
Comparison of PCR versus PCR-Free DNA Library Preparation for Characterising the Human Faecal Virome

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Faecal Sample Collection and Storage
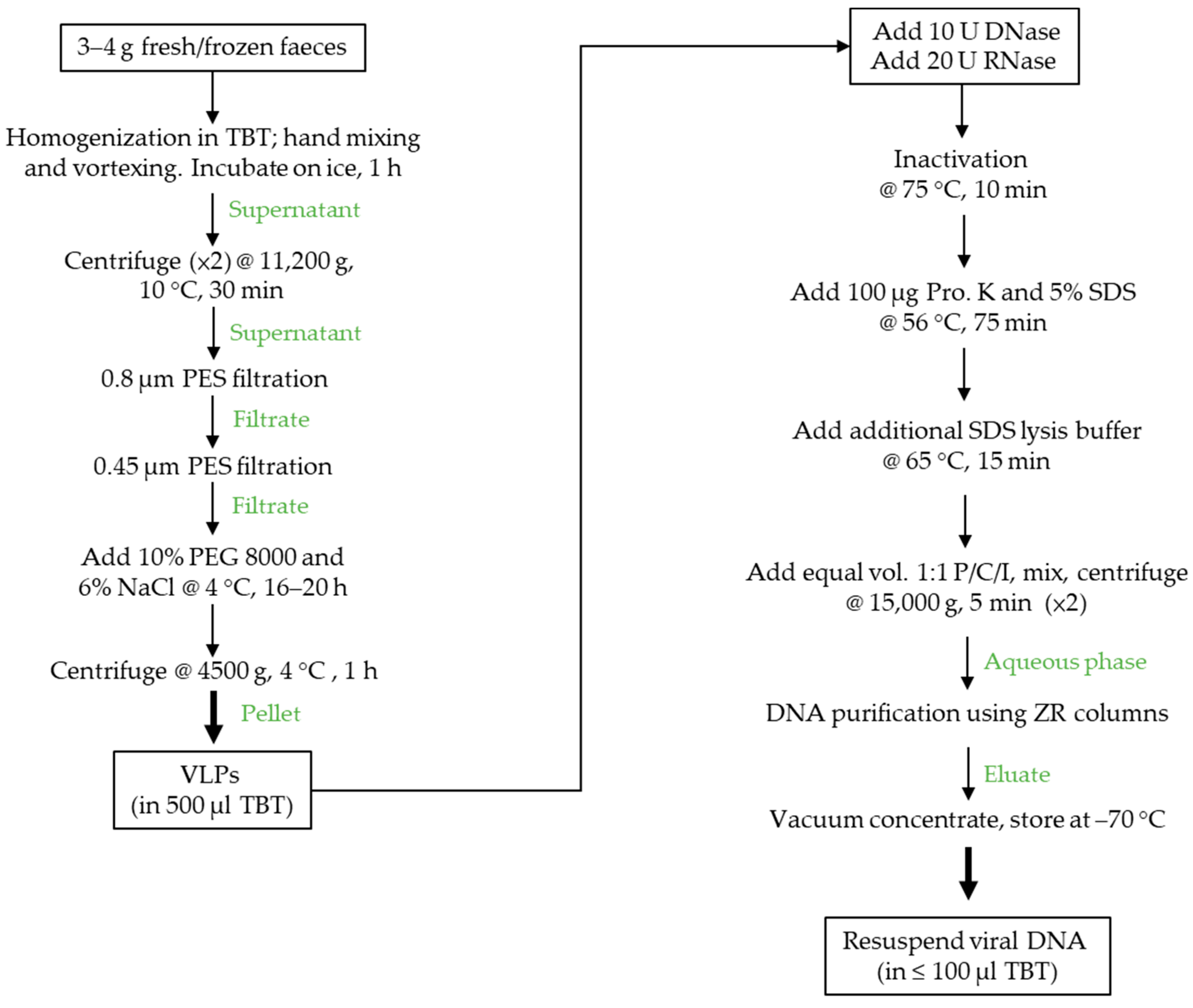
2.2. Faecal VLP and VLP DNA Isolation
2.3. Bacterial Culture and Preparation of Phage Stock
2.4. Plaque Assay-Based Phage Spiking and Recovery
2.5. Transmission Electron Microscopy (TEM)
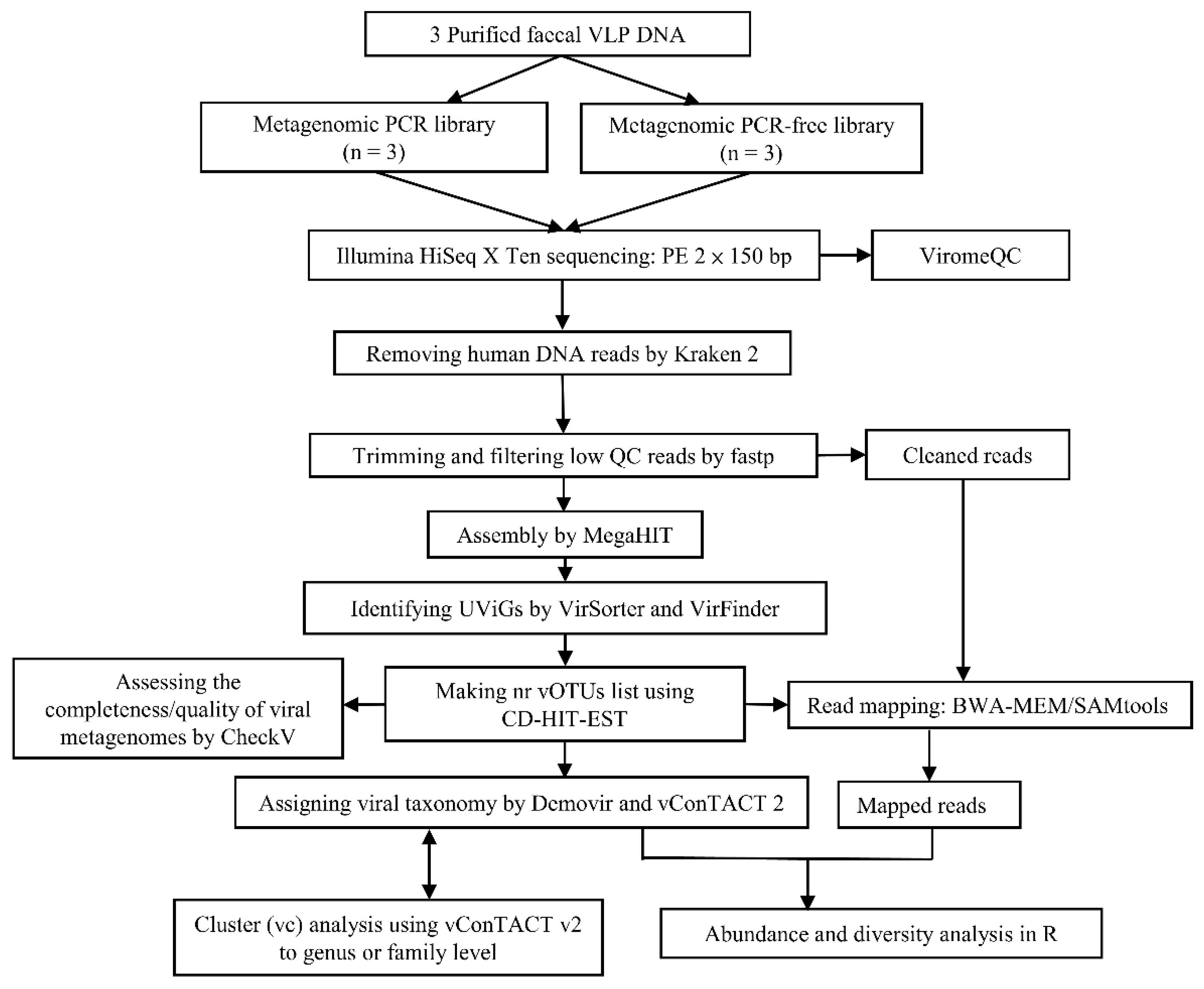
2.6. Library Preparation and Shotgun Metagenomic Sequencing
2.7. Genome Assembly and Viral Genome Detection
2.8. Read Mapping, Cluster Analysis and Taxonomic Annotation
2.9. Analysis of Relative Abundance, Alpha and Beta Diversity and Statistical Analysis
3. Results
3.1. VLP and DNA Extraction Protocol
3.2. Transmission Electron Microscopy (TEM)
3.3. Viromics Pipeline
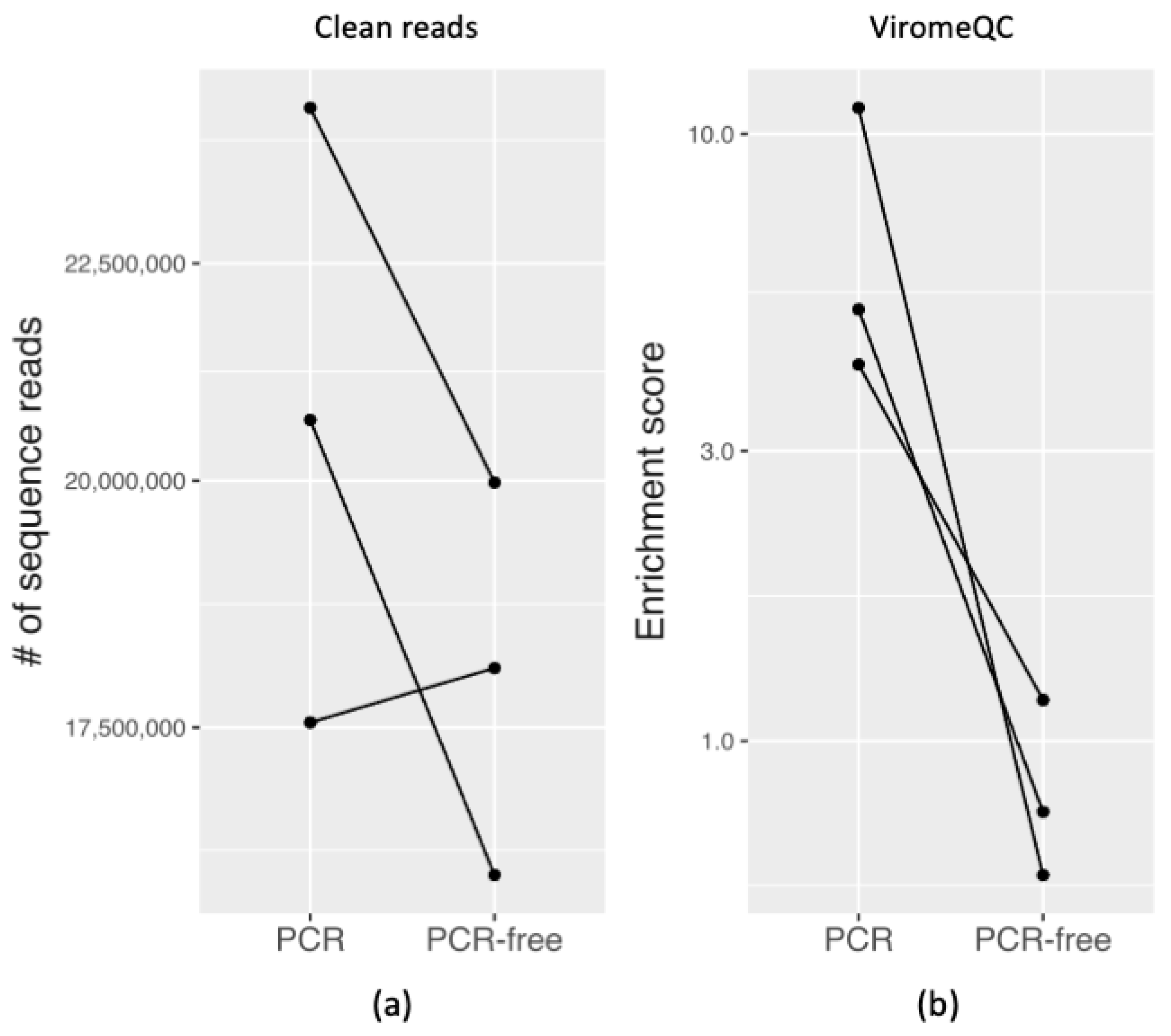
3.3.1. Quality and Quantity of Raw Sequencing Output
3.3.2. Evaluating VLP Enrichment
3.3.3. Quality and Quantity of Assembled Contigs
3.3.4. Identifying Putative Viruses
3.3.5. Read Mapping
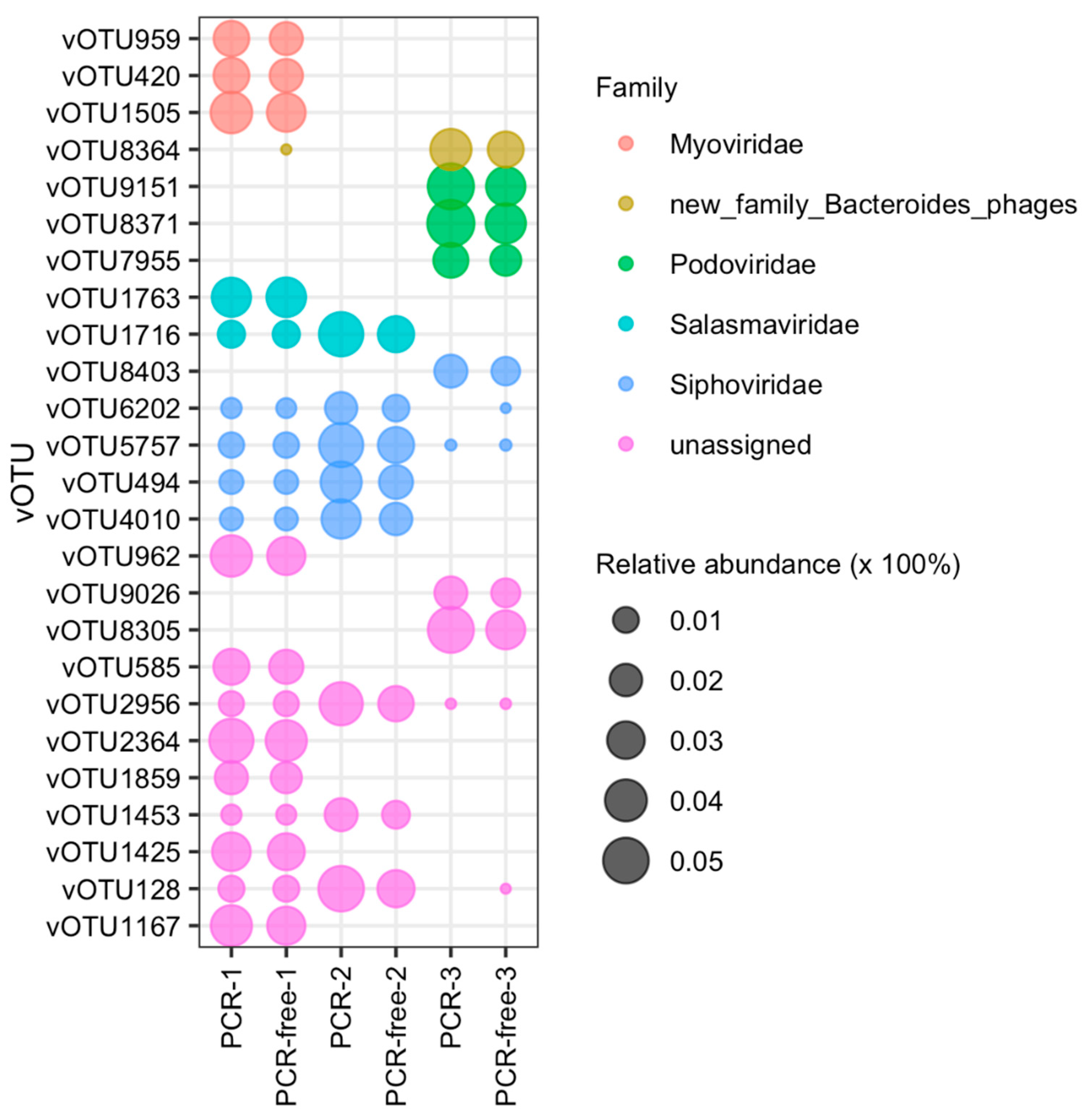
3.4. Comparing Virome-Derived PCR and PCR-Free Datasets: Relative Abundance Analysis
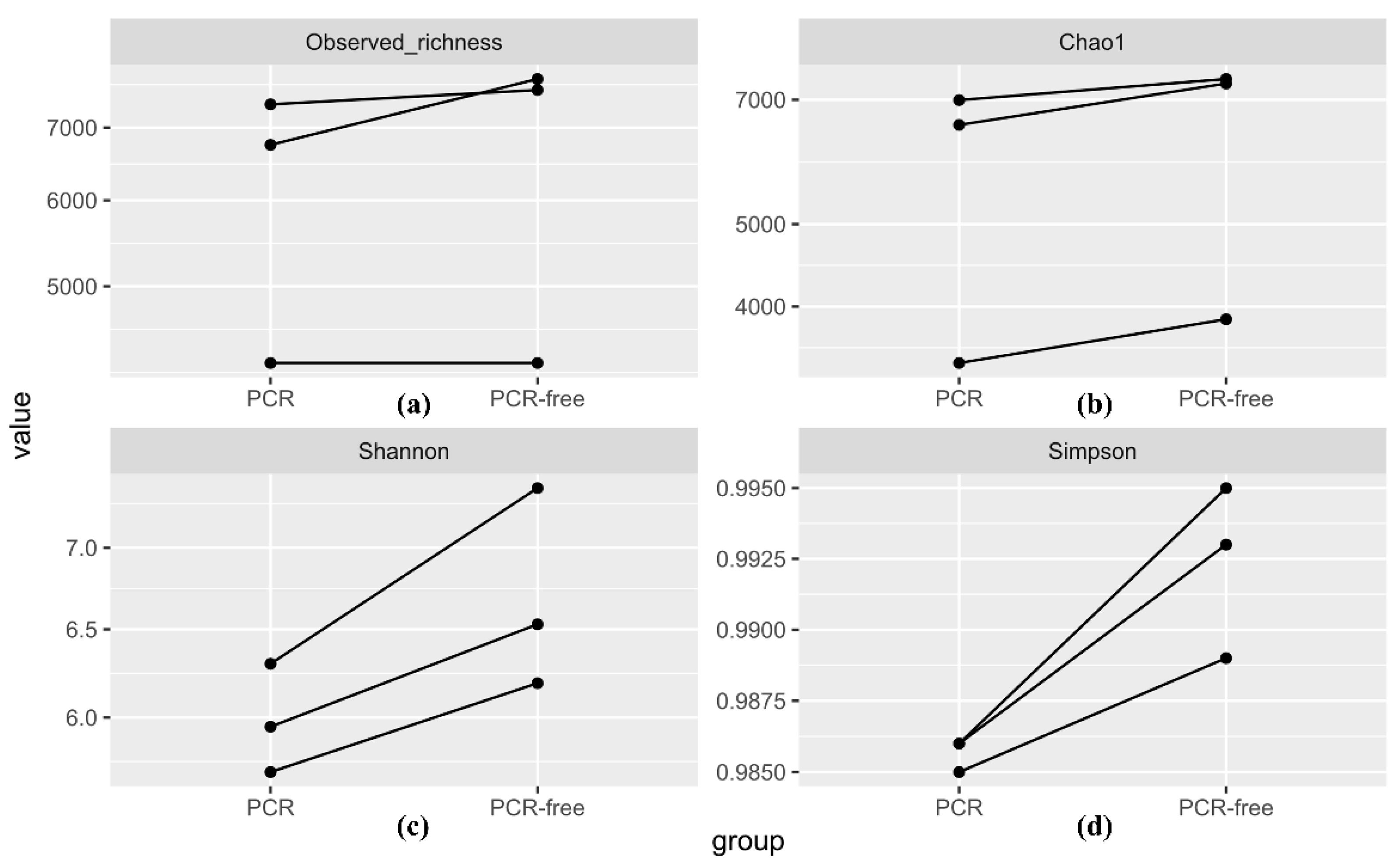
3.4.1. Alpha Diversity Analysis
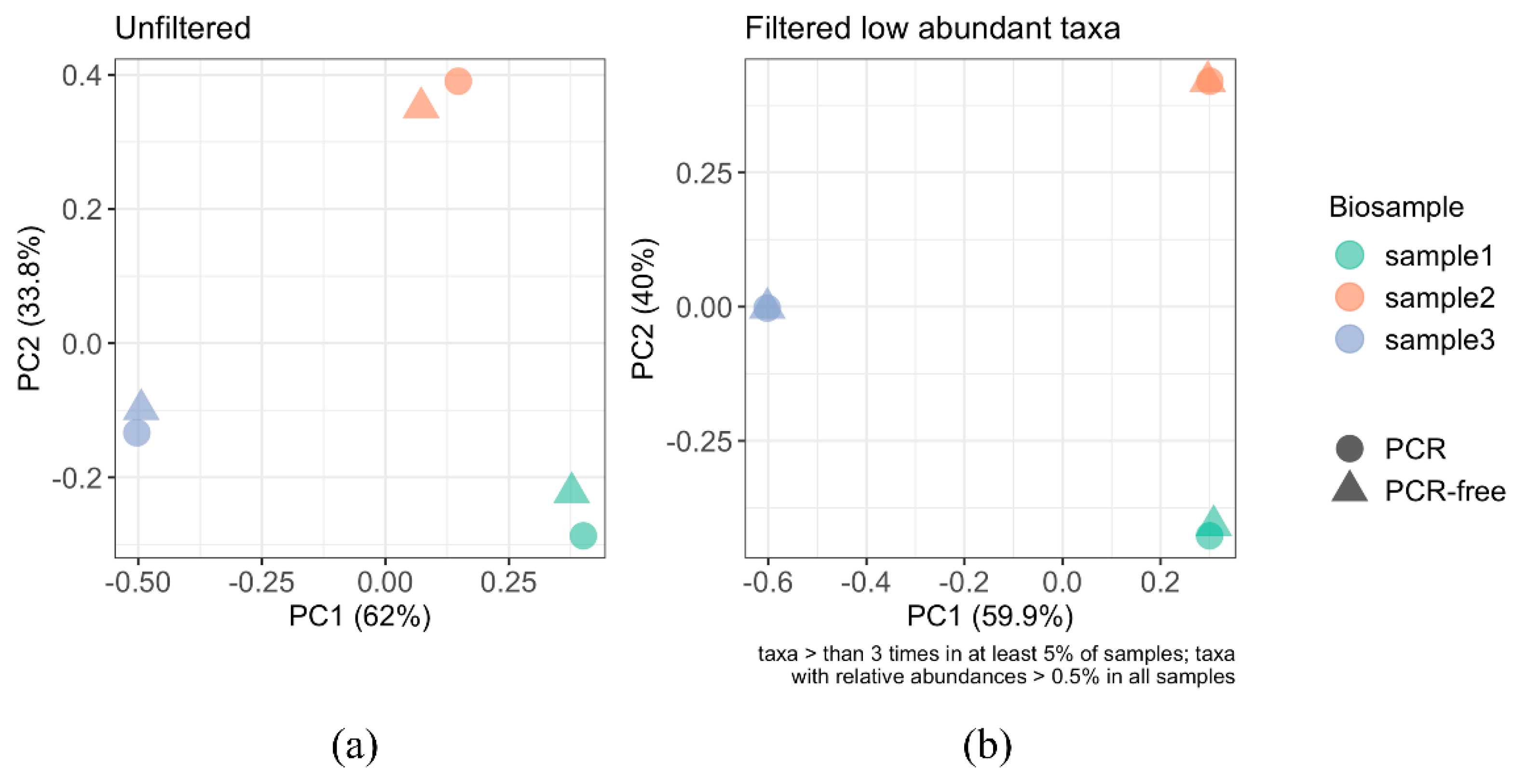
3.4.2. Beta Diversity Analysis
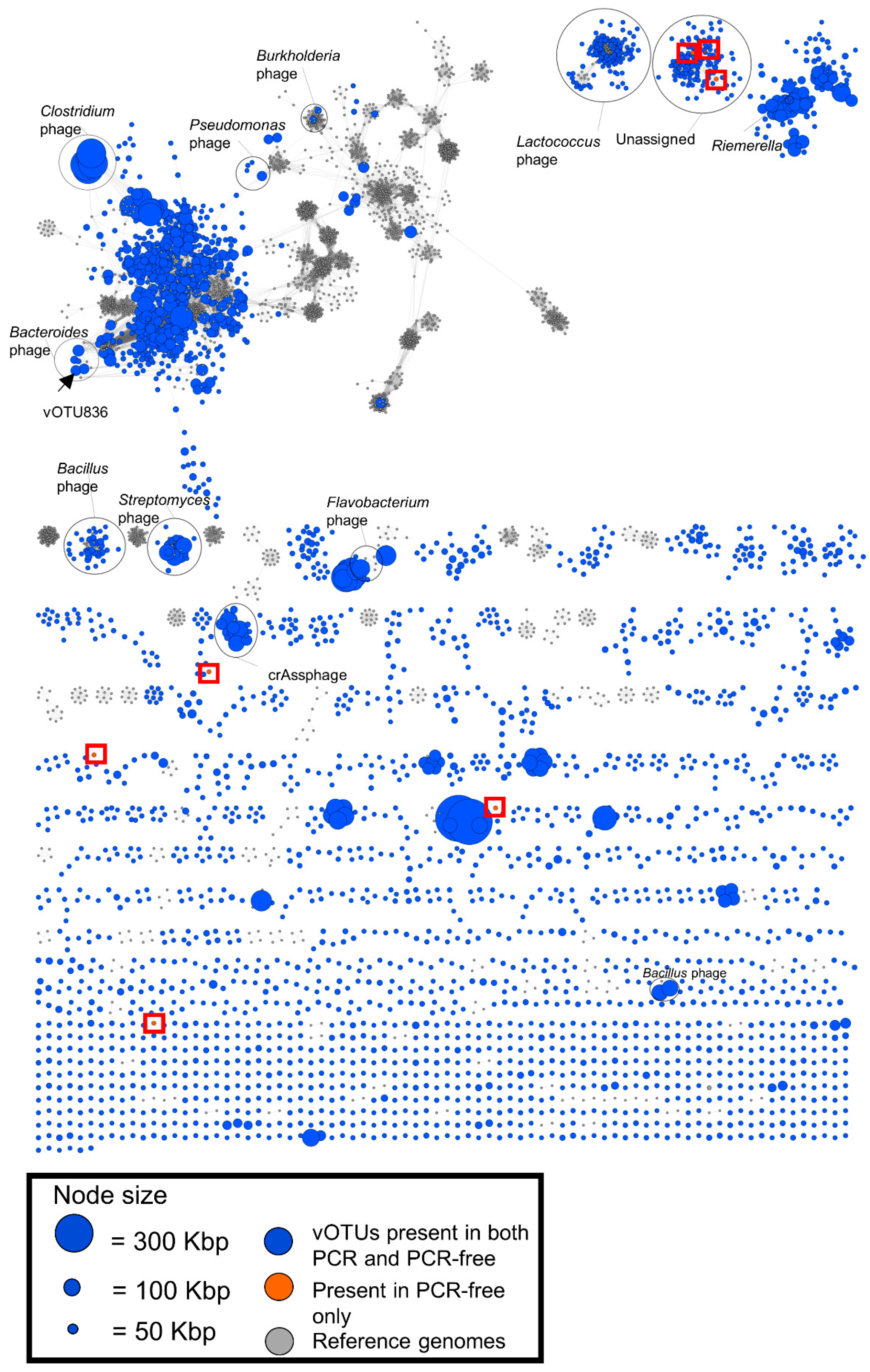
3.5. Viral Clustering and Taxonomy: Sequence Similarity Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Belkaid, Y.; Harrison, O.J. Homeostatic Immunity and the Microbiota. Immunity 2017, 46, 562–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Camarillo-Guerrero, L.F.; Almeida, A.; Rangel-Pineros, G.; Finn, R.D.; Lawley, T.D. Massive expansion of human gut bacteriophage diversity. Cell 2021, 184, 1098–1109. [Google Scholar] [CrossRef]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment. Pharm. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740.e8. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.; et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 2016, 19, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.Y.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Harkonen, T.; Hamalainen, A.M.; et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, M.A.; Verstraete, S.G.; Phan, T.G.; Deng, X.T.; Stekol, E.; LaMere, B.; Lynch, S.V.; Heyman, M.B.; Delwart, E. Enteric Virome and Bacterial Microbiota in Children with Ulcerative Colitis and Crohn Disease. J. Pediatr. Gastr Nutr 2019, 68, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, L.; McCartney, A.L.; Neve, H.; Gibson, G.R.; Sanderson, J.D.; Heller, K.J.; van Sinderen, D. Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol. 2014, 165, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Park, E.J.; Roh, S.W.; Bae, J.W. Diversity and Abundance of Single-Stranded DNA Viruses in Human Feces. Appl. Environ. Microb. 2011, 77, 8062–8070. [Google Scholar] [CrossRef] [Green Version]
- Conceicao-Neto, N.; Zeller, M.; Lefrere, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef] [Green Version]
- Marquet, M.; Hölzer, M.; Pletz, M.W.; Viehweger, A.; Makarewicz, O.; Ehricht, R.; Brandt, C. What the Phage: A scalable workflow for the identification and analysis of phage sequences. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sutton, T.D.S.; Clooney, A.G.; Ryan, F.J.; Ross, R.P.; Hill, C. Choice of assembly software has a critical impact on virome characterisation. Microbiome 2019, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Garmaeva, S.; Sinha, T.; Kurilshikov, A.; Fu, J.Y.; Wijmenga, C.; Zhernakova, A. Studying the gut virome in the metagenomic era: Challenges and perspectives. BMC Biol. 2019, 17, 84. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.M.; Clum, A.; Tice, H.; Lim, J.; Singh, K.; Ciobanu, D.; Ngan, C.Y.; Cheng, J.F.; Tringe, S.G.; Woyke, T. Impact of library preparation protocols and template quantity on the metagenomic reconstruction of a mock microbial community. BMC Genom. 2015, 16, 865. [Google Scholar] [CrossRef] [Green Version]
- Marine, R.; McCarren, C.; Vorrasane, V.; Nasko, D.; Crowgey, E.; Polson, S.W.; Wommack, K.E. Caught in the middle with multiple displacement amplification: The myth of pooling for avoiding multiple displacement amplification bias in a metagenome. Microbiome 2014, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Trubl, G.; Goudeau, D.; Nath, N.; Couradeau, E.; Ahlgren, N.A.; Zhan, Y.C.; Marsan, D.; Chen, F.; Fuhrman, J.A.; et al. Optimizing de novo genome assembly from PCR-amplified metagenomes. PeerJ 2019, 7, e6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parras-Molto, M.; Rodriguez-Galet, A.; Suarez-Rodriguez, P.; Lopez-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6, 119. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Ryan, F.J.; Draper, L.A.; Forde, A.; Stockdale, S.R.; Daly, K.M.; McDonnell, S.A.; Nolan, J.A.; Sutton, T.D.S.; Dalmasso, M.; et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 2018, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Ebdon, J.; Maite, M.; Taylor, H. The application of a recently isolated strain of Bacteroides (GB-124) to identify human sources of faecal pollution in a temperate river catchment. Water Res. 2007, 41, 3683–3690. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, L.A.; Caplin, J.; Dedi, C.; Diston, D.; Cheek, E.; Bowler, L.; Taylor, H.; Ebdon, J.; Jones, B.V. Comparative (Meta)genomic Analysis and Ecological Profiling of Human Gut-Specific Bacteriophage phi B124-14. PLoS ONE 2012, 7, e35053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Rio Deiros, D.; Pedersen, B.; Svensson, V. readfq. Available online: https://github.com/lh3/readfq (accessed on 1 August 2020).
- BGI-Zhenshen. FxTools. Available online: https://github.com/BGI-shenzhen/FxTools (accessed on 1 August 2020).
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Zolfo, M.; Pinto, F.; Asnicar, F.; Manghi, P.; Tett, A.; Bushman, F.D.; Segata, N. Detecting contamination in viromes using ViromeQC. Nat. Biotechnol. 2019, 37, 1408–1412. [Google Scholar] [CrossRef]
- Li, D.H.; Liu, C.M.; Luo, R.B.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F.Z. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W.; Wu, S.T.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Z.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef] [Green Version]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Feargalr. Demovir: Taxonomic Classification of Viruses at Order and Family Level. Available online: https://github.com/feargalr/Demovir (accessed on 20 August 2020).
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; Garcia, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Duhaime, M.B.; Sullivan, M.B. Ocean viruses: Rigorously evaluating the metagenomic sample-to-sequence pipeline. Virology 2012, 434, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solonenko, S.A.; Ignacio-Espinoza, J.C.; Alberti, A.; Cruaud, C.; Hallam, S.; Konstantinidis, K.; Tyson, G.; Wincker, P.; Sullivan, M.B. Sequencing platform and library preparation choices impact viral metagenomes. BMC Genom. 2013, 14, 320. [Google Scholar] [CrossRef] [Green Version]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [Green Version]
- Tisza, M.J.; Pastrana, D.V.; Welch, N.L.; Stewart, B.; Peretti, A.; Starrett, G.J.; Pang, Y.Y.S.; Krishnamurthy, S.R.; Pesavento, P.A.; McDermott, D.H.; et al. Discovery of several thousand highly diverse circular DNA viruses. eLife 2020, 9, e51971. [Google Scholar] [CrossRef]
- Chiang, Y.N.; Penades, J.R.; Chen, J. Genetic transduction by phages and chromosomal islands: The new and noncanonical. PLoS Pathog. 2019, 15, e1007878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez-Espino, D.; Chen, I.M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Szeto, E.; Pillay, M.; Huang, J.H.; Markowitz, V.M.; Nielsen, T.; et al. IMG/VR: A database of cultured and uncultured DNA Viruses and retroviruses. Nucleic Acids Res. 2017, 45, D457–D465. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Paez-Espino, D.; Chen, I.M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.B.K.; Nayfach, S.; Schulz, F.; Call, L.; et al. IMG/VR v3: An integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021, 49, D764–D775. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. Msphere 2018, 3, e00069-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. Phi CrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [Green Version]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Budinoff, C.R.; Loar, S.N.; LeCleir, G.R.; Wilhelm, S.W.; Buchan, A. A protocol for enumeration of aquatic viruses by epifluorescence microscopy using Anodisc (TM) 13 membranes. BMC Microbiol. 2011, 11, 168. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor # | PCR Library | PCR-Free Library | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | |
| # Raw reads | 17,618,631 | 20,715,201 | 24,640,272 | 18,647,093 | 16,540,559 | 20,897,792 |
| # Cleaned reads | 17,547,247 | 20,667,351 | 24,478,660 | 18,072,149 | 16,150,779 | 19,979,137 |
| Q30 (%) | 90.29 | 92.47 | 92.22 | 93.62 | 90.91 | 90.72 |
| % GC | 36.05 | 46.89 | 38.48 | 36.79 | 46.71 | 39.42 |
| # Contigs | 73,246 | 103,220 | 32,063 | 100,493 | 131,354 | 48,194 |
| # Contigs ≥ 1 kbp | 14,469 | 17,952 | 4427 | 17,951 | 26,414 | 6342 |
| N50 (bp) | 2011 | 2515 | 7049 | 2019 | 2331 | 2590 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, S.-Y.; Tariq, M.A.; Telatin, A.; Ansorge, R.; Adriaenssens, E.M.; Savva, G.M.; Booth, C.; Wileman, T.; Hoyles, L.; Carding, S.R. Comparison of PCR versus PCR-Free DNA Library Preparation for Characterising the Human Faecal Virome. Viruses 2021, 13, 2093. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102093
Hsieh S-Y, Tariq MA, Telatin A, Ansorge R, Adriaenssens EM, Savva GM, Booth C, Wileman T, Hoyles L, Carding SR. Comparison of PCR versus PCR-Free DNA Library Preparation for Characterising the Human Faecal Virome. Viruses. 2021; 13(10):2093. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102093
Chicago/Turabian StyleHsieh, Shen-Yuan, Mohammad A. Tariq, Andrea Telatin, Rebecca Ansorge, Evelien M. Adriaenssens, George M. Savva, Catherine Booth, Tom Wileman, Lesley Hoyles, and Simon R. Carding. 2021. "Comparison of PCR versus PCR-Free DNA Library Preparation for Characterising the Human Faecal Virome" Viruses 13, no. 10: 2093. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102093