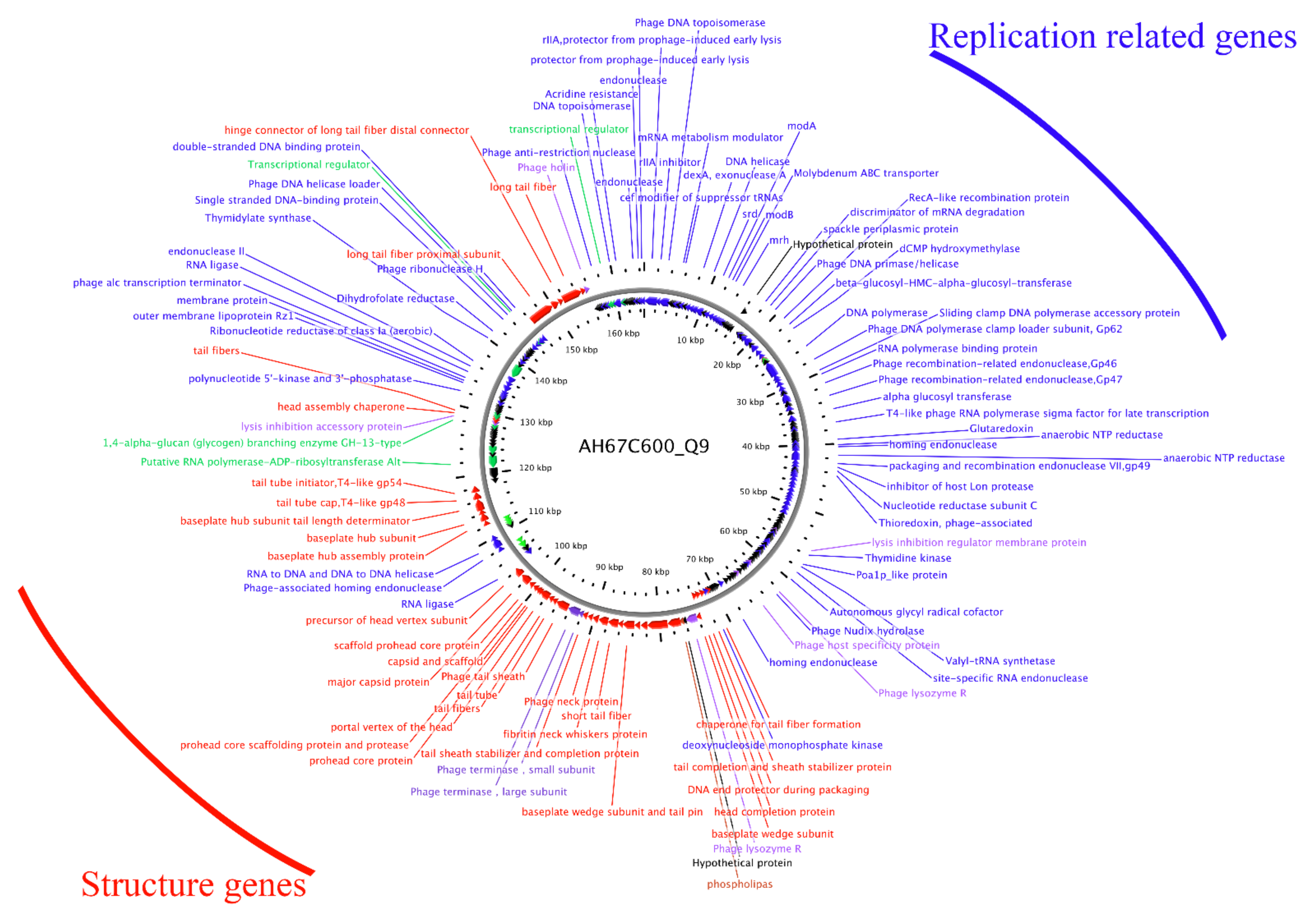
3.5. Sequencing and Bioinformatics Analysis of Phage AH67C600_Q9 Genome
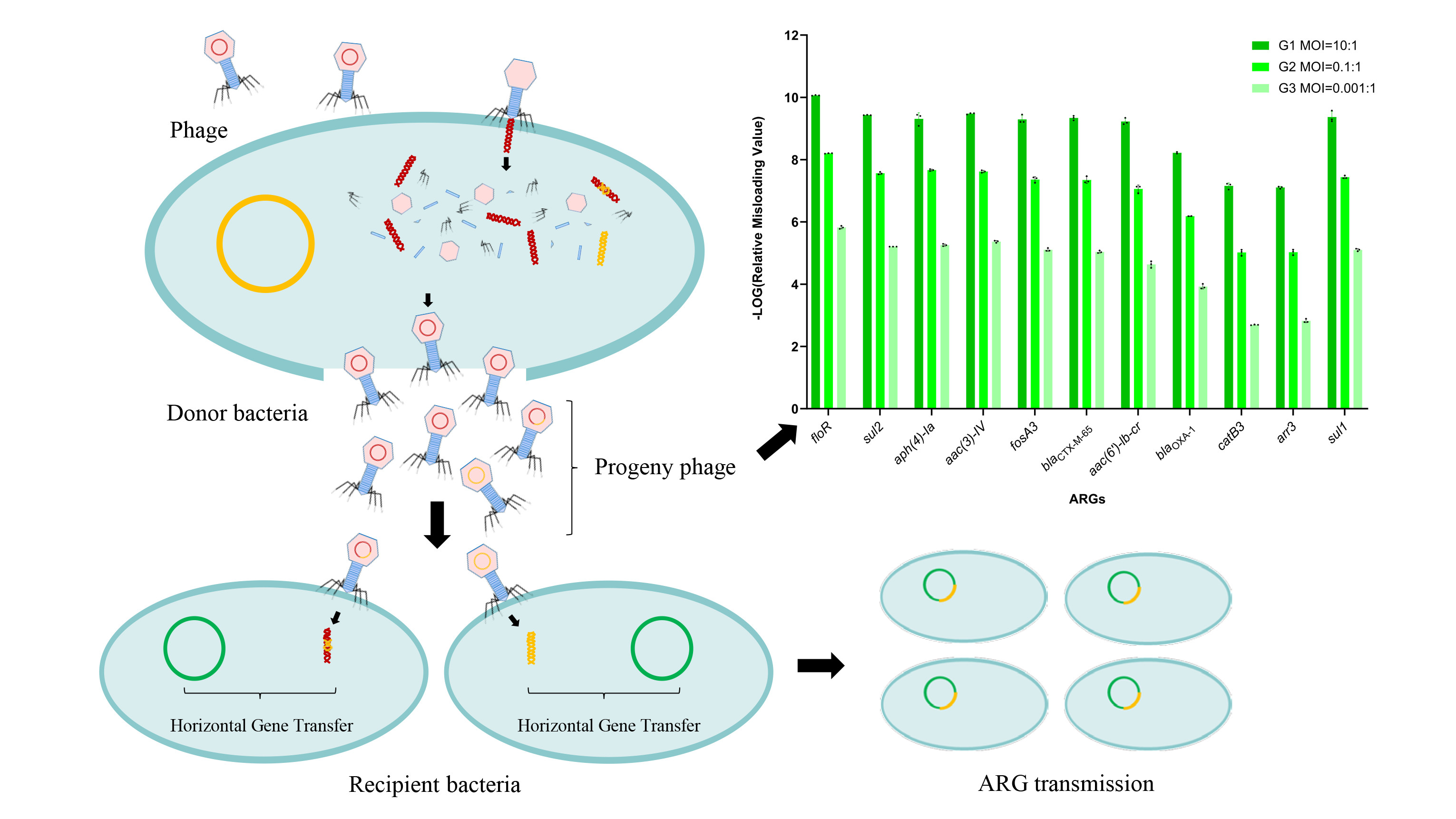
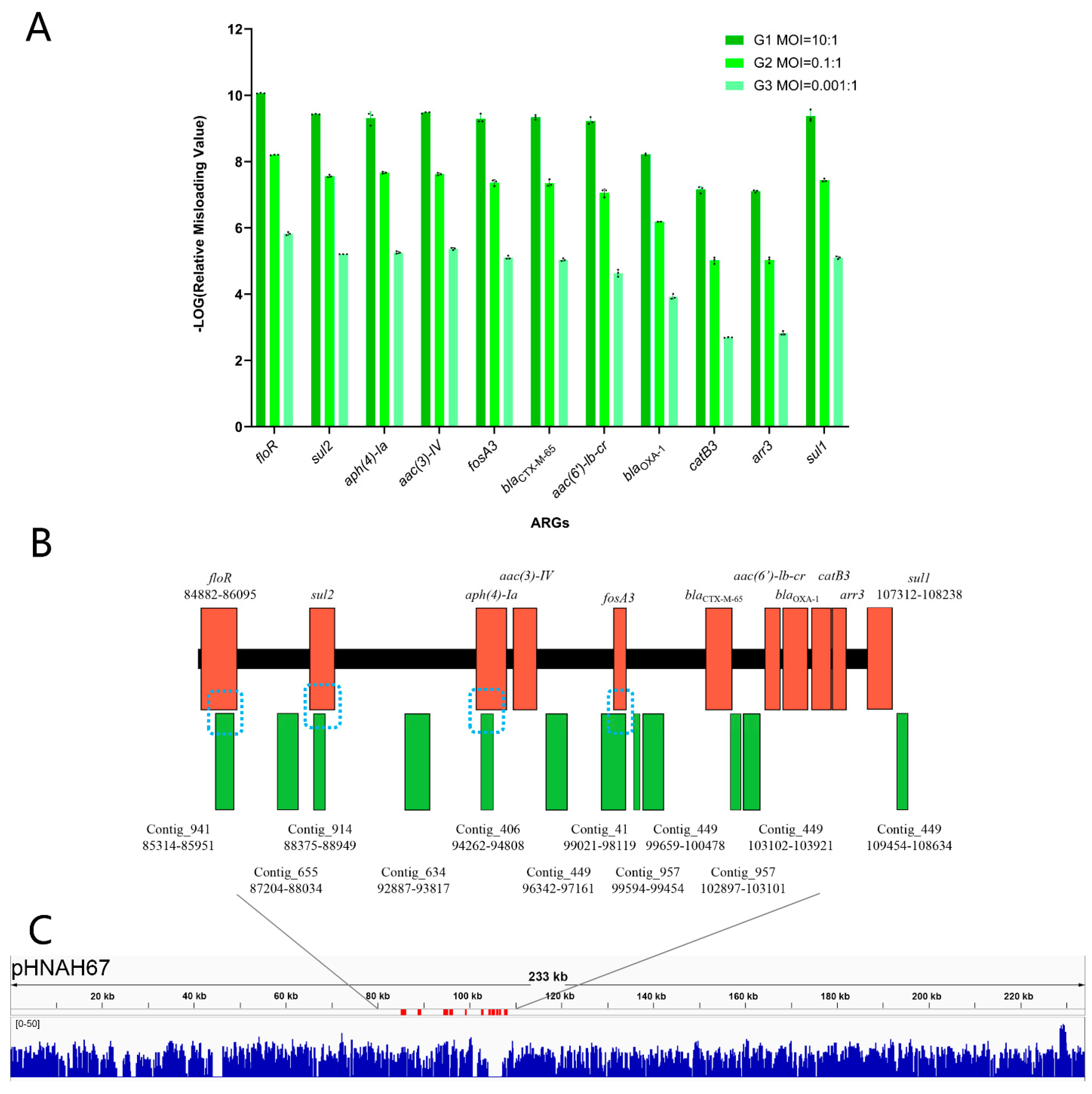
3.6. Lytic Phage Misloaded Plasmid-Borne ARGs by Generalized Transduction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in Aquatic Ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumby, N.; Edwards, A.M.; Davidson, A.R.; Maxwell, K.L. The Bacteriophage HK97 gp15 Moron Element Encodes a Novel Superinfection Exclusion Protein. J. Bacteriol. 2012, 194, 5012–5019. [Google Scholar] [CrossRef] [Green Version]
- Salmond, G.P.C.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Penadés, J.R.; Chen, J.; Quiles-Puchalt, N.; Carpena, N.; Novick, R. Bacteriophage-mediated spread of bacterial virulence genes. Curr. Opin. Microbiol. 2015, 23, 171–178. [Google Scholar] [CrossRef]
- Muniesa, M.; Imamovic, L.; Jofre, J. Bacteriophages and genetic mobilization in sewage and faecally polluted environments. Microb. Biotechnol. 2011, 4, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Calero-Caceres, W.; Ye, M.; Balcázar, J.L. Bacteriophages as Environmental Reservoirs of Antibiotic Resistance. Trends Microbiol. 2019, 27, 570–577. [Google Scholar] [CrossRef]
- Mavrich, T.; Hatfull, G.F. Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Arya, S.; Todman, H.; Baker, M.; Hooton, S.; Millard, A.; Kreft, J.-U.; Hobman, J.L.; Stekel, D.J. A generalised model for generalised transduction: The importance of co-evolution and stochasticity in phage mediated antimicrobial resistance transfer. FEMS Microbiol. Ecol. 2020, 96. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef]
- Whittle, G.; Shoemaker, N.B.; Salyers, A.A. The role of Bacteroides conjugative transposons in the dissemination of antibiotic resistance genes. Cell. Mol. Life Sci. 2002, 59, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.L.; Salyers, A.A. Regulation of CTnDOT Conjugative Transfer Is a Complex and Highly Coordinated Series of Events. mBio 2013, 4, e00569-13. [Google Scholar] [CrossRef] [Green Version]
- Monecke, S.; Coombs, G.; Shore, A.; Coleman, D.; Akpaka, P.E.; Borg, M.; Chow, H.; Ip, M.; Jatzwauk, L.; Jonas, D.; et al. A Field Guide to Pandemic, Epidemic and Sporadic Clones of Methicillin-Resistant Staphylococcus aureus. PLoS ONE 2011, 6, e17936. [Google Scholar] [CrossRef]
- Scharn, C.R.; Tenover, F.C.; Goering, R. Transduction of Staphylococcal Cassette ChromosomemecElements between Strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 5233–5238. [Google Scholar] [CrossRef] [Green Version]
- Goh, S.; Hussain, H.; Chang, B.J.; Emmett, W.; Riley, T.V.; Mullany, P. Phage varphiC2 Mediates Transduction of Tn 6215, Encoding Erythromycin Resistance, between Clostridium difficile Strains. mBio 2013, 4, e00840-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Novick, R.P. Phage-Mediated Intergeneric Transfer of Toxin Genes. Science 2009, 323, 139–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zeng, Z.; Jiang, F.; Zheng, Y.; Shen, H.; Macedo, N.; Sun, Y.; Sahin, O.; Li, G. Role of enterotoxigenic Escherichia coli prophage in spreading antibiotic resistance in a porcine-derived environment. Environ. Microbiol. 2020, 22, 4974–4984. [Google Scholar] [CrossRef]
- Wang, M.; Xiong, W.; Liu, P.; Xie, X.; Zeng, J.; Sun, Y.; Zeng, Z. Metagenomic Insights Into the Contribution of Phages to Antibiotic Resistance in Water Samples Related to Swine Feedlot Wastewater Treatment. Front. Microbiol. 2018, 9, 2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calero-Caceres, W.; Melgarejo, A.; Colomer-Lluch, M.; Stoll, C.; Lucena, F.; Jofre, J.; Muniesa, M. Sludge As a Potential Important Source of Antibiotic Resistance Genes in Both the Bacterial and Bacteriophage Fractions. Environ. Sci. Technol. 2014, 48, 7602–7611. [Google Scholar] [CrossRef] [PubMed]
- Lekunberri, I.; Villagrasa, M.; Balcázar, J.L.; Borrego, C. Contribution of bacteriophage and plasmid DNA to the mobilization of antibiotic resistance genes in a river receiving treated wastewater discharges. Sci. Total Environ. 2017, 601–602, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Sun, Y.; Zeng, Z.; Wang, Z. Metagenomics of wastewater phageome identifies an extensively cored antibiotic resistome in a swine feedlot water treatment environment. Ecotoxicol. Environ. Saf. 2021, 222, 112552. [Google Scholar] [CrossRef] [PubMed]
- Lekunberri, I.; Subirats, J.; Borrego, C.; Balcázar, J.L. Exploring the contribution of bacteriophages to antibiotic resistance. Environ. Pollut. 2017, 220, 981–984. [Google Scholar] [CrossRef]
- Blanco-Picazo, P.; Roscales, G.; Toribio-Avedillo, D.; Gómez-Gómez, C.; Avila, C.; Ballesté, E.; Muniesa, M.; Rodríguez-Rubio, L. Antibiotic Resistance Genes in Phage Particles from Antarctic and Mediterranean Seawater Ecosystems. Microorganisms 2020, 8, 1293. [Google Scholar] [CrossRef]
- Brown-Jaque, M.; Rodriguez Oyarzun, L.; Cornejo-Sánchez, T.; Martín-Gomez, M.T.; Gartner, S.; De Gracia, J.; Rovira, S.; Alvarez, A.; Jofre, J.; González-López, J.J.; et al. Detection of Bacteriophage Particles Containing Antibiotic Resistance Genes in the Sputum of Cystic Fibrosis Patients. Front. Microbiol. 2018, 9, 856. [Google Scholar] [CrossRef]
- Brown-Jaque, M.; Calero-Cáceres, W.; Espinal, P.; Rodríguez-Navarro, J.; Miró, E.; González-López, J.J.; Cornejo, T.; Hurtado, J.C.; Navarro, F.; Muniesa, M. Antibiotic resistance genes in phage particles isolated from human faeces and induced from clinical bacterial isolates. Int. J. Antimicrob. Agents 2018, 51, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Quiroz, R.C.; Dalia, T.N.; Camilli, A.; Dalia, A.B.; Silva-Valenzuela, C.A. Prophage-Dependent Neighbor Predation Fosters Horizontal Gene Transfer by Natural Transformation. mSphere 2020, 5. [Google Scholar] [CrossRef]
- Fillol-Salom, A.; Alsaadi, A.; De Sousa, J.A.M.; Zhong, L.; Foster, K.R.; Rocha, E.P.C.; Penadés, J.R.; Ingmer, H.; Haaber, J. Bacteriophages benefit from generalized transduction. PLoS Pathog. 2019, 15, e1007888. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.; Salabarria, A.-C.; Edwards, R.A.; Roach, D.R. Standardized bacteriophage purification for personalized phage therapy. Nat. Protoc. 2020, 15, 2867–2890. [Google Scholar] [CrossRef]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Topp, E. Abundance of Antibiotic Resistance Genes in Bacteriophage following Soil Fertilization with Dairy Manure or Municipal Biosolids, and Evidence for Potential Transduction. Appl. Environ. Microbiol. 2015, 81, 7905–7913. [Google Scholar] [CrossRef] [Green Version]
- Mocé-Llivina, L.; Lucena, F.; Jofre, J. Double-Layer Plaque Assay for Quantification of Enteroviruses. Appl. Environ. Microbiol. 2004, 70, 2801–2805. [Google Scholar] [CrossRef] [Green Version]
- Stalin, N.; Srinivasan, P. Efficacy of potential phage cocktails against Vibrio harveyi and closely related Vibrio species isolated from shrimp aquaculture environment in the south east coast of India. Vet. Microbiol. 2017, 207, 83–96. [Google Scholar] [CrossRef]
- Amarillas, L.; Rubí-Rangel, L.; Chaidez, C.; González-Robles, A.; Lightbourn-Rojas, L.; León-Félix, J. Isolation and Characterization of phiLLS, a Novel Phage with Potential Biocontrol Agent against Multidrug-Resistant Escherichia coli. Front. Microbiol. 2017, 8, 1355. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, P.; Feng, F.; Zhang, J.; Li, F.; Wang, M.; Sun, Y. Evaluation of Potential ARG Packaging by Two Environmental T7-Like Phage during Phage-Host Interaction. Viruses 2020, 12, 1060. [Google Scholar] [CrossRef]
- Clokie, M.R.J.; Kropinski, A.M. (Eds.) Bacteriophages; Methods in Molecular Biology Series; Humana Press: Totowa, NJ, USA, 1983. [Google Scholar]
- Gu, J.; Xu, W.; Lei, L.; Huang, J.; Feng, X.; Sun, C.; Du, C.; Zuo, J.; Li, Y.; Du, T.; et al. LysGH15, a Novel Bacteriophage Lysin, Protects a Murine Bacteremia Model Efficiently against Lethal Methicillin-Resistant Staphylococcus aureus Infection. J. Clin. Microbiol. 2010, 49, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhang, S.; Wang, J.; Li, C.; Li, N.; Yu, S.; Kong, L.; Zeng, H.; Yang, G.; Huang, Y.; et al. Isolation and characterization of a novel Escherichia coli Kayfunavirus phage DY1. Virus Res. 2021, 293, 198274. [Google Scholar] [CrossRef]
- Liang, J.; Qin, S.; Duan, R.; Zhang, H.; Wu, W.; Li, X.; Tang, D.; Fu, G.; Lu, X.; Lv, D.; et al. A Lytic Yersina pestis Bacteriophage Obtained From the Bone Marrow of Marmota himalayana in a Plague-Focus Area in China. Front. Cell. Infect. Microbiol. 2021, 11, 700322. [Google Scholar] [CrossRef]
- Chen, X.; Guo, J.; Liu, Y.; Chai, S.; Ma, R.; Munguntsetseg, B. Characterization and adsorption of a Lactobacillus plantarum virulent phage. J. Dairy Sci. 2019, 102, 3879–3886. [Google Scholar] [CrossRef]
- Feng, J.; Gao, L.; Li, L.; Zhang, Z.; Wu, C.; Li, F.; Tong, Y. Characterization and genome analysis of novel Klebsiella phage BUCT556A with lytic activity against carbapenemase-producing Klebsiella pneumoniae. Virus Res. 2021, 303, 198506. [Google Scholar] [CrossRef]
- Wang, M.; Liu, P.; Zhou, Q.; Tao, W.; Sun, Y.; Zeng, Z. Estimating the contribution of bacteriophage to the dissemination of antibiotic resistance genes in pig feces. Environ. Pollut. 2018, 238, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.; Gerdes, S.; Olsen, G.J.; Olson, R.J.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borodovsky, M.; Lomsadze, A. Gene Identification in Prokaryotic Genomes, Phages, Metagenomes, and EST Sequences with GeneMarkS Suite. Curr. Protoc. Microbiol. 2014, 32, 1E.7.1–1E.7.17. [Google Scholar] [CrossRef]
- Black, L.W. Old, new, and widely true: The bacteriophage T4 DNA packaging mechanism. Virology 2015, 479–480, 650–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, L.W.; Rao, V.B. Structure, Assembly, and DNA Packaging of the Bacteriophage T4 Head. Adv. Virus Res. 2012, 82, 119–153. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.; Alonso, J.C.; Tavares, P. A Defined in Vitro System for DNA Packaging by the Bacteriophage SPP1: Insights into the Headful Packaging Mechanism. J. Mol. Biol. 2005, 353, 529–539. [Google Scholar] [CrossRef]
- Yang, M.; Liang, Y.; Huang, S.; Zhang, J.; Wang, J.; Chen, H.; Ye, Y.; Gao, X.; Wu, Q.; Tan, Z. Isolation and Characterization of the Novel Phages vB_VpS_BA3 and vB_VpS_CA8 for Lysing Vibrio parahaemolyticus. Front. Microbiol. 2020, 11, 259. [Google Scholar] [CrossRef]
- Lu, L.; Cai, L.; Jiao, N.; Zhang, R. Isolation and characterization of the first phage infecting ecologically important marine bacteria Erythrobacter. Virol. J. 2017, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonak, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L.; et al. The real-time polymerase chain reaction. Mol. Asp. Med. 2006, 27, 95–125. [Google Scholar] [CrossRef] [PubMed]
- Botes, M.; De Kwaadsteniet, M.; Cloete, T. Application of quantitative PCR for the detection of microorganisms in water. Anal. Bioanal. Chem. 2013, 405, 91–108. [Google Scholar] [CrossRef]
- Kleiner, M.; Bushnell, B.; Sanderson, K.E.; Hooper, L.V.; Duerkop, B.A. Transductomics: Sequencing-based detection and analysis of transduced DNA in pure cultures and microbial communities. Microbiome 2020, 8, 158. [Google Scholar] [CrossRef]
- Nóbrega, F.L.; Vlot, M.; de Jonge, P.A.; Dreesens, L.L.; Beaumont, H.J.E.; Lavigne, R.; Dutilh, B.E.; Brouns, S.J.J. Targeting mechanisms of tailed bacteriophages. Nat. Rev. Microbiol. 2018, 16, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Brister, J.R. How to Name and Classify Your Phage: An Informal Guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Liu, C.; Zhang, Z.; Li, H.; Song, T.; Liang, T.; Li, B.; Li, L.; Feng, S.; Su, Q.; et al. Research and Technological Advances Regarding the Study of the Spread of Antimicrobial Resistance Genes and Antimicrobial-Resistant Bacteria Related to Animal Husbandry. Int. J. Environ. Res. Public Health 2019, 16, 4896. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.; Rodríguez, A.; García, P. Phage or foe: An insight into the impact of viral predation on microbial communities. ISME J. 2018, 12, 1171–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Barceló, C. The disparate effects of bacteriophages on antibiotic-resistant bacteria. Emerg. Microbes Infect. 2018, 7, 168. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.; Tavares, P.; Alonso, J.C. Headful DNA packaging: Bacteriophage SPP1 as a model system. Virus Res. 2013, 173, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, P.; Reichard, P. Ribonucleotide Reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef]
- Krieger, I.V.; Kuznetsov, V.; Chang, J.-Y.; Zhang, J.; Moussa, S.H.; Young, R.F.; Sacchettini, J.C. The Structural Basis of T4 Phage Lysis Control: DNA as the Signal for Lysis Inhibition. J. Mol. Biol. 2020, 432, 4623–4636. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.; São-José, C. Probing the function of the two holin-like proteins of bacteriophage SPP1. Virology 2017, 500, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, J.M.; Brzozowski, R.S.; Faith, D.; Round, J.L.; Secor, P.R.; Duerkop, B.A. Bacteriophage-Bacteria Interactions in the Gut: From Invertebrates to Mammals. Annu. Rev. Virol. 2021, 8, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.S.; Koefoed, A.K.; Jakobsen, R.R.; Deng, L.; Castro-Mejía, J.L.; Brunse, A.; Neve, H.; Vogensen, F.K.; Nielsen, D.S. Bacteriophage-mediated manipulation of the gut microbiome—Promises and presents limitations. FEMS Microbiol. Rev. 2020, 44, 507–521. [Google Scholar] [CrossRef]
- Varga, M.; Kuntová, L.; Pantucek, R.; Maslanova, I.; Růžičková, V.; Doškař, J. Efficient transfer of antibiotic resistance plasmids by transduction within methicillin-resistant Staphylococcus aureus USA300 clone. FEMS Microbiol. Lett. 2012, 332, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Yap, M.L.; Rossmann, M.G. Structure and function of bacteriophage T4. Future Microbiol. 2014, 9, 1319–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; He, X.; Shen, S.; Shi, M.; Zhou, Q.; Liu, J.; Wang, M.; Sun, Y. Effects of the Newly Isolated T4-like Phage on Transmission of Plasmid-Borne Antibiotic Resistance Genes via Generalized Transduction. Viruses 2021, 13, 2070. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102070
Zhang J, He X, Shen S, Shi M, Zhou Q, Liu J, Wang M, Sun Y. Effects of the Newly Isolated T4-like Phage on Transmission of Plasmid-Borne Antibiotic Resistance Genes via Generalized Transduction. Viruses. 2021; 13(10):2070. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102070
Chicago/Turabian StyleZhang, Junxuan, Xiaolu He, Shuqing Shen, Mengya Shi, Qin Zhou, Junlin Liu, Mianzhi Wang, and Yongxue Sun. 2021. "Effects of the Newly Isolated T4-like Phage on Transmission of Plasmid-Borne Antibiotic Resistance Genes via Generalized Transduction" Viruses 13, no. 10: 2070. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102070