
Molecular Characterization and Taxonomic Assignment of Three Phage Isolates from a Collection Infecting Pseudomonas syringae pv. actinidiae and P. syringae pv. phaseolicola from Northern Italy

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation
2.2. Sample Preparation, Phage Isolation, Amplification and Purification
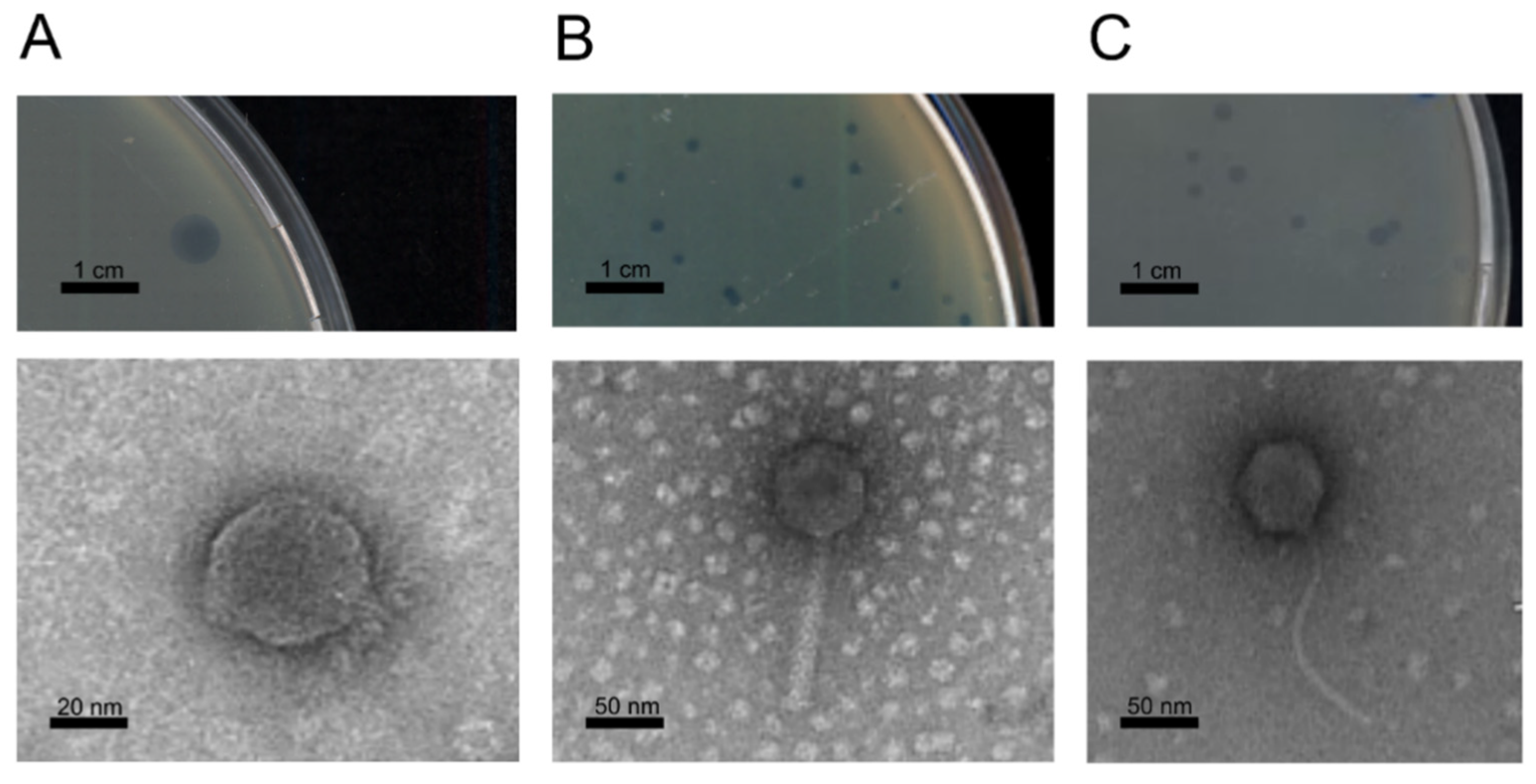
2.3. Transmission Electron Microscopy
2.4. Host Range Assay
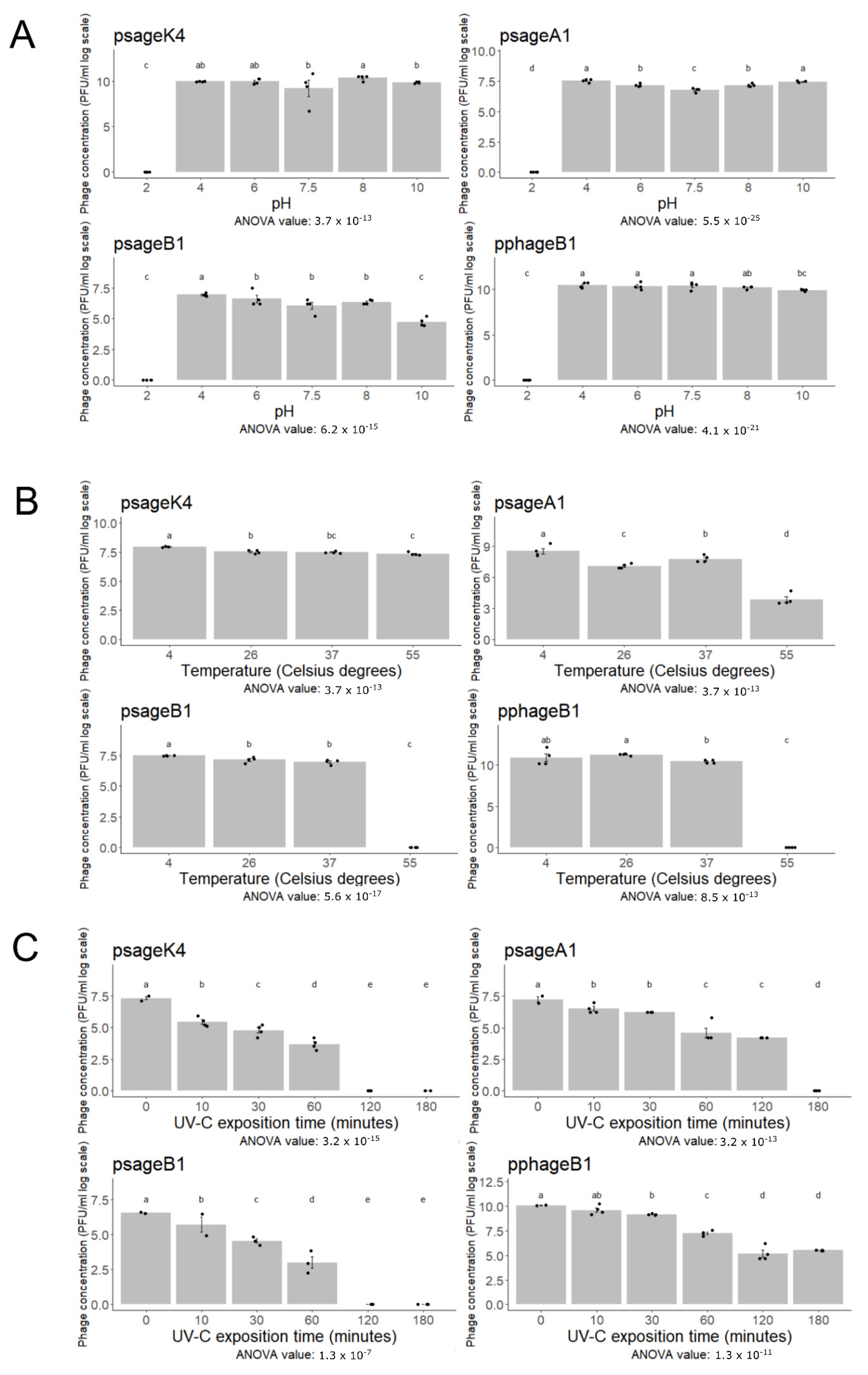
2.5. Influence of pH, Temperature, and UV Radiation to Phage Viability
2.6. Phage Genome Sequencing, Assembly, and Annotation
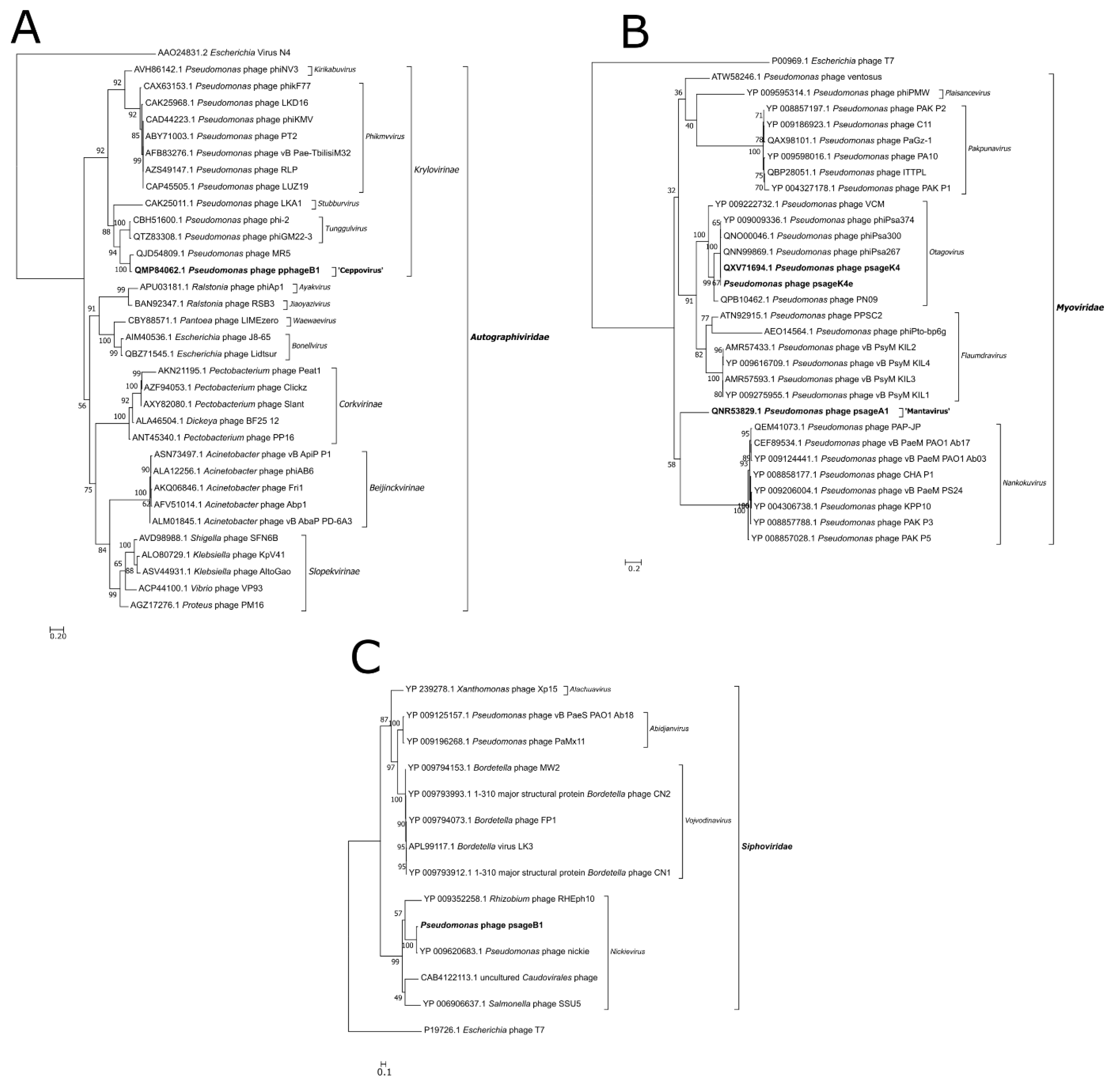
2.7. Phylogenetic Analysis
3. Results
3.1. Bacterial Isolation
3.2. Phage Isolation and Morphology
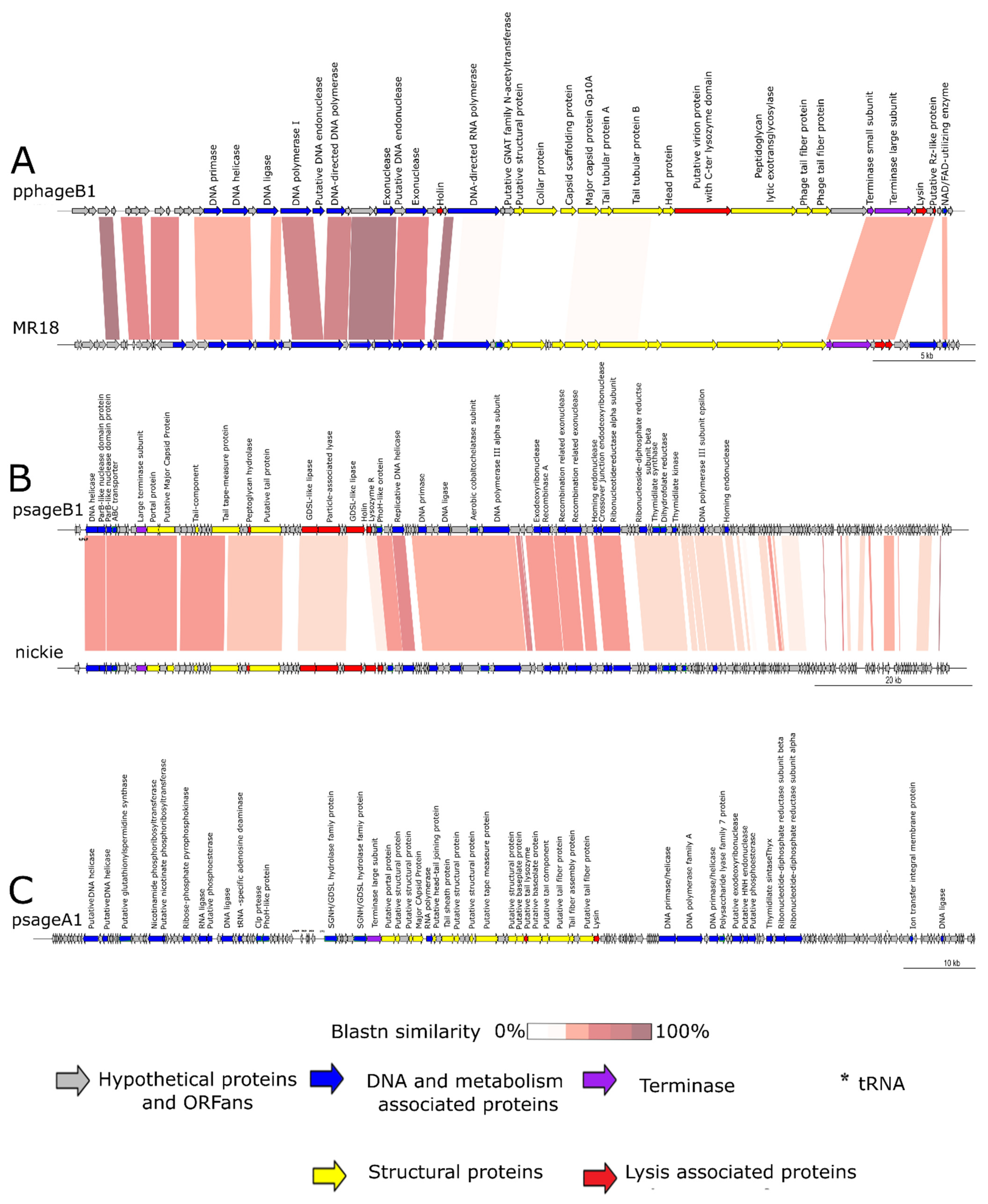
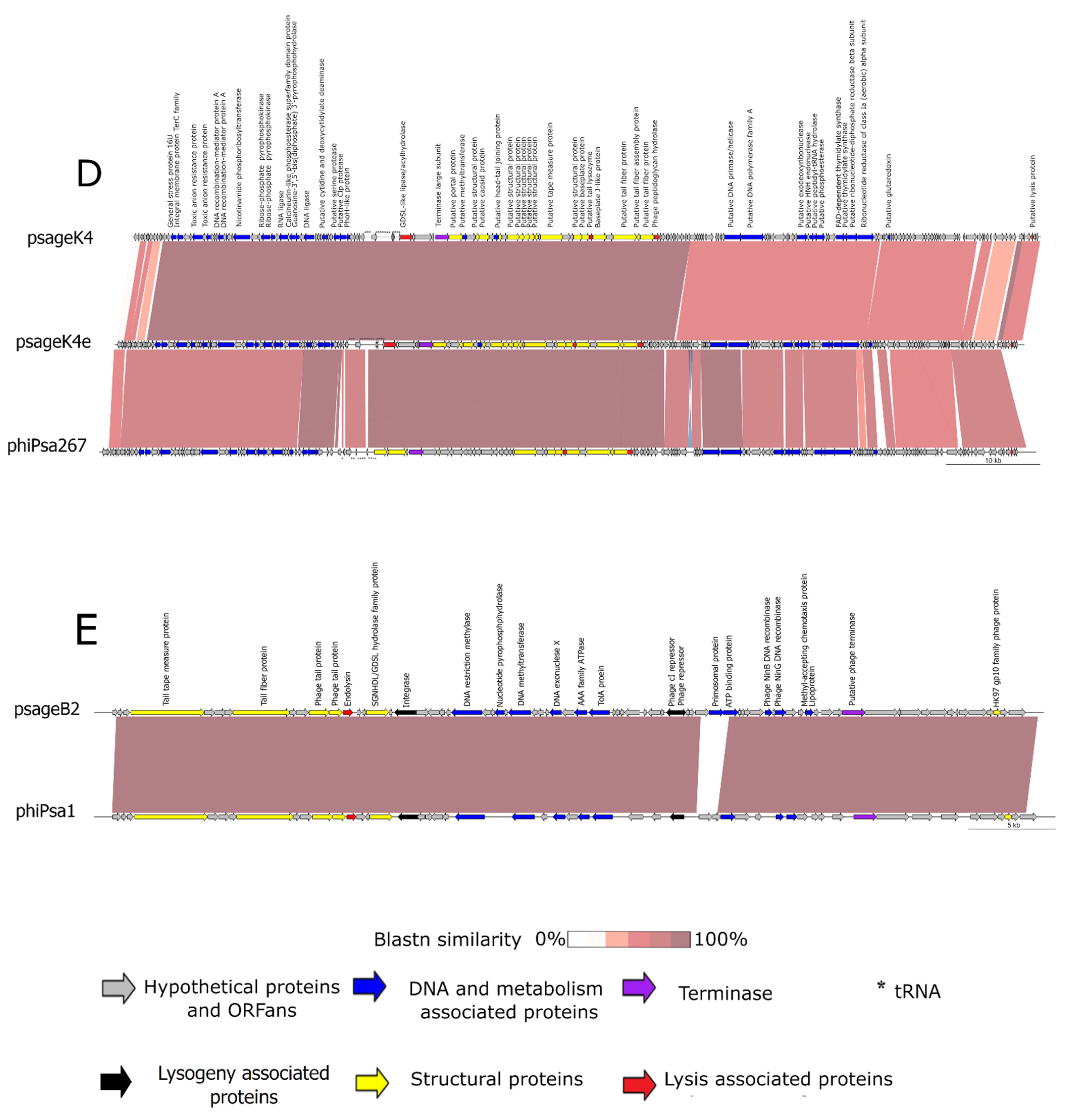
3.3. Phage Sequencing and Annotation
3.3.1. Genomic Characteristics of pphageB1
3.3.2. Genomic Characteristics of psageB1
3.3.3. Genomic Characteristics of psageA1
3.3.4. Genomic Characteristics of psageB2 and psageK9
3.3.5. Genomic Characteristics of psageK4
3.4. Phage Host Ranges
3.5. Phage Resistance to Abiotic Stresses
3.6. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mansfield, J.; Genin, S.; Magori, S.; Citovsky, V.; Sriariyanum, M.; Ronald, P.; Dow, M.; Verdier, V.; Beer, S.V.; Machado, M.A.; et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, I.; Cellini, A.; Sangiorgio, D.; Vanneste, J.L.; Scortichini, M.; Balestra, G.M.; Spinelli, F. Pseudomonas syringae pv. actinidiae: Ecology, infection dynamics and disease epidemiology. Microb. Ecol. 2020, 80, 81–102. [Google Scholar] [CrossRef] [PubMed]
- Froud, K.J.; Everett, K.R.; Tyson, J.L.; Beresford, R.M.; Cogger, N. Review of the risk factors associated with kiwifruit bacterial canker caused by Pseudomonas syringae pv actinidiae. N. Z. Plant Prot. 2015, 68, 313–327. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.D.; Teverson, D.M.; Davis, J.H.C. Sources of resistance to Pseudomonas syringae pv phaseolicola races in Phaseolus vulgaris. Plant Pathol. 1996, 45, 479–485. [Google Scholar] [CrossRef]
- Arnold, D.L.; Lovell, H.C.; Jackson, R.W.; Mansfield, J.W. Pseudomonas syringae pv. phaseolicola: From ‘has bean’ to supermodel. Mol. Plant Pathol. 2011, 12, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sanz, A.M.; Rodicio, M.R.; González, A.J. Pseudomonas syringae pv. phaseolicola isolated from weeds in bean crop fields. Lett. Appl. Microbiol. 2016, 62, 344–348. [Google Scholar] [CrossRef] [Green Version]
- Terán, H.; Lema, M.; Webster, D.; Singh, S.P. 75 years of breeding pinto bean for resistance to diseases in the United States. Euphytica 2009, 167, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Colombi, E.; Straub, C.; Künzel, S.; Templeton, M.; McCann, H.; Rainey, P. Evolution of copper resistance in the kiwifruit pathogen Pseudomonas syringae pv. actinidiae through acquisition of integrative conjugative elements and plasmids. Environ. Microbiol. 2017, 19, 070391. [Google Scholar] [CrossRef] [PubMed]
- Svircev, A.; Roach, D.; Castle, A. Framing the Future with Bacteriophages in Agriculture. Viruses 2018, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Holtappels, D.; Fortuna, K.; Lavigne, R.; Wagemans, J. The future of phage biocontrol in integrated plant protection for sustainable crop production. Curr. Opin. Biotechnol. 2020, 68, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, S.; Volckaert, A.; Venneman, S.; Devlercq, B.; Vandenheuvel, D.; Allonsius, C.N.; Van Malderghem, C.; Jang, H.B.; Briers, Y.; Noben, J.P.; et al. Characterization of noval bacteriophages for biocontrol of bacterial blight in leek caused by Pseudomonas syringae pv. porri. Front. Microbiol. 2016, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- James, S.; Rabiey, M.; Neuman, B.; Percival, G.; Jackson, R. Isolation, Characterisation and experimental evolution of phage that infect the horse chestnut tree pathogen, Pseudomonas syringae pv. aesculi. Curr. Microbiol. 2020, 77, 1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabiey, M.; Roy, S.R.; Holtappels, D.; Franceschetti, L.; Quilty, B.J.; Creeth, R.; Sundin, G.W.; Wagemans, J.; Lavigne, R.; Jackson, R.W. Phage biocontrol to combat Pseudomonas syringae pathogens causing disease in cherry. Microb. Biotechnol. 2020, 13, 1428–1445. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.; Pereira, C.; Frazão, C.; Balcão, V.; Almeida, A. Efficiency of phage φ6 for biocontrol of Pseudomonas syringae pv. syringae: An in vitro preliminary study. Microorganisms 2019, 7, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lallo, G.; Evangelisti, M.; Mancuso, F.; Ferrante, P.; Marcelletti, S.; Tinari, A.; Superti, F.; Migliore, L.; D’Addabbo, P.; Frezza, D.; et al. Isolation and partial characterization of bacteriophages infecting Pseudomonas syringae pv. actinidiae, causal agent of kiwifruit bacterial canker. J. Basic Microbiol. 2014, 54, 1210–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, R.; Taylor, C.; Holguín, M.A.; Visnovsky, S.; Petty, N.; Pitman, A.; Fineran, P. Identification of bacteriophages for biocontrol of the kiwifruit canker phytopathogen Pseudomonas syringae pv. actinidiae. Appl. Environ. Microbiol. 2014, 80, 2216–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, R.A.; Acedo, E.L.; Young, V.L.; Chen, D.N.; Tong, B.; Taylor, C.; Easingwood, R.A.; Pitman, A.R.; Kleffmann, T.; Bostina, M.; et al. Genome, proteome and structure of a T7-like bacteriophage of the kiwifruit canker phytopathogen Pseudomonas syringae pv. actinidiae. Viruses 2015, 7, 3361–3379. [Google Scholar] [CrossRef]
- Yu, J.G.; Lim, J.A.; Song, Y.R.; Heu, S.; Kim, G.H.; Koh, Y.J.; Oh, C.S. Isolation and characterization of bacteriophages against Pseudomonas syringae pv. actinidiae causing bacterial canker disease in kiwifruit. J. Microbiol. Biotechnol. 2016, 26, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, L.; Pereira, C.; Barreal, M.; Gallego, P.; Balcão, V.; Almeida, A. Use of phage ϕ6 to inactivate Pseudomonas syringae pv. actinidiae in kiwifruit plants: In vitro and ex vivo experiments. Appl. Microbiol. Biotechnol. 2020, 104, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Ni, P.; Wang, L.; Deng, B.; Jiu, S.; Ma, C.; Zhang, C.; Almeida, A.; Wang, D.; Xu, W.; Wang, S. Combined application of bacteriophages and carvacrol in the control of Pseudomonas syringae pv. actinidiae planktonic and biofilm forms. Microorganisms 2020, 8, 837. [Google Scholar] [CrossRef]
- Flores, O.; Retamales, J.; Nunez, M.; Leon, M.; Salinas, P.; Besoain, X.; Yanez, C.; Bastias, R. Characterization of bacteriophages against Pseudomonas syringae pv. actinidiae with potential use as natural antimicrobials in kiwifruit plants. Microorganisms 2020, 8, 974. [Google Scholar] [CrossRef]
- Yin, Y.; Ni, P.E.; Deng, B.; Wang, S.; Xu, W.; Wang, D. Isolation and characterisation of phages against Pseudomonas syringae pv. actinidiae. Acta Agric. Scand. Sect. B Soil Plant Sci. 2019, 69, 199–208. [Google Scholar] [CrossRef]
- Vidaver, A.; Koski, R.; Van Etten, J. Bacteriophage phi6: A lipid-containing virus of Pseudomonas phaseolicola. J. Virol. 1973, 11, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Eman, O.H.; Afaf, Z.A.E. Biocontrol of halo blight of bean caused by Pseudomonas Phaseolicola. Int. J. Virol. 2014, 10, 235–242. [Google Scholar]
- Sistrom, M.; Park, D.; O’Brien, H.; Wang, Z.; Guttman, D.; Townsend, J.; Turner, P. Genomic and gene-expression comparisons among phage-resistant type-IV pilus mutants of Pseudomonas syringae pathovar phaseolicola. PLoS ONE 2015, 10, e1005510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diallo, M.D.; Monteil, C.L.; Vinatzer, B.A.; Clarke, C.R.; Glaux, C.; Guilbaud, C.; Desbiez, C.; Morris, C.E. Pseudomonas syringae naturally lacking the canonical type III secretion system are ubiquitous in nonagricultural habitats, are phylogenetically diverse and can be pathogenic. ISME J. 2012, 6, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.; Sands, D.; Vanneste, J.; Montarry, J.; Oakley, B.; Guilbaud, C.; Glaux, C. Inferring the evolutionary history of the plant pathogen Pseudomonas syringae from its biogeography in headwaters of rivers in North America, Europe, and New Zealand. mBio 2010, 1, e00107-10. [Google Scholar] [CrossRef] [Green Version]
- Gallelli, A.; L’Aurora, A.; Loreti, S. Gene sequence analysis for the molecular detection of Pseudomonas syringae pv. actinidiae: Developing diagnostic protocols. J. Plant Path. 2011, 93, 425–435. [Google Scholar]
- Koh, Y.J.; Nou, I.S. DNA markers for identification of Pseudomonas syringae pv. actinidiae. Mol. Cells 2002, 13, 309–314. [Google Scholar]
- Audy, P.; Braat, C.E.; Saindon, G.; Huang, H.C.; Laroche, A. A rapid and sensitive PCR-based assay for concurrent detection of bacteria causing common and halo blights in bean seed. Phytopathology 1996, 86, 361–366. [Google Scholar] [CrossRef]
- Sanders, E. Aseptic laboratory techniques: Plating methods. J. Visual. Exp. 2012, 63, e3064. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Rasband, W.; Eliceiri, K. NIH Image to ImageJ: 25 years of image analysis. Nat. Meth. 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Abedon, S.T. Detection of bacteriophages: Phage plaques. In Bacteriophages: Biology, Technology, Therapy; Springer: Cham, Switzerland, 2021; pp. 507–538. [Google Scholar]
- Ginestet, C. ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. Ser. A 2011, 174, 245. [Google Scholar] [CrossRef]
- De Mendiburu, F.; Reinhard, S. Agricolae—Ten years of an open source statistical tool for experiments in breeding, agriculture and biology. Peer J. 2015, 3, e1404v1. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D. Molecular Cloning: A Laboratory Manual (3-Volume Set) 3 Lab Edition; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Bolger, A.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acid. Res. 2016, 45, D535–D542. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.; Lesin, V.; Nikolenko, S.; Pham, S.; Prjibelski, A.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Brettin, T.; Davis, J.; Disz, T.; Edwards, R.; Gerdes, S.; Olsen, G.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.; Chan, P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acid. Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kelley, L.A.; Sternberg, M.J.E. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Prot. 2009, 4, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.; Wootton, J.; Gertz, E.; Agarwala, R.; Morgulis, A.; Schäffer, A.; Yu, Y. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef]
- Joensen, K.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.; Nielsen, E.; Aarestrup, F. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Myers, E.; Miller, W. Optimal alignments in linear space. Comput. Appl. Biosci. 1988, 4, 11–17. [Google Scholar] [CrossRef]
- Tynecki, P.; Guziński, A.; Kazimierczak, J.; Jadczuk, M.; Dastych, J.; Onisko, A. PhageAI—Bacteriophage life cycle recognition with machine learning and natural language processing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Guy, L.; Roat Kultima, J.; Andersson, S.G.E. genoPlotR: Comparative gene and genome visualization in R. Bioinformatics 2020, 26, 2334–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruening, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acid. Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, P.; Siddell, S.; Lefkowitz, E.; Mushegian, A.; Adriaenssens, E.; Dempsey, D.; Dutilh, B.; Harrach, B.; Harrison, R.; Hendrickson, R.; et al. Changes to virus taxonomy and the statutes ratified by the international committee on taxonomy of viruses (2020). Arch. Virol. 2020, 165, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.; Potter, S.; Finn, R.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acid. Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifinopoulos, J.; Lam-Tung, N.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acid. Res 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Merrill, B.; Ward, A.; Grose, J.; Hope, S. Software-based analysis of bacteriophage genomes, physical ends, and packaging strategies. BMC Genom. 2016, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.; Kropinski, A.; Adriaenssens, E. A roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K. λ recombination and recombineering. EcoSal Plus 2016, 7, 1–70. [Google Scholar] [CrossRef] [Green Version]
- Calendar, R. The Bacteriophages: Volume 1; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Monteiro, R.; Pires, D.; Costa, A.; Azeredo, J. Phage therapy: Going temperate? Trends Microbiol. 2019, 27, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, H.; Li, L.; Liu, Y.; Li, D.; Pan, H.; Zhong, C.; Rikkerink, E.; Templeton, M.; Straub, C.; Colombi, E.; et al. Origin and evolution of the kiwifruit canker pandemic. Genome Biol. Evol. 2017, 9, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Wojtus, J.; Frampton, R.; Warring, S.; Hendrickson, H.; Fineran, P. Genome sequence of a jumbo bacteriophage that infects the kiwifruit phytopathogen Pseudomonas syringae pv. actinidiae. Microbiol. Resour. Announc. 2019, 8, e00224-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iriarte, F.; Obradović, A.; Wernsing, M.; Jackson, L.; Balogh, B.; Hong, J.; Momol, M.; Jones, J.; Vallad, G. Soil-based systemic delivery and phyllosphere in vivo propagation of bacteriophages: Two possible strategies for improving bacteriophage persistence for plant disease control. Bacteriophage 2012, 2, e23530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyssens, P.; Lavigne, R. Bacteriophages of Pseudomonas. Future Microbiol. 2010, 5, 1041–1055. [Google Scholar] [CrossRef]
- Ferrante, P.; Scortichini, M. Redefining the global populations of Pseudomonas syringae pv. actinidiae based on pathogenic, molecular and phenotypic characteristics. Plant Path. 2015, 64, 51–62. [Google Scholar] [CrossRef]
- Hofstatter, P.; Tice, A.; Kang, S.; Brown, M.; Lahr, D. Evolution of bacterial recombinase A (recA) in eukaryotes explained by addition of genomic data of key microbial lineages. Proc. Biol. Sci. 2016, 283, 20161453. [Google Scholar] [CrossRef] [Green Version]
- Kupczok, A.; Neve, H.; Huang, K.; Hoeppner, M.; Heller, K.; Franz, C.; Dagan, T. Rates of Mutation and Recombination in Siphoviridae phage genome evolution over three decades. Mol. Biol. Evol. 2018, 35, 1147–1159. [Google Scholar] [CrossRef]
- Harper, D.R.; Abedon, S.T.; Burrowes, B.H.; McConville, M.L. Bacteriophages: Biology, Technology, Therapy; Springer International Publishing: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Ercolani, G.L.; Crosse, J.E. The growth of Pseudomonas phaseolicola and related plant pathogens in vivo. Microbiology 1966, 45, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Dion, M.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lin, Y.R.; Lu, M.W.; Sung, P.J.; Wang, W.H.; Lin, C.S. Genome sequences characterizing five mutations in RNA polymerase and major capsid of phages ϕA318 and ϕAs51 of Vibrio alginolyticus with different burst efficiencies. BMC Genom. 2014, 15, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavigne, R.; Burkal’tseva, M.; Robben, J.; Sykilinda, N.; Kurochkina, L.; Grymonprez, B.; Jonckx, B.; Krylov, V.; Mesyanzhinov, V.; Volckaert, G. The genome of bacteriophage phiKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology 2003, 312, 4959. [Google Scholar] [CrossRef] [Green Version]
- Masschalck, B.; Van Houdt, R.; Van Haver, E.; Michiels, C. Inactivation of gram-negative bacteria by lysozyme, denatured lysozyme, and lysozyme-derived peptides under high hydrostatic pressure. Appl. Environ. Microbiol. 2001, 67, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, J.B.; Djurhuus, A.M.; Carstens, A.B.; Kot, W.; Neve, H.; Morris, C.E.; Hansen, L.H. Presentation of three novel tailed phages targeting multiple strains of Pseudomonas syringae. PHAGE 2020, 1, 245–250. [Google Scholar] [CrossRef]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, G.; Askora, A.; Blanc-Mathieu, R.; Kawasaki, T.; Li, Y.; Nakano, M.; Ogata, H.; Yamada, T. Xanthomonas citri jumbo phage XacN1 exhibits a wide host range and high complement of tRNA genes. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, T.; Sawada, H. Genome analysis of Pseudomonas syringae pv. actinidiae biovar 6, which produces the phytotoxins, phaseolotoxin and coronatine. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Palleroni, N.J. Pseudomonas. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 67–68. [Google Scholar]
- Balogh, B.; Jones, J.B.; Iriarte, F.B.; Momol, M.T. Phage therapy for plant disease control. Curr. Pharma. Biotechnol. 2010, 11, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E.; Sulakvelidze, A. Bacteriophages: Biology and Applications; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Ni, P.; Wang, L.; Deng, B.; Jiu, S.; Ma, C.; Zhang, C.; Almeida, A.; Wang, D.; Xu, W.; Wang, S. Characterization of a lytic bacteriophage against Pseudomonas syringae pv. actinidiae and its endolysin. Viruses 2021, 13, 641. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Lobocka, M.; Tong, Y.G.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018-2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Ruiz, I.; Coutinho, F.; Rodriguez-Valera, F. Thousands of novel endolysins discovered in uncultured phage genomes. Front. Microbiol. 2018, 9, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, M.; Thakur, S.; Almeida, R.; Wang, P.; Weir, B.; Guttman, D. Recombination of ecologically and evolutionarily significant loci maintains genetic cohesion in the Pseudomonas syringae species complex. Genome Biol. 2019, 20, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Schmerer, M.; Molineux, I.J.; Bull, J.J. Synergy as a rationale for phage therapy using phage cocktails. Peer J. 2014, 2, e590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.R.W.; Zamze, S.E.; Hignett, R.C. Morphology and hydrolytic activity of A7, a typing phage of Pseudomonas syringae pv. morsprunorum. Microbiology 1994, 140, 905–913. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification | Origin | Geographical Origin | Source | Bacterial Strains | Year | pphageB1 | pphageB21 | pphageBV72 | pphage T12 | pphage T21 | psage A1 | psage K4 | psage K4e | psage B1 | psage B2 | MR8 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | Agrion | K7#1 | 2019 | - | - | - | - | - | EOP | |||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | Agrion | K7#8 | 2019 | - | - | - | - | - | <0.01 | |||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | Agrion | K4#3 | 2018 | - | - | - | - | - | 0.01–0.1 | |||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | Agrion | K4#6 | 2018 | - | - | - | - | - | - | - | 0.1–1 | |||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | Agrion | K4#7 | 2018 | - | - | - | - | - | 1 | |||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | Agrion | K4#9 | 2018 | - | - | - | - | - | - | 1–10 | ||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | Agrion | K4#10 | 2018 | - | - | - | - | - | - | - | 10–100 | |||||
| P.s. pv. actinidiae | Actinidia chinensis * | Veneto | PIS $ | #5712.19 | 2019 | - | - | - | - | - | >100 | |||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Veneto | PIS $ | #5726.19 | 2019 | - | - | - | - | - | - | - | ||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Veneto | PIS $ | #5747.19 | 2019 | - | - | - | - | - | ||||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Veneto | PIS $ | #5846.19 | 2019 | - | - | - | - | - | - | - | ||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Veneto | PIS $ | #5847.19 | 2019 | - | - | - | - | - | ||||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Veneto | PIS $ | #5850.19 | 2019 | - | - | - | - | - | ||||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #298a | 2019 | - | - | - | - | - | - | - | ||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #391a | 2018 | - | - | - | - | - | ||||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #392a | 2018 | - | - | - | - | - | - | |||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #453d | 2016 | - | - | - | - | - | - | - | ||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #453e | 2016 | - | - | - | - | - | - | |||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #454d | 2018 | - | - | - | - | - | - | - | ||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #454e | 2018 | - | - | - | - | - | - | - | ||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #509c | 2018 | - | - | - | - | - | ||||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #509d | 2018 | - | - | - | - | - | ||||||||
| P.s. pv. actinidiae | Actinidia chinensis * | Piedmont | PIS $ | #509e | 2018 | - | - | - | - | - | ||||||||
| P.s. pv. phaseolicola | Phaseolus vulgaris | Piedmont | PIS $ | Pph PSS | 2015 | - | - | - | - | |||||||||
| P.s. pv. phaseolicola | Phaseolus vulgaris | China | Private company | Pph #9 | 2015 | - | ||||||||||||
| P.s. pv. phaseolicola | Phaseolus vulgaris | China | Private company | Pph #13 | 2015 | - | - | |||||||||||
| P.s. pv. phaseolicola | Phaseolus vulgaris | China | Private company | Pph #14 | 2015 | - | - | |||||||||||
| P.s. pv. phaseolicola | Phaseolus vulgaris | Piedmont | IPSP | Pph Cuneo | 2016 | - | - | - | ||||||||||
| P.s. pv. phaseolicola | Phaseolus vulgaris | Piedmont | Agrion | Pph Cuneo #6_17 | 2017 | - | - | - | ||||||||||
| P.s. pv. phaseolicola | Phaseolus vulgaris | Piedmont | Agrion | Pph Cuneo #6_18 | 2018 | - | - | |||||||||||
| P.s. pv. phaseolicola | Phaseolus vulgaris | Piedmont | Agrion | Pph Cuneo #7 | 2018 | - | - | |||||||||||
| P.s. pv. phaseolicola | Phaseolus vulgaris | Piedmont | Agrion | Pph Cuneo #13 | 2017 | - | - | - | ||||||||||
| P. syringae | Water | France | INRAe | UB197 | 2010 | - | - | - | - | - | - | - | - | - | - | - | ||
| P. syringae | Water | France | INRAe | UB210 | 2010 | - | - | - | - | - | - | - | - | - | - | |||
| P. syringae | Water | France | INRAe | UB246 | 2010 | - | - | - | - | - | - | - | - | - | - | |||
| P. syringae | Water | United States | INRAe | USA052 | 2010 | - | - | - | - | - | - | - | - | - | - | |||
| P. syringae | Water | France | INRAe | CC1504 | 2010 | - | - | - | - | - | - | - | - | - | - | - | ||
| P. syringae | Water | France | INRAe | SZ030 | 2010 | - | - | - | - | - | - | - | - | - | - | - | ||
| P. syringae | Water | France | INRAe | SZ122 | 2010 | - | - | - | - | - | - | - | - | - | - | - | ||
| P. syringae | Water | France | INRAe | SZ131 | 2010 | - | - | - | - | - | - | - | - | - | - | |||
| P.s. pv. syringae | Prunus armeniaca | Lombardia | UNIMI | Pss ML#1 | 2018 | - | - | - | - | - | - | - | - | - | - | - |
| Phage Name | Source | Origin a | Field Species and Cultivar | Source | Region | Year | Plaque Morphology (LBLS 0.6%) b | Classification c |
|---|---|---|---|---|---|---|---|---|
| psageK4 | Soil | Manta (CN) | Actinidia chinensis var. deliciosa | Agrion | Piedmont | 2018 | Clear, 1 mm | Myoviridae |
| psageK4e | Soil | Manta (CN) | Actinidia chinensis var. deliciosa | Agrion | Piedmont | 2019 | Clear, 1 mm | Myoviridae |
| psageK9 | Soil | Borgo d’Ale (VC) | Actinidia chinensis var. deliciosa | IPSP | Piedmont | 2019 | clear or turbid 2 mm | Siphoviridae |
| psageA1 | Soil | Manta (CN) | Actinidia chinensis var. deliciosa | Agrion | Piedmont | 2018 | Clear, 1–2 mm | Myoviridae |
| psageA2 | Soil | Manta (CN) | Actinidia chinensis var. deliciosa | Agrion | Piedmont | 2018 | Clear, 1–2 mm | Myoviridae |
| psageB1 | Soil | Manta (CN) | Actinidia chinensis var. deliciosa | Agrion | Piedmont | 2018 | Clear, 2 mm | Siphoviridae |
| psageB2 | Soil | Manta (CN) | Actinidia chinensis var. deliciosa | Agrion | Piedmont | 2018 | Clear or turbid, 2 mm | Siphoviridae |
| pphageB1 | Soil | Boves (CN) | Phaseolus vulgaris cv. Billò | Agrion | Piedmont | 2018 | Clear, 5 mm | Autographiviridae |
| pphageB12 | Soil | Boves (CN) | Phaseolus vulgaris cv. Billò | Agrion | Piedmont | 2019 | Clear, 5 mm | Autographiviridae |
| pphageB13 | Soil | Boves (CN) | Phaseolus vulgaris cv. Billò | Agrion | Piedmont | 2020 | Clear, 5 mm | Autographiviridae |
| pphageB21 | Soil | Boves (CN) | Phaseolus vulgaris cv. Billò | Agrion | Piedmont | 2018 | Clear, 5 mm | Autographiviridae |
| pphageB51 | Soil | Boves (CN) | Phaseolus vulgaris cv. Billò | Agrion | Piedmont | 2018 | Clear, 5 mm | Autographiviridae |
| pphageB101 | Soil | Boves (CN) | Phaseolus vulgaris cv. Billò | Agrion | Piedmont | 2018 | Clear, 5 mm | Autographiviridae |
| pphageT11 | Soil | Tetti Pesio (CN) | Phaseolus vulgaris cv. Borlotto nano | Agrion | Piedmont | 2018 | Clear, 5 mm | Autographiviridae |
| pphageT12 | Soil | Tetti Pesio (CN) | Phaseolus vulgaris cv. Borlotto nano | Agrion | Piedmont | 2019 | Clear, 5 mm | Autographiviridae |
| pphageT13 | Soil | Tetti Pesio (CN) | Phaseolus vulgaris cv. Borlotto nano | Agrion | Piedmont | 2020 | Clear, 5 mm | Autographiviridae |
| pphageT21 | Soil | Tetti Pesio (CN) | Phaseolus vulgaris cv. Borlotto nano | Agrion | Piedmont | 2018 | Clear, 5 mm | Autographiviridae |
| pphageT22 | Soil | Tetti Pesio (CN) | Phaseolus vulgaris cv. Borlotto nano | Agrion | Piedmont | 2018 | Clear, 5 mm | Autographiviridae |
| pphageT23 | Soil | Tetti Pesio (CN) | Phaseolus vulgaris cv. Borlotto nano | Agrion | Piedmont | 2018 | Clear, 5 mm | Autographiviridae |
| pphageBV2 | Soil | Verona (VR) | Phaseolus vulgaris cv. Spagnolet (Lamon) | Phytosanitary Inspection Service | Veneto | 2019 | Clear, 5 mm | Autographiviridae |
| pphageBV4 | Soil | Verona (VR) | Phaseolus vulgaris cv. Spagnolet (Lamon) | Phytosanitary Inspection Service | Veneto | 2019 | Clear, 5 mm | Autographiviridae |
| pphageBV71 | Soil | Verona (VR) | Phaseolus vulgaris cv. Canalino (Lamon) | Phytosanitary Inspection Service | Veneto | 2019 | Clear, 5 mm | Autographiviridae |
| pphageBV72 | Soil | Verona (VR) | Phaseolus vulgaris cv. Canalino (Lamon) | Phytosanitary Inspection Service | Veneto | 2019 | Clear, 5 mm | Autographiviridae |
| Phage Name | Accession Number | Bacterial Host | Genome Length | GC Content (%) | ORFs | Hypothetical Proteins | Proteins with Predicted Function | ORFans | tRNAs |
|---|---|---|---|---|---|---|---|---|---|
| psageA1 | MT740307 | Psa K7 #8 | 98,780 bp | 48.79% | 176 | 76 | 51 | 49 | 14 |
| psageB1 | MT354569 | Psa K7 #8 | 112,269 bp | 56.47% | 161 | 105 | 40 | 16 | 4 |
| pphageB1 | MT354570 | Pph Cuneo #6_18 | 41,714 bp | 56.63% | 52 | 23 | 25 | 4 | 0 |
| psageK4 | MZ348426 | Psa K7 #8 | 98,440 bp | 60.44% | 179 | 112 | 51 | 16 | 18 |
| psageB2 | MZ348425 | Psa K7 #8 | 50,739 bp | 58.51% | 77 | 47 | 26 | 4 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martino, G.; Holtappels, D.; Vallino, M.; Chiapello, M.; Turina, M.; Lavigne, R.; Wagemans, J.; Ciuffo, M. Molecular Characterization and Taxonomic Assignment of Three Phage Isolates from a Collection Infecting Pseudomonas syringae pv. actinidiae and P. syringae pv. phaseolicola from Northern Italy. Viruses 2021, 13, 2083. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102083
Martino G, Holtappels D, Vallino M, Chiapello M, Turina M, Lavigne R, Wagemans J, Ciuffo M. Molecular Characterization and Taxonomic Assignment of Three Phage Isolates from a Collection Infecting Pseudomonas syringae pv. actinidiae and P. syringae pv. phaseolicola from Northern Italy. Viruses. 2021; 13(10):2083. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102083
Chicago/Turabian StyleMartino, Gabriele, Dominique Holtappels, Marta Vallino, Marco Chiapello, Massimo Turina, Rob Lavigne, Jeroen Wagemans, and Marina Ciuffo. 2021. "Molecular Characterization and Taxonomic Assignment of Three Phage Isolates from a Collection Infecting Pseudomonas syringae pv. actinidiae and P. syringae pv. phaseolicola from Northern Italy" Viruses 13, no. 10: 2083. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102083