
Evolutionary Dynamics of H5 Highly Pathogenic Avian Influenza Viruses (Clade 2.3.4.4B) Circulating in Bulgaria in 2019–2021

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Whole Genome Sequencing
2.3. Phylogenetic and Evolutionary Analyses
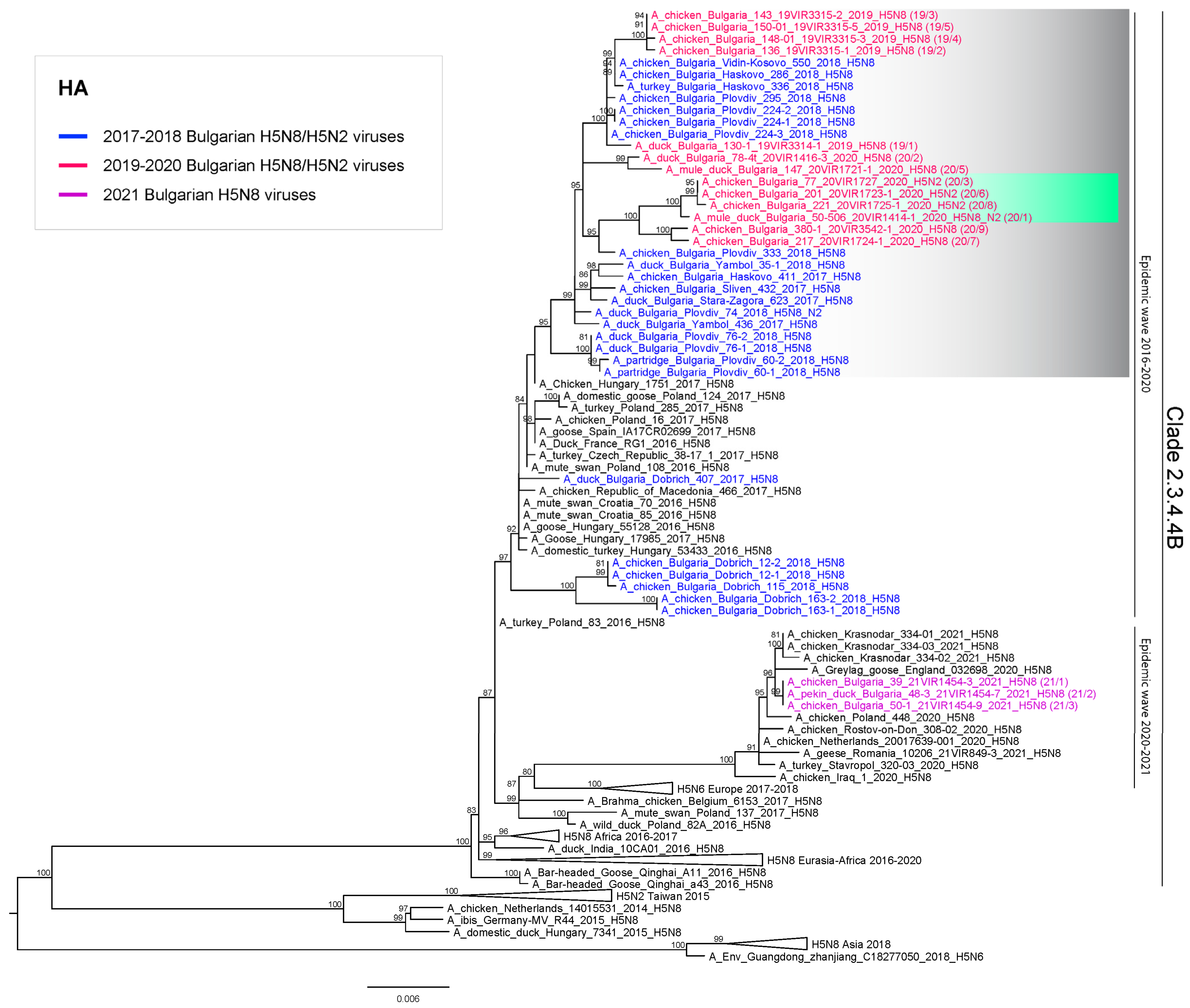
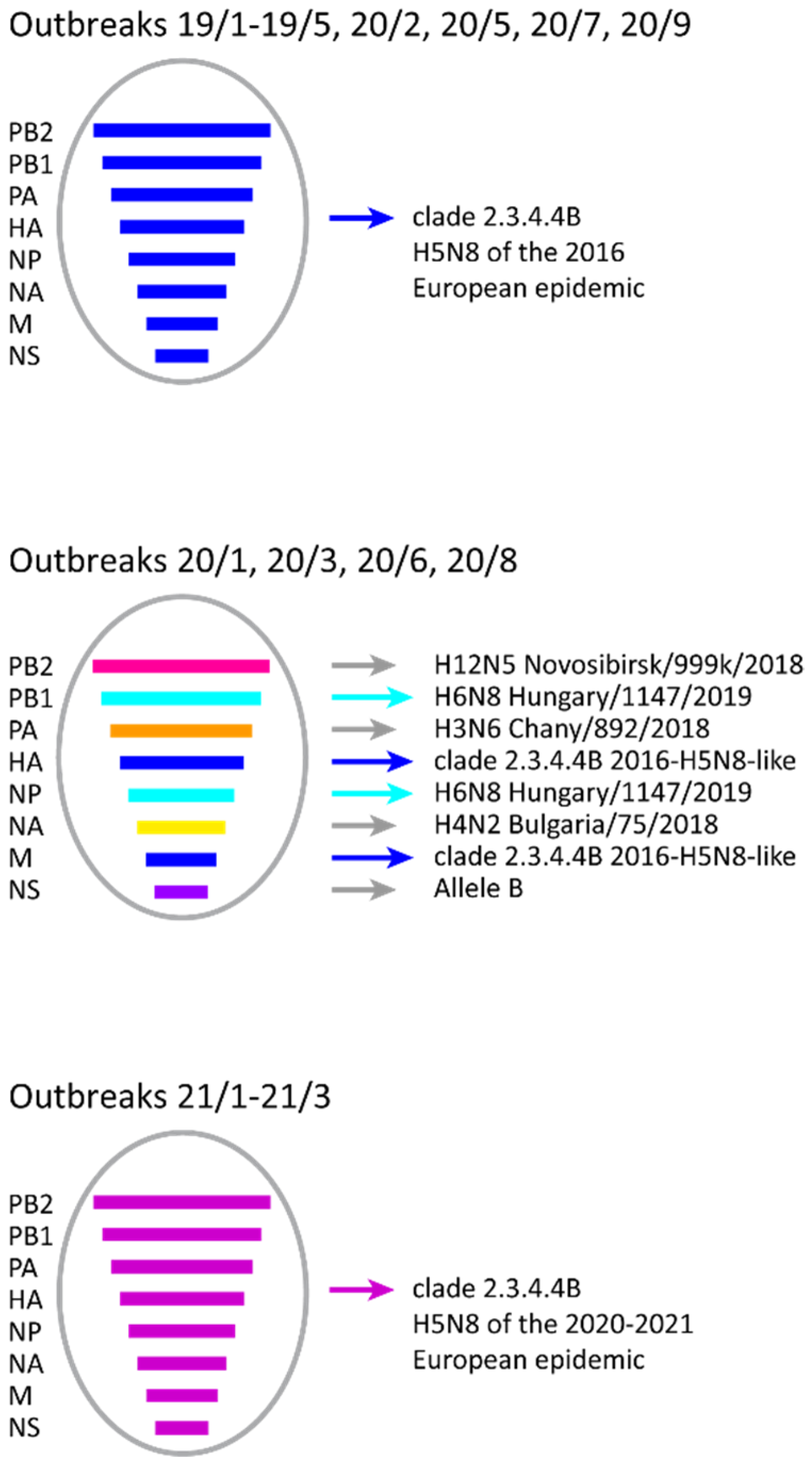
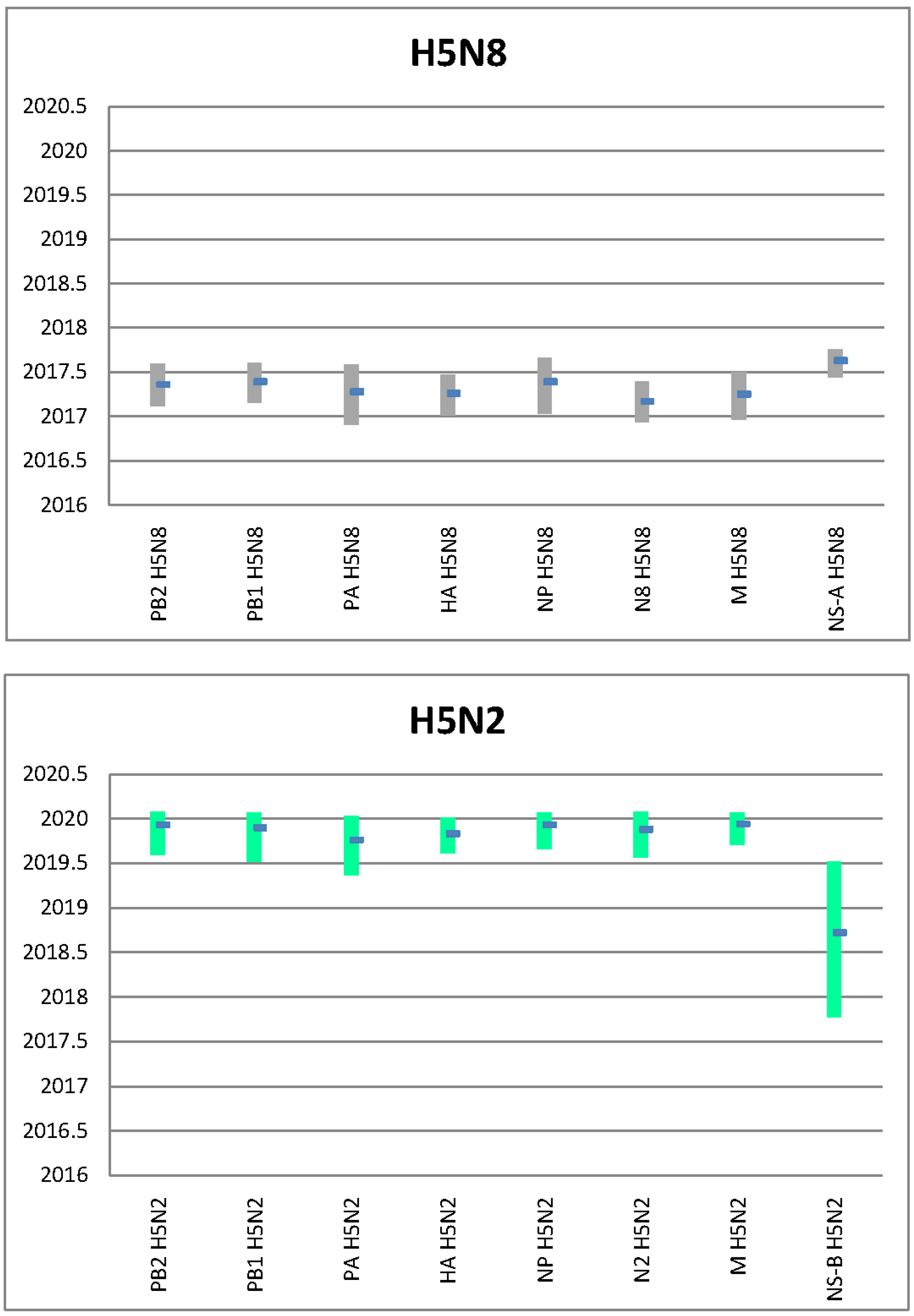
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alarcon, P.; Brouwer, A.; Venkatesh, D.; Duncan, D.; Dovas, C.I.; Georgiades, G.; Monne, I.; Fusaro, A.; Dan, A.; Śmietanka, K.; et al. Comparison of 2016–17 and Previous Epizootics of Highly Pathogenic Avian Influenza H5 Guangdong Lineage in Europe. Emerg. Infect. Dis. 2018, 24, 2270–2283. [Google Scholar] [CrossRef] [PubMed]
- Baldinelli, F.; Papanikolaou, A.; Stoicescu, A.; Van der Stede, Y.; Aznar, I. Annual Report on surveillance for Avian Influenza in poultry and wild birds in Member States of the European Union in 2019. EFSA J. 2020, 18, 6349. [Google Scholar] [CrossRef]
- Brouwer, A.; Gonzales, J.; Huneau, A.; Mulatti, P.; Kuiken, T.; Staubach, C.; Stegeman, A.; Antoniou, S.E.; Baldinelli, F.; Van der Stede, Y.; et al. Annual Report on surveillance for avian influenza in poultry and wild birds in Member States of the European Union in 2018. EFSA J. 2019, 17. [Google Scholar] [CrossRef]
- Śmietanka, K.; Świętoń, E.; Kozak, E.; Wyrostek, K.; Tarasiuk, K.; Tomczyk, G.; Konopka, B.; Welz, M.; Domańska-Blicharz, K.; Niemczuk, K. Highly pathogenic avian influenza H5N8 in Poland in 2019–2020. J. Vet. Res. 2020, 64, 469–476. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Kuiken, T.; Niqueux, E.; Staubach, C.; Terregino, C.; Guajardo, I.M.; Baldinelli, F. Avian influenza overview November 2019–February2020. EFSA J. 2020, 18. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Kuiken, T.; Niqueux, E.; Staubach, C.; Terregino, C.; Guajardo, I.M.; Baldinelli, F. Avian influenza overview February–May 2020. EFSA J. 2020, 18. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Kuiken, T.; Niqueux, É.; Staubach, C.; Terregino, C.; Muñoz Guajardo, I.; Baldinelli, F. Avian influenza overview May–August 2020. EFSA J. 2020, 18. [Google Scholar] [CrossRef]
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; Meruyert Akberovna, S.; King, J.; Harder, T.; et al. Emergence and spread of novel H5N8, H5N5 and H5N1 clade 2.3.4.4 highly pathogenic avian influenza in 2020. Emerg. Microbes. Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef]
- Venkatesh, D.; Brouwer, A.; Ellis, R.; Goujgoulova, G.; Seekings, J.; Brown, I.; Lewis, N. Regional transmission and reassortment of 2.3.4.4b highly pathogenic avian influenza (HPAI) viruses in Bulgarian poultry 2017/18. Viruses 2020, 12, 605. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; Smietanka, K.; Terregino, C.; Van der Stede, Y.; et al. Avian influenza overview—update on 19 November 2020, EU/EEA and the UK. EFSA J. 2020, 18, e06341. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; Terregino, C.; Baldinelli, F. Avian influenza overview August–December 2020. EFSA J. 2020, 18, 6379. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; Terregino, C.; Muñoz Guajardo, I.; Lima, E.; et al. Avian influenza overview December 2020–February 2021. EFSA J. 2021, 19, e06497. [Google Scholar] [CrossRef]
- Stoimenov, G.M.; Goujgoulova, G.V.; Nikolov, B.; Hristov, K.; Teneva, A. Pathological changes in natural infection of pheasants with highly pathogenic avian influenza A (H5N8) in Bulgaria. J. Vet. Res. 2019, 63, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St. George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-Reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza A Viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Auwera, G.; Carneiro, M.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinforma. 2013, 43, 10.1–10.33. [Google Scholar] [CrossRef]
- Depristo, M.A.; Banks, E.; Poplin, R.E.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, B.; Rambaut, A.; Drummond, A.J. Choosing Appropriate Substitution Models for the Phylogenetic Analysis of Protein-Coding Sequences. Mol. Biol. Evol. 2006, 23, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Matrosovich, M.; Zhou, N.; Kawaoka, Y.; Webster, R. The Surface Glycoproteins of H5 Influenza Viruses Isolated from Humans, Chickens, and Wild Aquatic Birds Have Distinguishable Properties. J. Virol. 1999, 73, 1146. [Google Scholar] [CrossRef] [Green Version]
- Adlhoch, C.; Brouwer, A.; Kuiken, T.; Mulatti, P.; Smietanka, K.; Staubach, C.; Muñoz Guajardo, I.; Verdonck, F.; Amato, L.; Baldinelli, F. Avian influenza overview February–May 2018. EFSA J. 2018, 16, e05358. [Google Scholar] [CrossRef]
- World Organisation for Animal Health (OIE). OIE Situation Report for Highly Pathogenic Avian Influenza; 2018; Available online: https://www.oie.int/fileadmin/Home/eng/Animal_Health_in_the_World/docs/pdf/OIE_AI_situation_report/OIE_SituationReport_AI_August2018.pdf (accessed on 10 October 2021).
- Kim, J.-K.; Negovetich, N.J.; Forrest, H.L.; Webster, R.G. Ducks: The “Trojan Horses” of H5N1 influenza. Influenza Other Respi. Viruses 2009, 3, 121–128. [Google Scholar] [CrossRef]
- Webster, R.G.; Webby, R.J.; Hoffmann, E.; Rodenberg, J.; Kumar, M.; Chu, H.J.; Seiler, P.; Krauss, S.; Songserm, T. The immunogenicity and efficacy against H5N1 challenge of reverse genetics-derived H5N3 influenza vaccine in ducks and chickens. Virology 2006, 351, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Marinova-Petkova, A.; Georgiev, G.; Petkov, T.; Darnell, D.; Franks, J.; Kayali, G.; Walker, D.; Seiler, P.; Danner, A.; Graham, A.; et al. Influenza surveillance on “foie gras” duck farms in Bulgaria, 2008–2012. Influenza Other Respi. Viruses 2016, 10, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Henning, J.; Wibawa, H.; Morton, J.; Usman, T.B.; Junaidi, A.; Meers, J. Scavenging Ducks and Transmission of Highly Pathogenic Avian Influenza, Java, Indonesia. Emerg. Infect. Dis. 2010, 16, 1244. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.; Xiao, X.; Pfeiffer, D.U.; Epprecht, M.; Boles, S.; Czarnecki, C.; Chaitaweesub, P.; Kalpravidh, W.; Minh, P.Q.; Otte, M.J.; et al. Mapping H5N1 highly pathogenic avian influenza risk in Southeast Asia. Proc. Natl. Acad. Sci. USA 2008, 105, 4769–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.; Chaitaweesub, P.; Parakamawongsa, T.; Premashthira, S.; Tiensin, T.; Kalpravidh, W.; Wagner, H.; Slingenbergh, J. Free-grazing Ducks and Highly Pathogenic Avian Influenza, Thailand. Emerg. Infect. Dis. 2006, 12, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.-H.; Lee, D.-H.; Criado, M.F.; Killmaster, L.; Ali, M.Z.; Giasuddin, M.; Samad, M.A.; Karim, M.R.; Hasan, M.; Brum, E.; et al. Genetic evolution and transmission dynamics of clade 2.3.2.1a highly pathogenic avian influenza A/H5N1 viruses in Bangladesh. Virus Evol. 2020, 6, veaa046. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.; Marinova-Petkova, A.; Hasan, M.K.; Akhtar, S.; El-Shesheny, R.; Turner, J.C.; Franks, J.; Walker, D.; Seiler, J.; Friedman, K.; et al. Role of domestic ducks in the emergence of a new genotype of highly pathogenic H5N1 avian influenza A viruses in Bangladesh. Emerg. Microbes Infect. 2017, 6, e72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoimenov, G.M. Review Highly Pathogenic Avian Influenza in Bulgaria A Review. Bulg. J. Vet. Med. 2020. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; Terregino, C.; Aznar, I.; Guajardo, I.M.; et al. Avian influenza overview February–May 2021. EFSA J. 2021. Available online: https://www.ecdc.europa.eu/en/publications-data/avian-influenza-overview-february-may-2021#no-link (accessed on 10 October 2021).




{kind=link}
{kind=link}
{kind=link}
| Outbreak | Virus | Subtype | Region | District | Collection Date (dd/mm/yyyy) | Species | Production Category | Accession Number (GISAID EpiFlu™) |
|---|---|---|---|---|---|---|---|---|
| 19/1 | A/duck/Bulgaria/130-1_19VIR3314-1/2019 | H5N8 | Lovech | Yoglav | 27/03/2019 | Mule duck | Fattening | EPI1807349-EPI1807356 |
| 19/2 | A/chicken/Bulgaria/136_19VIR3315-1/2019 | H5N8 | Plovdiv | Krumovo | 01/04/2019 | Chicken | Backyard | EPI1807268-EPI1807275 |
| 19/3 | A/chicken/Bulgaria/143_19VIR3315-2/2019 | H5N8 | Plovdiv | Asenograd | 04/04/2019 | Chicken | Laying hens | EPI1807276-EPI1807283 |
| 19/4 | A/chicken/Bulgaria/148-01_19VIR3315-3/2019 | H5N8 | Plovdiv | Asenograd | 05/04/2019 | Chicken | Laying hens | EPI1807284-EPI1807291 |
| 19/5 | A/chicken/Bulgaria/150-01_19VIR3315-5/2019 | H5N8 | Plovdiv | Asenograd | 05/04/2019 | Chicken | Laying hens | EPI1807292-EPI1807299 |
| 20/1 | A/mule_duck/Bulgaria/50-506_20VIR1414-1/2020 | H5N2/N8 | Plovdiv | Rakovski | 11/02/2020 | Mule duck | Fattening | EPI1807332-EPI1807339, EPI1807348 |
| 20/2 | A/duck/Bulgaria/78-4t_20VIR1416-3/2020 | H5N8 | Plovdiv | Padarsko | 21/02/2020 | Duck | Foie gras | EPI1807300-EPI1807307 |
| 20/3 | A/chicken/Bulgaria/77_20VIR1727/2020 | H5N2 | Plovdiv | Trilistnik | 21/02/2020 | Chicken | Layer | EPI1780067, EPI1780069, EPI1780071, EPI1780073, EPI1780075, EPI1780077, EPI1780079, EPI1780081 |
| 20/5 | A/mule_duck/Bulgaria/147_20VIR1721-1/2020 | H5N8 | Plovdiv | Bolyarino | 21/02/2020 | Mule duck | Foie gras | EPI1807340-EPI1807347 |
| 20/6 | A/chicken/Bulgaria/201_20VIR1723-1/2020 | H5N2 | Plovdiv | Trilistnik | 02/03/2020 | Chicken | Layer | EPI1807308-EPI1807315 |
| 20/7 | A/chicken/Bulgaria/217_20VIR1724-1/2020 | H5N8 | Kurdzhali | Perperek | 09/03/2020 | Chicken | Laying hens | EPI1780066, EPI1780068, EPI1780070, EPI1780072, EPI1780074, EPI1780076, EPI1780078, EPI1780080 |
| 20/8 | A/chicken/Bulgaria/221_20VIR1725-1/2020 | H5N2 | Plovdiv | Manole | 11/03/2020 | Chicken | Laying hens | EPI1807316-EPI1807323 |
| 20/9 | A/chicken/Bulgaria/380-1_20VIR3542-1/2020 | H5N8 | Plovdiv | Asenograd | 03/06/2020 | Chicken | Laying hens | EPI1807324-EPI1807331 |
| 21/1 | A/chicken/Bulgaria/39_21VIR1454-3/2021 | H5N8 | Pleven | Slavyanovo | 01/02/2021 | Chicken | Laying hens | EPI1858599-EPI1858606 |
| 21/2 | A/pekin_duck/Bulgaria/48-3_21VIR1454-7/2021 | H5N8 | Pleven | Slavyanovo | 04/02/2021 | Pekin duck | Fattening | EPI1858607-EPI1858614 |
| 21/3 | A/chicken/Bulgaria/50-1_21VIR1454-9/2021 | H5N8 | Pleven | Slavyanovo | 08/02/2021 | Chicken | Breeding hens | EPI1858615- EPI1858622 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zecchin, B.; Goujgoulova, G.; Monne, I.; Salviato, A.; Schivo, A.; Slavcheva, I.; Pastori, A.; Brown, I.H.; Lewis, N.S.; Terregino, C.; et al. Evolutionary Dynamics of H5 Highly Pathogenic Avian Influenza Viruses (Clade 2.3.4.4B) Circulating in Bulgaria in 2019–2021. Viruses 2021, 13, 2086. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102086
Zecchin B, Goujgoulova G, Monne I, Salviato A, Schivo A, Slavcheva I, Pastori A, Brown IH, Lewis NS, Terregino C, et al. Evolutionary Dynamics of H5 Highly Pathogenic Avian Influenza Viruses (Clade 2.3.4.4B) Circulating in Bulgaria in 2019–2021. Viruses. 2021; 13(10):2086. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102086
Chicago/Turabian StyleZecchin, Bianca, Gabriela Goujgoulova, Isabella Monne, Annalisa Salviato, Alessia Schivo, Iskra Slavcheva, Ambra Pastori, Ian H. Brown, Nicola S. Lewis, Calogero Terregino, and et al. 2021. "Evolutionary Dynamics of H5 Highly Pathogenic Avian Influenza Viruses (Clade 2.3.4.4B) Circulating in Bulgaria in 2019–2021" Viruses 13, no. 10: 2086. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102086