
The Biogenesis of Dengue Virus Replication Organelles Requires the ATPase Activity of Valosin-Containing Protein

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, DNAs, Viruses, and Reagents
2.2. Lentiviral Transduction
2.3. Cell Viability Assays
2.4. Plaque Assays
2.5. Luciferase Assays
2.6. Transfection
2.7. Immunofluorescence-Based Confocal Microscopy
2.8. Co-Immunoprecipitation Assays
2.9. Transmission Electron Microscopy
3. Statistical Analysis
4. Results
4.1. VCP ATPase Activity Is Required for Efficient DENV Replication
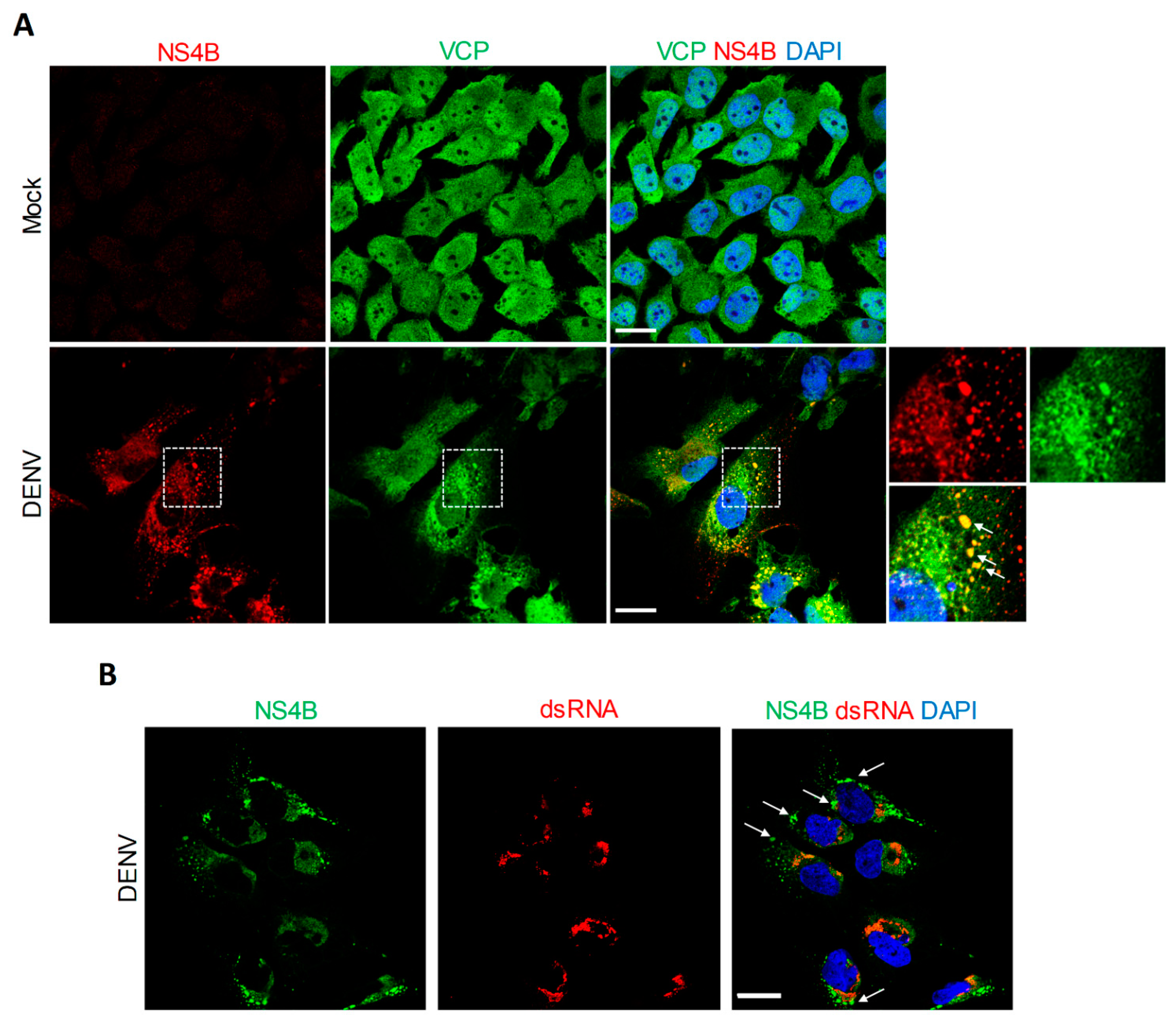
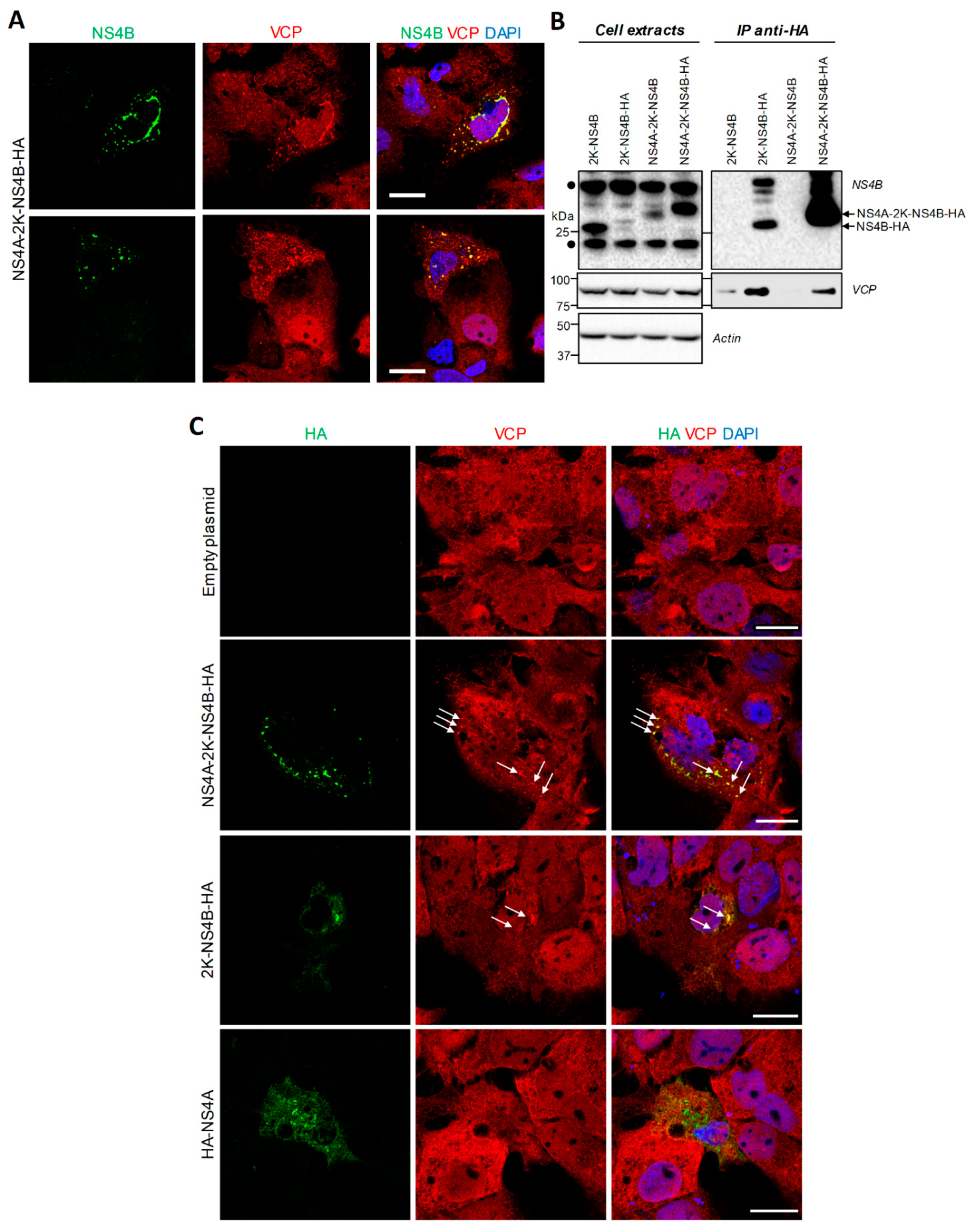
4.2. VCP Associates with DENV NS4B Independently of Other Viral Proteins
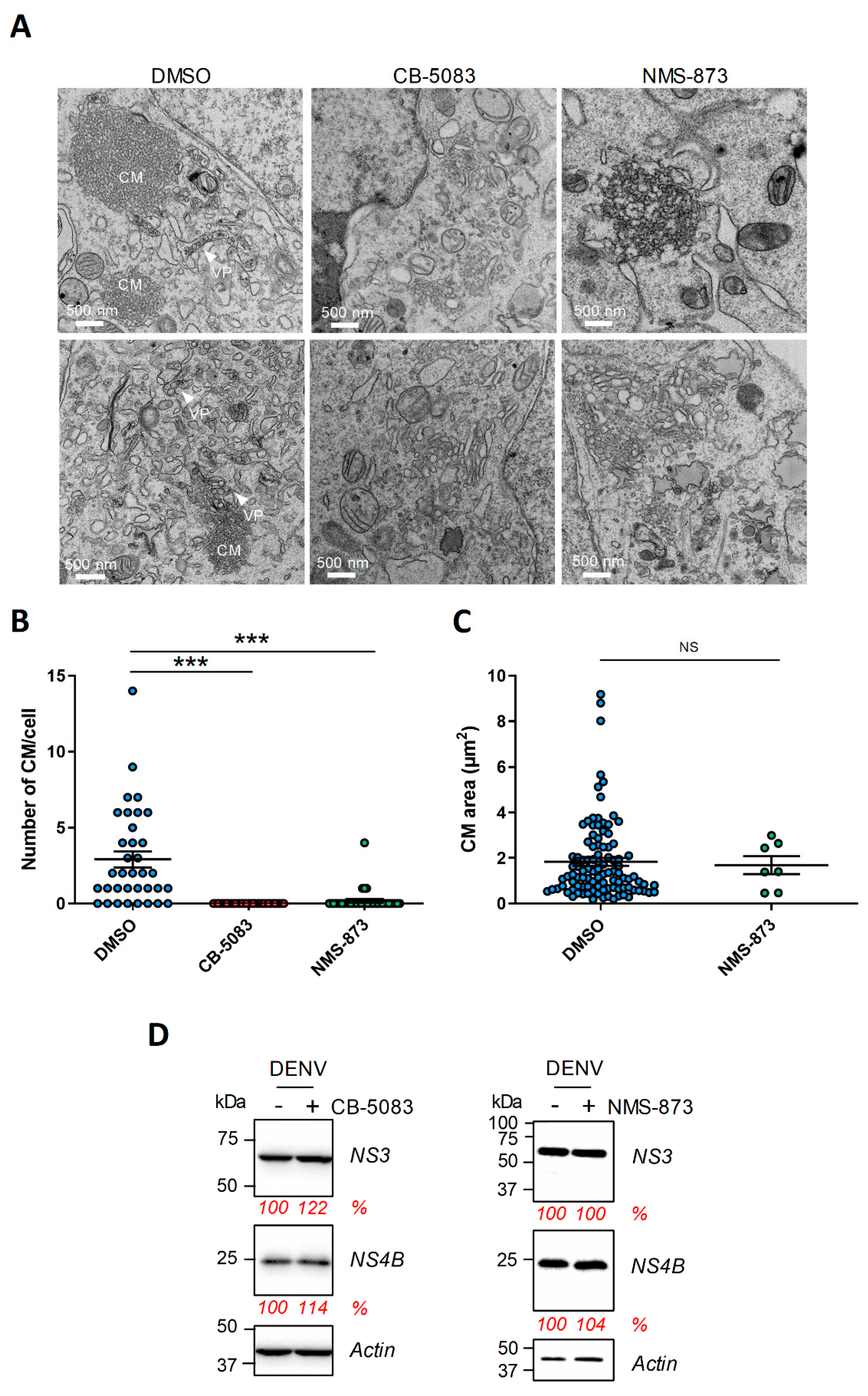
4.3. The Abundance of DENV Convoluted Membranes in Infected Cells Depends on VCP ATPase Activity
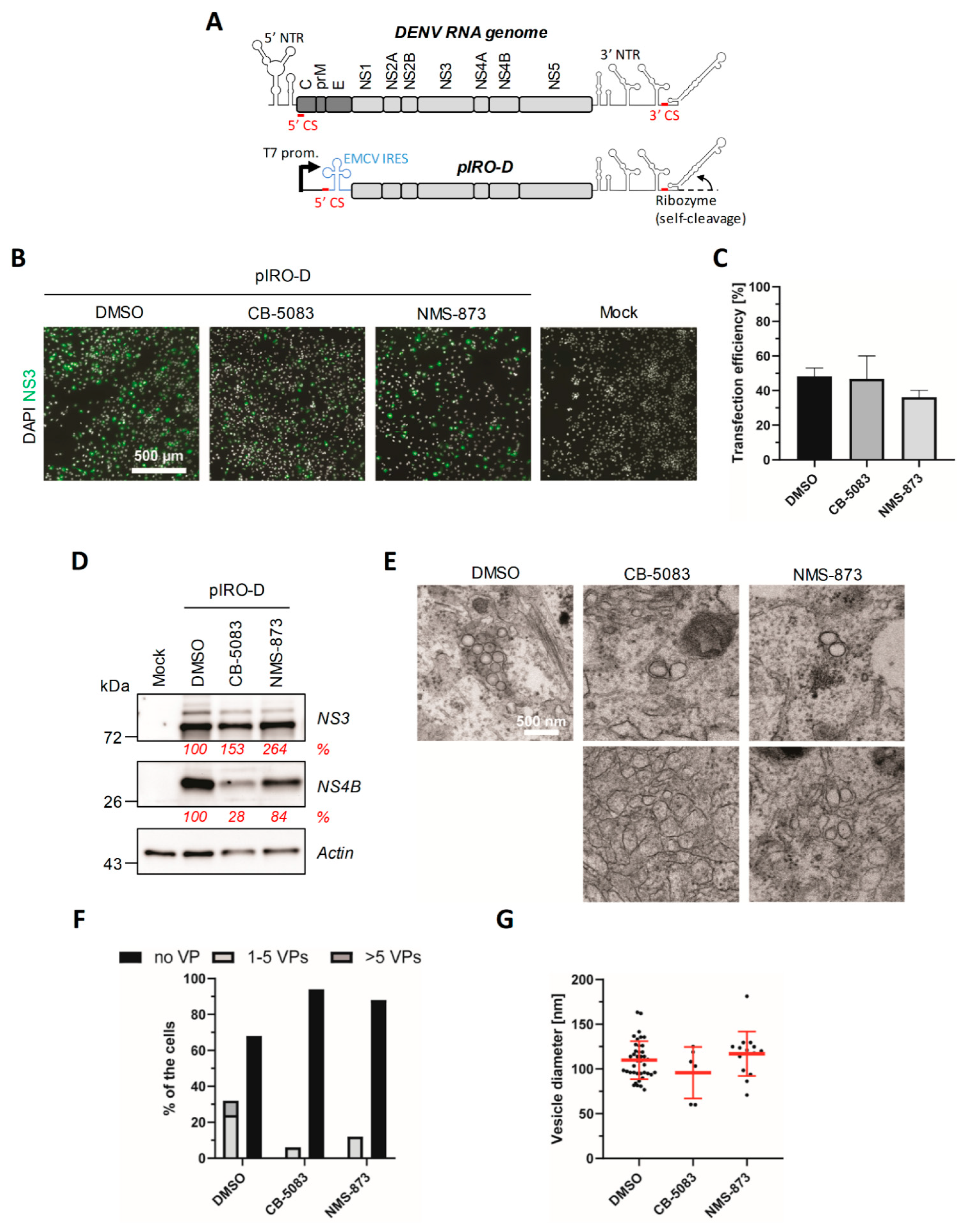
4.4. VCP Inhibition Impedes the Biogenesis of the DENV VPs
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, S. Dengue shock. J. Emergencies Trauma Shock. 2011, 4, 120–127. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Shepard, D.S.; Undurraga, E.A.; Halasa, Y.A.; Coffeng, L.E.; Brady, O.J.; Hay, S.I.; Bedi, N.; Bensenor, I.M.; Castaneda-Orjuela, C.A.; et al. The global burden of dengue: An analysis from the Global Burden of Disease Study 2013. Lancet Infect. Dis. 2016, 16, 712–723. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.; Luedtke, A.; Langevin, E.; Zhu, M.; Bonaparte, M.; Machabert, T.; Savarino, S.; Zambrano, B.; Moureau, A.; Khromava, A.; et al. Effect of Dengue Serostatus on Dengue Vaccine Safety and Efficacy. N. Engl. J. Med. 2018, 379, 327–340. [Google Scholar] [CrossRef]
- Mazeaud, C.; Freppel, W.; Chatel-Chaix, L. The Multiples Fates of the Flavivirus RNA Genome During Pathogenesis. Front. Genet. 2018, 9, 595. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef]
- Junjhon, J.; Pennington, J.G.; Edwards, T.J.; Perera, R.; Lanman, J.; Kuhn, R.J. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J. Virol. 2014, 88, 4687–4697. [Google Scholar] [CrossRef] [Green Version]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westaway, E.G.; Mackenzie, J.M.; Kenney, M.T.; Jones, M.K.; Khromykh, A.A. Ultrastructure of Kunjin virus-infected cells: Colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 1997, 71, 6650–6661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anton, A.; Mazeaud, C.; Freppel, W.; Gilbert, C.; Tremblay, N.; Sow, A.A.; Roy, M.; Rodrigue-Gervais, I.G.; Chatel-Chaix, L. Valosin-containing protein ATPase activity regulates the morphogenesis of Zika virus replication organelles and virus-induced cell death. Cell. Microbiol. 2021, 23, e13302. [Google Scholar] [CrossRef]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [Green Version]
- Bily, T.; Palus, M.; Eyer, L.; Elsterova, J.; Vancova, M.; Ruzek, D. Electron Tomography Analysis of Tick-Borne Encephalitis Virus Infection in Human Neurons. Sci. Rep. 2015, 5, 10745. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [Green Version]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabata, K.; Arimoto, M.; Arakawa, M.; Nara, A.; Saito, K.; Omori, H.; Arai, A.; Ishikawa, T.; Konishi, E.; Suzuki, R.; et al. Unique Requirement for ESCRT Factors in Flavivirus Particle Formation on the Endoplasmic Reticulum. Cell Rep. 2016, 16, 2339–2347. [Google Scholar] [CrossRef] [Green Version]
- Chatel-Chaix, L.; Fischl, W.; Scaturro, P.; Cortese, M.; Kallis, S.; Bartenschlager, M.; Fischer, B.; Bartenschlager, R. A Combined Genetic-Proteomic Approach Identifies Residues within Dengue Virus NS4B Critical for Interaction with NS3 and Viral Replication. J. Virol. 2015, 89, 7170–7186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Lee, L.T.; Wang, Q.Y.; Xie, X.; Lu, S.; Yau, Y.H.; Yuan, Z.; Geifman Shochat, S.; Kang, C.; Lescar, J.; et al. Mapping the Interactions between the NS4B and NS3 proteins of dengue virus. J. Virol. 2015, 89, 3471–3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Xie, X.; Lee, L.T.; Chandrasekaran, R.; Reynaud, A.; Yap, L.; Wang, Q.Y.; Dong, H.; Kang, C.; Yuan, Z.; et al. Dimerization of flavivirus NS4B protein. J. Virol. 2014, 88, 3379–3391. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Sparacio, S.; Bartenschlager, R. Subcellular localization and membrane topology of the Dengue virus type 2 Non-structural protein 4B. J. Biol. Chem. 2006, 281, 8854–8863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moquin, S.A.; Simon, O.; Karuna, R.; Lakshminarayana, S.B.; Yokokawa, F.; Wang, F.; Saravanan, C.; Zhang, J.; Day, C.W.; Chan, K.; et al. NITD-688, a pan-serotype inhibitor of the dengue virus NS4B protein, shows favorable pharmacokinetics and efficacy in preclinical animal models. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- van Cleef, K.W.; Overheul, G.J.; Thomassen, M.C.; Kaptein, S.J.; Davidson, A.D.; Jacobs, M.; Neyts, J.; van Kuppeveld, F.J.; van Rij, R.P. Identification of a new dengue virus inhibitor that targets the viral NS4B protein and restricts genomic RNA replication. Antivir. Res. 2013, 99, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Y.; Dong, H.; Zou, B.; Karuna, R.; Wan, K.F.; Zou, J.; Susila, A.; Yip, A.; Shan, C.; Yeo, K.L.; et al. Discovery of Dengue Virus NS4B Inhibitors. J. Virol. 2015, 89, 8233–8244. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Wang, Q.Y.; Xu, H.Y.; Qing, M.; Kramer, L.; Yuan, Z.; Shi, P.Y. Inhibition of dengue virus by targeting viral NS4B protein. J. Virol. 2011, 85, 11183–11195. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Zou, J.; Wang, Q.Y.; Shi, P.Y. Targeting dengue virus NS4B protein for drug discovery. Antivir. Res. 2015, 118, 39–45. [Google Scholar] [CrossRef]
- Shah, P.S.; Link, N.; Jang, G.M.; Sharp, P.P.; Zhu, T.; Swaney, D.L.; Johnson, J.R.; Von Dollen, J.; Ramage, H.R.; Satkamp, L.; et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175, 1931–1945.e1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beskow, A.; Grimberg, K.B.; Bott, L.C.; Salomons, F.A.; Dantuma, N.P.; Young, P. A conserved unfoldase activity for the p97 AAA-ATPase in proteasomal degradation. J. Mol. Biol. 2009, 394, 732–746. [Google Scholar] [CrossRef]
- Bodnar, N.O.; Rapoport, T.A. Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 2017, 169, 722–735.e729. [Google Scholar] [CrossRef] [Green Version]
- DeLaBarre, B.; Christianson, J.C.; Kopito, R.R.; Brunger, A.T. Central pore residues mediate the p97/VCP activity required for ERAD. Mol. Cell 2006, 22, 451–462. [Google Scholar] [CrossRef]
- Jarosch, E.; Taxis, C.; Volkwein, C.; Bordallo, J.; Finley, D.; Wolf, D.H.; Sommer, T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 2002, 4, 134–139. [Google Scholar] [CrossRef]
- Ju, J.S.; Miller, S.E.; Hanson, P.I.; Weihl, C.C. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J. Biol. Chem. 2008, 283, 30289–30299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.C.; Tresse, E.; Kolaitis, R.M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 2013, 78, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojcik, C.; Rowicka, M.; Kudlicki, A.; Nowis, D.; McConnell, E.; Kujawa, M.; DeMartino, G.N. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell 2006, 17, 4606–4618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Halawani, D.; LeBlanc, A.C.; Rouiller, I.; Michnick, S.W.; Servant, M.J.; Latterich, M. Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol. Cell. Biol. 2009, 29, 4484–4494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, H.; Ewens, C.A.; Tsang, C.; Yeung, H.O.; Zhang, X.; Freemont, P.S. The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J. Biol. Chem. 2012, 287, 8561–8570. [Google Scholar] [CrossRef] [Green Version]
- Weihl, C.C.; Dalal, S.; Pestronk, A.; Hanson, P.I. Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet. 2006, 15, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Phongphaew, W.; Kobayashi, S.; Sasaki, M.; Carr, M.; Hall, W.W.; Orba, Y.; Sawa, H. Valosin-containing protein (VCP/p97) plays a role in the replication of West Nile virus. Virus Res. 2017, 228, 114–123. [Google Scholar] [CrossRef]
- Ramanathan, H.N.; Zhang, S.; Douam, F.; Mar, K.B.; Chang, J.; Yang, P.L.; Schoggins, J.W.; Ploss, A.; Lindenbach, B.D. A Sensitive Yellow Fever Virus Entry Reporter Identifies Valosin-Containing Protein (VCP/p97) as an Essential Host Factor for Flavivirus Uncoating. mBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Tabata, K.; Arakawa, M.; Ishida, K.; Kobayashi, M.; Nara, A.; Sugimoto, T.; Okada, T.; Mori, K.; Morita, E. Endoplasmic reticulum-associated degradation controls virus protein homeostasis that is required for the flavivirus propagation. J. Virol. 2021. [Google Scholar] [CrossRef]
- Gestuveo, R.J.; Royle, J.; Donald, C.L.; Lamont, D.J.; Hutchinson, E.C.; Merits, A.; Kohl, A.; Varjak, M. Analysis of Zika virus capsid-Aedes aegypti mosquito interactome reveals pro-viral host factors critical for establishing infection. Nat. Commun. 2021, 12, 2766. [Google Scholar] [CrossRef]
- Sehrawat, S.; Khasa, R.; Deb, A.; Prajapat, S.K.; Mallick, S.; Basu, A.; Surjit, M.; Kalia, M.; Vrati, S. Valosin-containing protein/p97 plays critical roles in the Japanese encephalitis virus life cycle. J. Virol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cerikan, B.; Goellner, S.; Neufeldt, C.J.; Haselmann, U.; Mulder, K.; Chatel-Chaix, L.; Cortese, M.; Bartenschlager, R. A Non-Replicative Role of the 3′ Terminal Sequence of the Dengue Virus Genome in Membranous Replication Organelle Formation. Cell Rep. 2020, 32, 107859. [Google Scholar] [CrossRef] [PubMed]
- Goellner, S.; Cerikan, B.; Cortese, M.; Neufeldt, C.J.; Haselmann, U.; Bartenschlager, R. Replication-Independent Generation and Morphological Analysis of Flavivirus Replication Organelles. STAR Protoc. 2020, 1, 100173. [Google Scholar] [CrossRef] [PubMed]
- Fischl, W.; Bartenschlager, R. High-throughput screening using dengue virus reporter genomes. Methods Mol. Biol. 2013, 1030, 205–219. [Google Scholar] [CrossRef]
- Anderson, D.J.; Le Moigne, R.; Djakovic, S.; Kumar, B.; Rice, J.; Wong, S.; Wang, J.; Yao, B.; Valle, E.; Kiss von Soly, S.; et al. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 2015, 28, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Le Moigne, R.; Aftab, B.T.; Djakovic, S.; Dhimolea, E.; Valle, E.; Murnane, M.; King, E.M.; Soriano, F.; Menon, M.K.; Wu, Z.Y.; et al. The p97 Inhibitor CB-5083 Is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2375–2386. [Google Scholar] [CrossRef] [Green Version]
- Magnaghi, P.; D’Alessio, R.; Valsasina, B.; Avanzi, N.; Rizzi, S.; Asa, D.; Gasparri, F.; Cozzi, L.; Cucchi, U.; Orrenius, C.; et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 2013, 9, 548–556. [Google Scholar] [CrossRef]
- Zhou, H.J.; Wang, J.; Yao, B.; Wong, S.; Djakovic, S.; Kumar, B.; Rice, J.; Valle, E.; Soriano, F.; Menon, M.K.; et al. Discovery of a First-in-Class, Potent, Selective, and Orally Bioavailable Inhibitor of the p97 AAA ATPase (CB-5083). J. Med. Chem. 2015, 58, 9480–9497. [Google Scholar] [CrossRef] [Green Version]
- Tresse, E.; Salomons, F.A.; Vesa, J.; Bott, L.C.; Kimonis, V.; Yao, T.P.; Dantuma, N.P.; Taylor, J.P. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010, 6, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umareddy, I.; Chao, A.; Sampath, A.; Gu, F.; Vasudevan, S.G. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J. Gen. Virol. 2006, 87, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Kingsolver, M.B.; Huang, Z.; Hardy, R.W. Insect antiviral innate immunity: Pathways, effectors, and connections. J. Mol. Biol. 2013, 425, 4921–4936. [Google Scholar] [CrossRef] [Green Version]
- Scaturro, P.; Cortese, M.; Chatel-Chaix, L.; Fischl, W.; Bartenschlager, R. Dengue Virus Non-structural Protein 1 Modulates Infectious Particle Production via Interaction with the Structural Proteins. PLoS Pathog. 2015, 11, e1005277. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Xie, X.; Wang, Q.Y.; Dong, H.; Lee, M.Y.; Kang, C.; Yuan, Z.; Shi, P.Y. Characterization of dengue virus NS4A and NS4B protein interaction. J. Virol. 2015, 89, 3455–3470. [Google Scholar] [CrossRef] [Green Version]
- Scaturro, P.; Stukalov, A.; Haas, D.A.; Cortese, M.; Draganova, K.; Plaszczyca, A.; Bartenschlager, R.; Gotz, M.; Pichlmair, A. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature 2018, 561, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.H.; Schneider, W.M.; Rozen-Gagnon, K.; Miles, L.A.; Schuster, F.; Razooky, B.; Jacobson, E.; Wu, X.; Yi, S.; Rudin, C.M.; et al. TMEM41B Is a Pan-flavivirus Host Factor. Cell 2021, 184, 133–148.e120. [Google Scholar] [CrossRef]
- Tian, J.N.; Yang, C.C.; Chuang, C.K.; Tsai, M.H.; Wu, R.H.; Chen, C.T.; Yueh, A. A Dengue Virus Type 2 (DENV-2) NS4B-Interacting Host Factor, SERP1, Reduces DENV-2 Production by Suppressing Viral RNA Replication. Viruses 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Doroshow, J.H.; Parchment, R.; Moscow, J. NCI Experimental Therapeutics (NExT) Program May 8. Available online: https://deainfo.nci.nih.gov/advisory/fac/0518/Doroshow.pdf. (accessed on 1 July 2021).
- Huryn, D.M.; Kornfilt, D.J.P.; Wipf, P. p97: An Emerging Target for Cancer, Neurodegenerative Diseases, and Viral Infections. J. Med. Chem. 2019. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazeaud, C.; Anton, A.; Pahmeier, F.; Sow, A.A.; Cerikan, B.; Freppel, W.; Cortese, M.; Bartenschlager, R.; Chatel-Chaix, L. The Biogenesis of Dengue Virus Replication Organelles Requires the ATPase Activity of Valosin-Containing Protein. Viruses 2021, 13, 2092. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102092
Mazeaud C, Anton A, Pahmeier F, Sow AA, Cerikan B, Freppel W, Cortese M, Bartenschlager R, Chatel-Chaix L. The Biogenesis of Dengue Virus Replication Organelles Requires the ATPase Activity of Valosin-Containing Protein. Viruses. 2021; 13(10):2092. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102092
Chicago/Turabian StyleMazeaud, Clément, Anaïs Anton, Felix Pahmeier, Aïssatou Aïcha Sow, Berati Cerikan, Wesley Freppel, Mirko Cortese, Ralf Bartenschlager, and Laurent Chatel-Chaix. 2021. "The Biogenesis of Dengue Virus Replication Organelles Requires the ATPase Activity of Valosin-Containing Protein" Viruses 13, no. 10: 2092. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102092