
The HIV-1 Nucleocapsid Regulates Its Own Condensation by Phase-Separated Activity-Enhancing Sequestration of the Viral Protease during Maturation

 , , , , ,
, , , , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Proteins, Nucleic Acids, and Reagents
2.2. NP Complex Assembly and Electrophoretic Mobility Shift Assay
2.3. Dynamic Light Scattering
2.4. AFM Imaging
2.5. Proteolysis Assays
2.6. Electron Microscopy of HIV-1 Particles
2.7. Molecular Dynamics Simulations and Analysis
3. Results
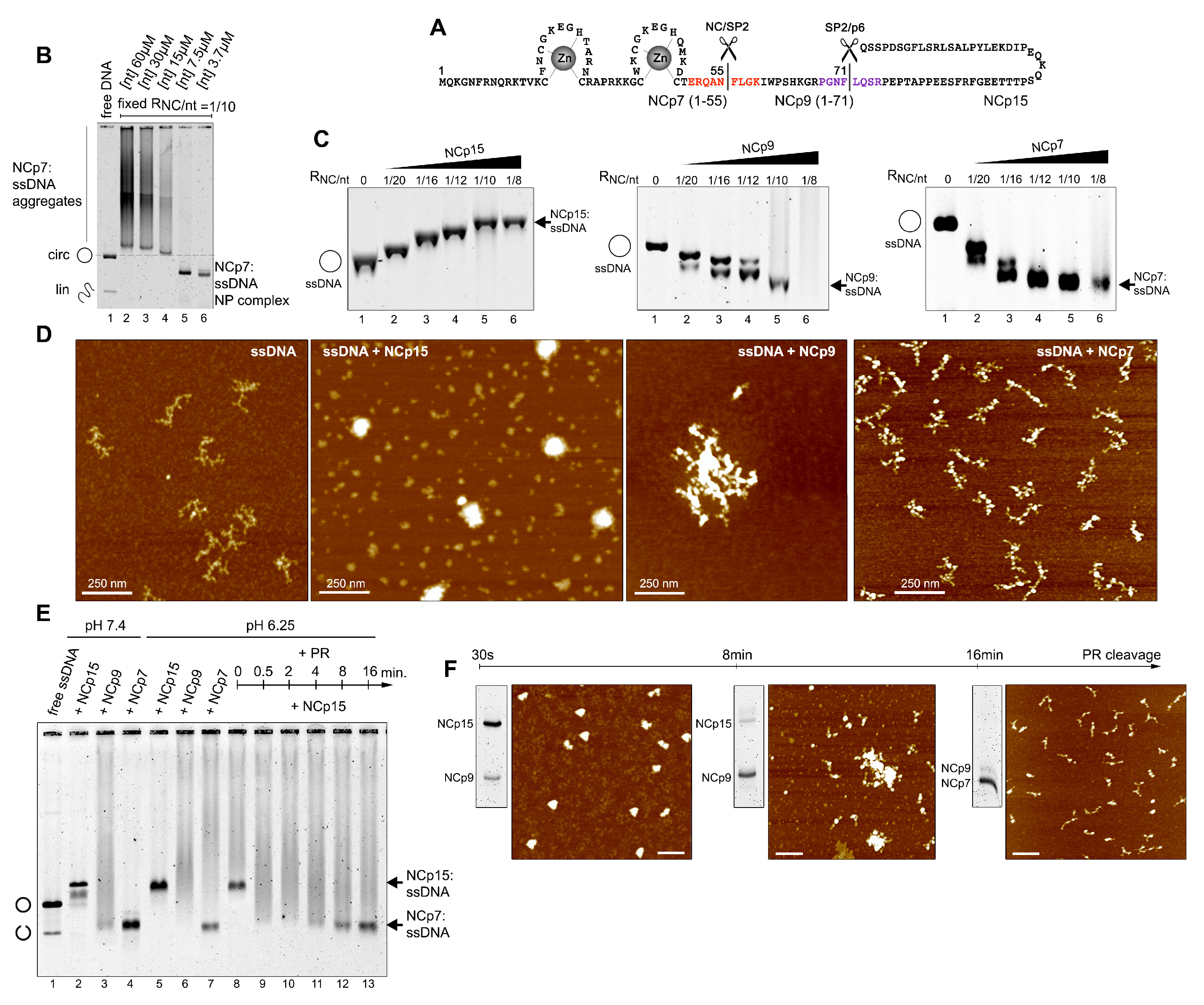
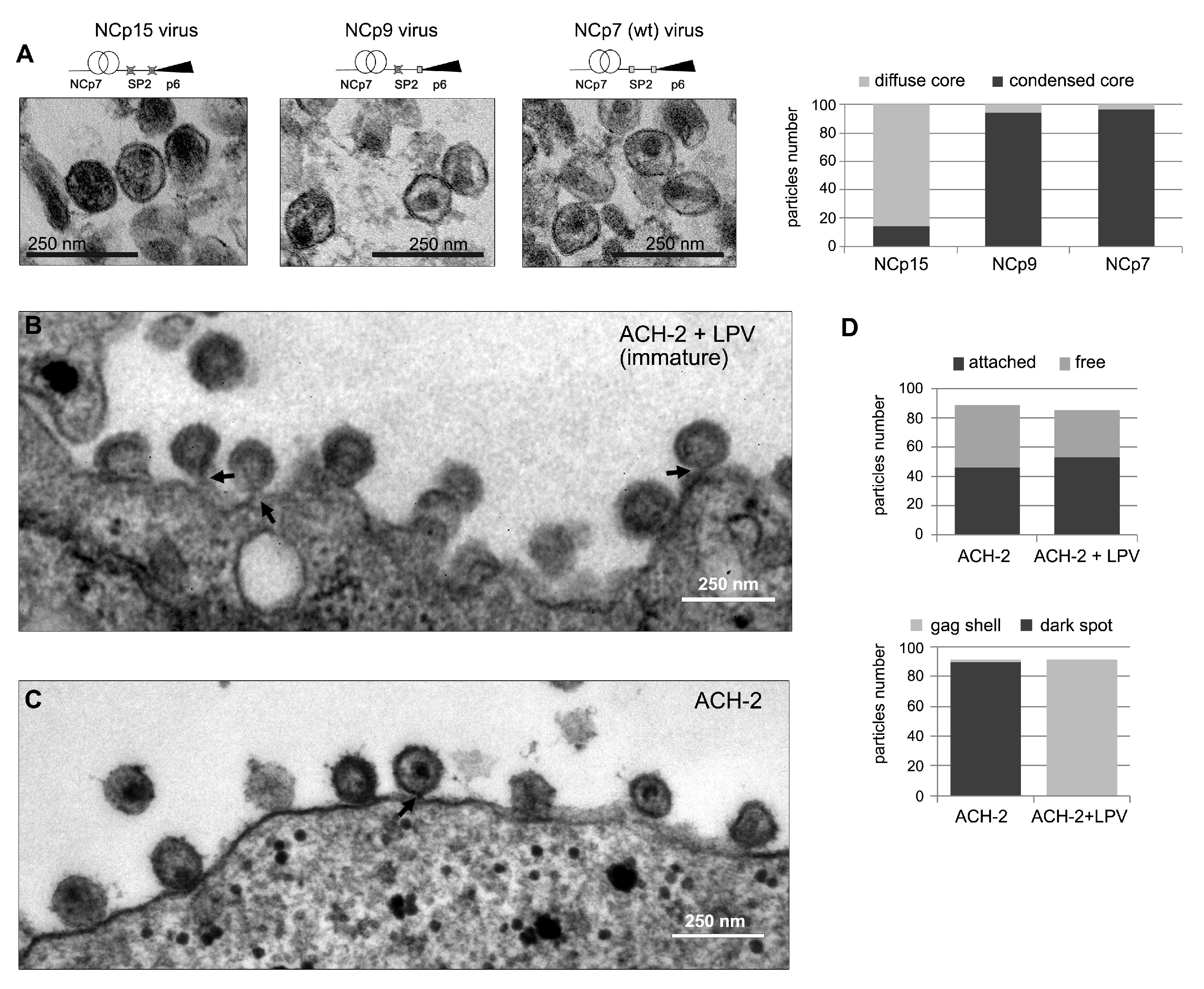
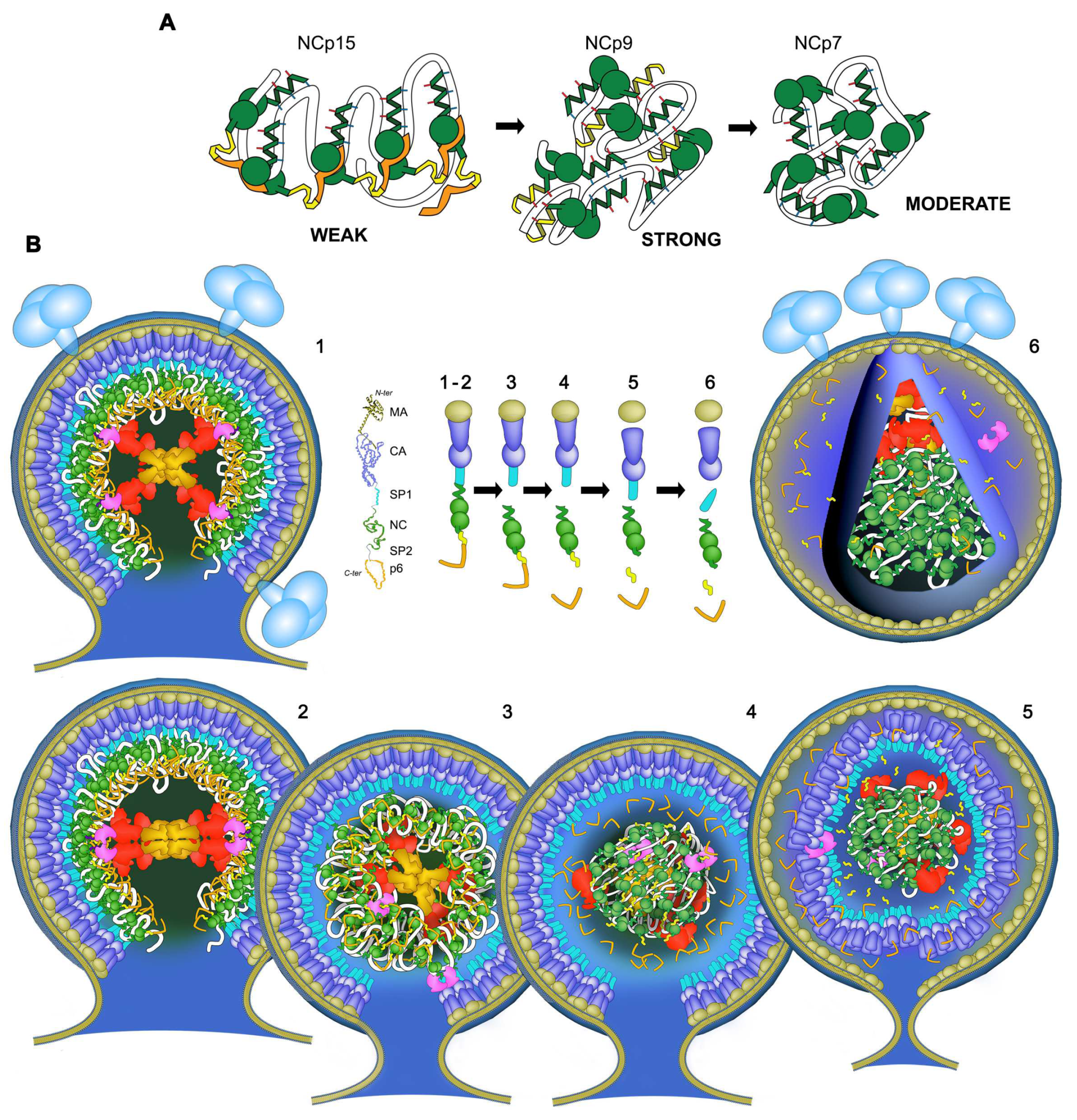
3.1. Cleavage of NCp15 to NCp9 and NCp7 Underpins Weak-Strong-Moderate Quinary Condensate Properties
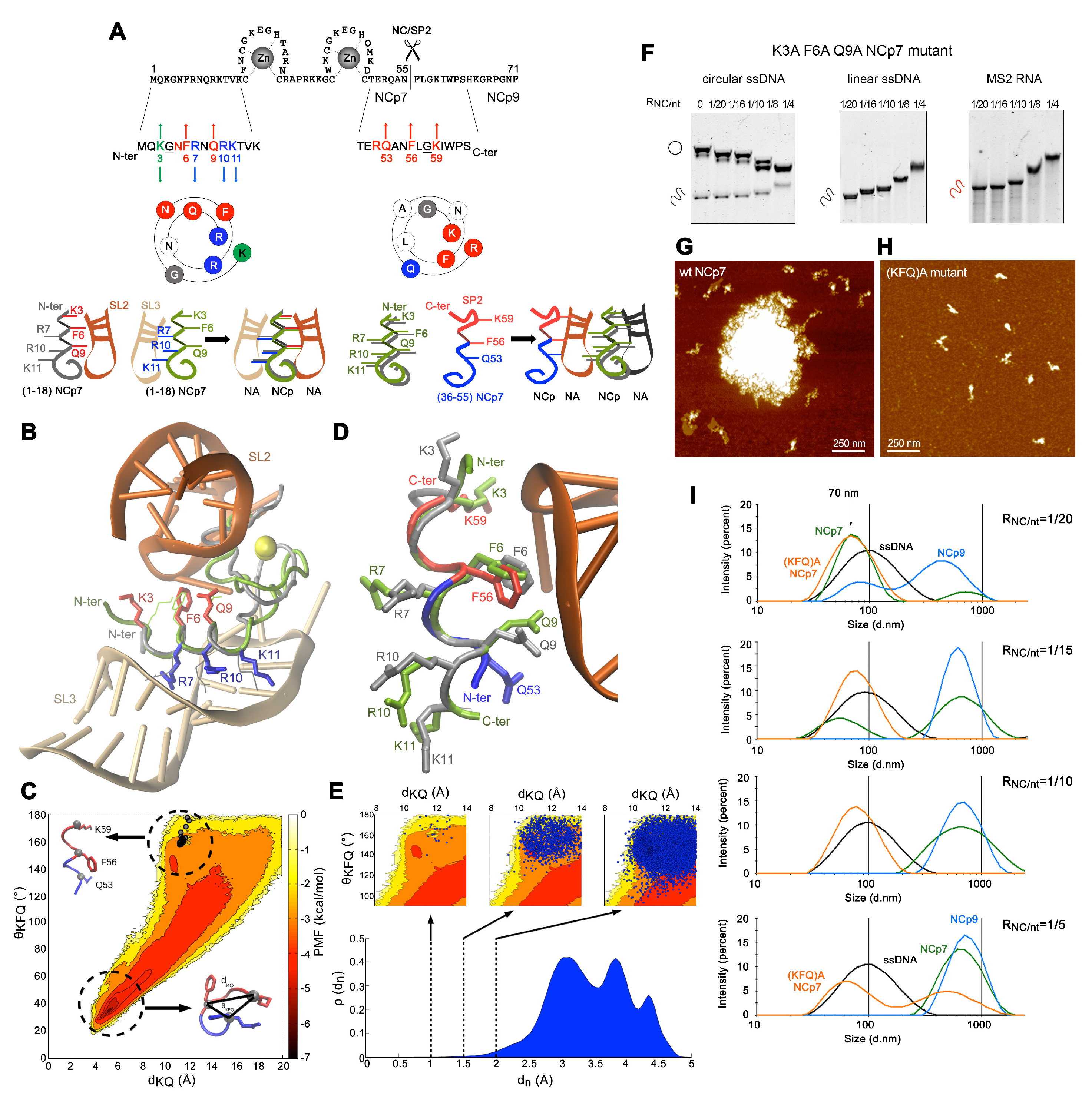
3.2. Transiently Unmasked NC Binding Sites Enable Modulation of NC:NA Molecular Interactions
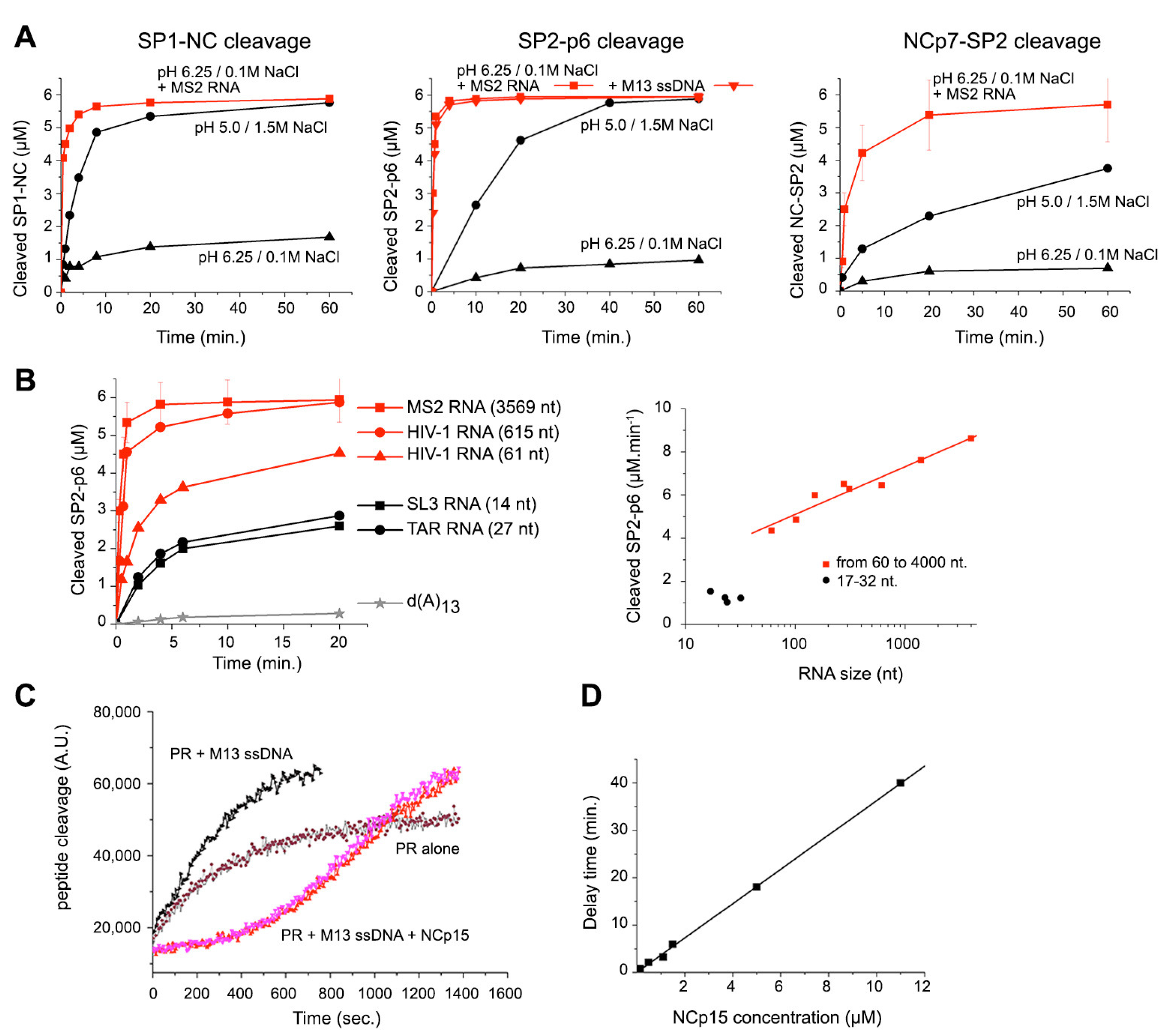
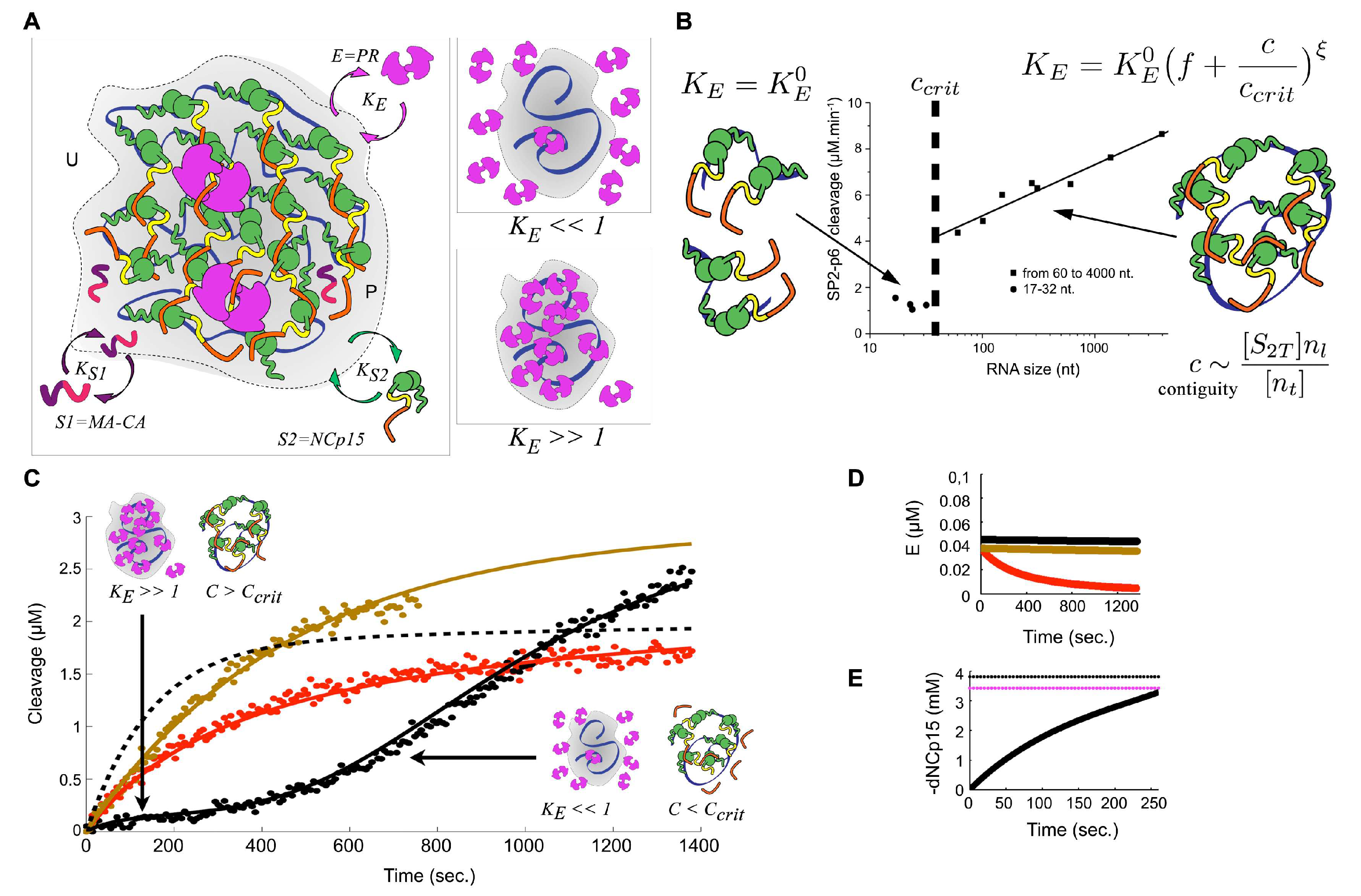
3.3. Quinary Cooperation between NC and RNA Drives PR Sequestration and RNA-Length-Dependent Catalytic Acceleration
3.4. Condensate-Driven Accelerated PR Processing Temporally Couples Budding to Maturation
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HIV | Human immunodeficiency virus |
| RNA | Ribonucleic acid |
References
- André, A.A.; Spruijt, E. Liquid–liquid phase separation in crowded environments. Int. J. Mol. Sci. 2020, 21, 5908. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Nozawa, R.S.; Jia, T.Z.; Saio, T.; Mori, E. Biological phase separation: Cell biology meets biophysics. Biophys. Rev. 2020, 12, 519–539. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Chen, X.; Wu, X.; Zhang, M. Formation of biological condensates via phase separation: Characteristics, analytical methods, and physiological implications. J. Biol. Chem. 2019, 294, 14823–14835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, E.W.; Kear-Scott, J.L.; Pilipenko, E.V.; Schwartz, M.H.; Laskowski, P.R.; Rojek, A.E.; Katanski, C.D.; Riback, J.A.; Dion, M.F.; Franks, A.M.; et al. Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 2015, 162, 1286–1298. [Google Scholar] [CrossRef] [Green Version]
- Franzmann, T.M.; Alberti, S. Protein phase separation as a stress survival strategy. Cold Spring Harb. Perspect. Biol. 2019, 11, a034058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panas, M.D.; Ivanov, P.; Anderson, P. Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 2016, 215, 313–323. [Google Scholar] [CrossRef]
- Riback, J.A.; Katanski, C.D.; Kear-Scott, J.L.; Pilipenko, E.V.; Rojek, A.E.; Sosnick, T.R.; Drummond, D.A. Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell 2017, 168, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef]
- Cai, D.; Liu, Z.; Lippincott-Schwartz, J. Biomolecular condensates and their links to cancer progression. Trends Biochem. Sci. 2021, 46, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.K.; Vibhute, M.A.; Spruijt, E. Biomolecular chemistry in liquid phase separated compartments. Front. Mol. Biosci. 2019, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Flynn, B.G.; Mittag, T. The role of liquid–liquid phase separation in regulating enzyme activity. Curr. Opin. Cell Biol. 2021, 69, 70–79. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, G.; Mekhail, K. Biomolecular condensates as arbiters of biochemical reactions inside the nucleus. Commun. Biol. 2020, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Narlikar, G.J.; Kutateladze, T.G. Enzymatic reactions inside biological condensates. J. Mol. Biol. 2021, 433, 166624. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R. mRNP granules: Assembly, function, and connections with disease. RNA Biol. 2014, 11, 1019–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 2021, 22, 183–195. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing protein-RNA interaction networks control multiphase intracellular organization. Cell 2020, 181, 306–324. [Google Scholar] [CrossRef] [PubMed]
- Sanulli, S.; Narlikar, G.J. Generation and biochemical characterization of phase-separated droplets formed by nucleic acid binding proteins: Using HP1 as a model system. Curr. Protoc. 2021, 1, e109. [Google Scholar] [CrossRef]
- Luo, J.; Qu, L.; Gao, F.; Lin, J.; Liu, J.; Lin, A. LncRNAs: Architectural scaffolds or more potential roles in phase separation. Front. Genet. 2021, 12, 369. [Google Scholar] [CrossRef]
- Louka, A.; Zacco, E.; Temussi, P.A.; Tartaglia, G.G.; Pastore, A. RNA as the stone guest of protein aggregation. Nucleic Acids Res. 2020, 48, 11880–11889. [Google Scholar] [CrossRef] [PubMed]
- Wiedner, H.J.; Giudice, J. It’s not just a phase: Function and characteristics of RNA-binding proteins in phase separation. Nat. Struct. Mol. Biol. 2021, 28, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Gotor, N.L.; Armaos, A.; Calloni, G.; Torrent Burgas, M.; Vabulas, R.M.; De Groot, N.S.; Tartaglia, G.G. RNA-binding and prion domains: The Yin and Yang of phase separation. Nucleic Acids Res. 2020, 48, 9491–9504. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Shorter, J. It’s raining liquids: RNA tunes viscoelasticity and dynamics of membraneless organelles. Mol. Cell 2015, 60, 189–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodruff, J.B.; Hyman, A.A.; Boke, E. Organization and function of non-dynamic biomolecular condensates. Trends Biochem. Sci. 2018, 43, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, P.; Gierasch, L.M. Challenges and dreams: Physics of weak interactions essential to life. Mol. Biol. Cell 2014, 25, 3474–3477. [Google Scholar] [CrossRef] [Green Version]
- McConkey, E.H. Molecular evolution, intracellular organization, and the quinary structure of proteins. Proc. Natl. Acad. Sci. USA 1982, 79, 3236–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteith, W.B.; Cohen, R.D.; Smith, A.E.; Guzman-Cisneros, E.; Pielak, G.J. Quinary structure modulates protein stability in cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1739–1742. [Google Scholar] [CrossRef] [Green Version]
- Guin, D.; Gruebele, M. Weak chemical interactions that drive protein evolution: Crowding, sticking, and quinary structure in folding and function. Chem. Rev. 2019, 119, 10691–10717. [Google Scholar] [CrossRef]
- Rickard, M.M.; Zhang, Y.; Gruebele, M.; Pogorelov, T.V. In-cell protein–protein contacts: Transient interactions in the crowd. J. Phys. Chem. Lett. 2019, 10, 5667–5673. [Google Scholar] [CrossRef] [PubMed]
- Gopi, S.; Naganathan, A.N. Non-specific DNA-driven quinary interactions promote structural transitions in proteins. Phys. Chem. Chem. Phys. 2020, 22, 12671–12677. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.J.; Mallinson, S.J.; John, P.C.S.; Bomble, Y.J. Advances in integrative structural biology: Towards understanding protein complexes in their cellular context. Comput. Struct. Biotechnol. J. 2020, 19, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Choi, J.M.; Holehouse, A.S.; Pappu, R.V. Physical principles underlying the complex biology of intracellular phase transitions. Annu. Rev. Biophys. 2020, 49, 107–133. [Google Scholar] [CrossRef] [Green Version]
- Dignon, G.L.; Best, R.B.; Mittal, J. Biomolecular phase separation: From molecular driving forces to macroscopic properties. Annu. Rev. Phys. Chem. 2020, 71, 53–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Mazarakos, K.; Zhou, H.X. Three archetypical classes of macromolecular regulators of protein liquid–liquid phase separation. Proc. Natl. Acad. Sci. USA 2019, 116, 19474–19483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.X.; Nguemaha, V.; Mazarakos, K.; Qin, S. Why do disordered and structured proteins behave differently in phase separation? Trends Biochem. Sci. 2018, 43, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Bratek-Skicki, A.; Pancsa, R.; Meszaros, B.; Van Lindt, J.; Tompa, P. A guide to regulation of the formation of biomolecular condensates. FEBS J. 2020, 287, 1924–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lindt, J.; Bratek-Skicki, A.; Nguyen, P.N.; Pakravan, D.; Durán-Armenta, L.F.; Tantos, A.; Pancsa, R.; Van Den Bosch, L.; Maes, D.; Tompa, P. A generic approach to study the kinetics of liquid–liquid phase separation under near-native conditions. Commun. Biol. 2021, 4, 77. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Burgos, I.; Espinosa, J.R.; Joseph, J.A.; Collepardo-Guevara, R. Valency and binding affinity variations can regulate the multilayered organization of protein condensates with many components. Biomolecules 2021, 11, 278. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, J.R.; Joseph, J.A.; Sanchez-Burgos, I.; Garaizar, A.; Frenkel, D.; Collepardo-Guevara, R. Liquid network connectivity regulates the stability and composition of biomolecular condensates with many components. Proc. Natl. Acad. Sci. USA 2020, 117, 13238–13247. [Google Scholar] [CrossRef]
- Dar, F.; Pappu, R. Phase separation: Restricting the sizes of condensates. eLife 2020, 9, e59663. [Google Scholar] [CrossRef] [PubMed]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef] [PubMed]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 2018, 173, 706–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qamar, S.; Wang, G.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Ströhl, F.; et al. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell 2018, 173, 720–734. [Google Scholar] [CrossRef] [Green Version]
- Etibor, T.A.; Yamauchi, Y.; Amorim, M.J. Liquid biomolecular condensates and viral lifecycles: Review and perspectives. Viruses 2021, 13, 366. [Google Scholar] [CrossRef] [PubMed]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid–liquid phase separation by intrinsically disordered protein regions of viruses: Roles in viral life cycle and control of virus–host interactions. Int. J. Mol. Sci. 2020, 21, 9045. [Google Scholar] [CrossRef] [PubMed]
- Iserman, C.; Roden, C.A.; Boerneke, M.A.; Sealfon, R.S.; McLaughlin, G.A.; Jungreis, I.; Fritch, E.J.; Hou, Y.J.; Ekena, J.; Weidmann, C.A.; et al. Genomic RNA elements drive phase separation of the SARS-CoV-2 nucleocapsid. Mol. Cell 2020, 80, 1078–1091. [Google Scholar] [CrossRef]
- Chen, H.; Cui, Y.; Han, X.; Hu, W.; Sun, M.; Zhang, Y.; Wang, P.H.; Song, G.; Chen, W.; Lou, J. Liquid–liquid phase separation by SARS-CoV-2 nucleocapsid protein and RNA. Cell Res. 2020, 30, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Savastano, A.; de Opakua, A.I.; Rankovic, M.; Zweckstetter, M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat. Commun. 2020, 11, 6041. [Google Scholar] [CrossRef] [PubMed]
- Perdikari, T.M.; Murthy, A.C.; Ryan, V.H.; Watters, S.; Naik, M.T.; Fawzi, N.L. SARS-CoV-2 nucleocapsid protein phase-separates with RNA and with human hnRNPs. EMBO J. 2020, 39, e106478. [Google Scholar] [CrossRef] [PubMed]
- Monette, A.; Niu, M.; Chen, L.; Rao, S.; Gorelick, R.J.; Mouland, A.J. Pan-retroviral nucleocapsid-mediated phase separation regulates genomic RNA positioning and trafficking. Cell Rep. 2020, 31, 107520. [Google Scholar] [CrossRef] [PubMed]
- Monette, A.; Mouland, A.J. Zinc and copper ions differentially regulate prion-like phase separation dynamics of pan-virus nucleocapsid biomolecular condensates. Viruses 2020, 12, 1179. [Google Scholar] [CrossRef] [PubMed]
- Mirambeau, G.; Lyonnais, S.; Coulaud, D.; Hameau, L.; Lafosse, S.; Jeusset, J.; Justome, A.; Delain, E.; Gorelick, R.J.; Le Cam, E. Transmission electron microscopy reveals an optimal HIV-1 nucleocapsid aggregation with single-stranded nucleic acids and the mature HIV-1 nucleocapsid protein. J. Mol. Biol. 2006, 364, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Mirambeau, G.; Lyonnais, S.; Coulaud, D.; Hameau, L.; Lafosse, S.; Jeusset, J.; Borde, I.; Reboud-Ravaux, M.; Restle, T.; Gorelick, R.J.; et al. HIV-1 protease and reverse transcriptase control the architecture of their nucleocapsid partner. PLoS ONE 2007, 2, e669. [Google Scholar] [CrossRef]
- Mirambeau, G.; Lyonnais, S.; Gorelick, R.J. Features, processing states, and heterologous protein interactions in the modulation of the retroviral nucleocapsid protein function. RNA Biol. 2010, 7, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Naiyer, N.; Mitra, M.; Li, J.; Williams, M.C.; Rouzina, I.; Gorelick, R.J.; Wu, Z.; Musier-Forsyth, K. Distinct nucleic acid interaction properties of HIV-1 nucleocapsid protein precursor NCp15 explain reduced viral infectivity. Nucleic Acids Res. 2014, 42, 7145–7159. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Kräusslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Konvalinka, J.; Kräusslich, H.G.; Müller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmukh, L.; Ghirlando, R.; Clore, G.M. Conformation and dynamics of the Gag polyprotein of the human immunodeficiency virus 1 studied by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 3374–3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potempa, M.; Nalivaika, E.; Ragland, D.; Lee, S.K.; Schiffer, C.A.; Swanstrom, R. A direct interaction with RNA dramatically enhances the catalytic activity of the HIV-1 protease in vitro. J. Mol. Biol. 2015, 427, 2360–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marco, A.; Heuser, A.M.; Glass, B.; Kräusslich, H.G.; Müller, B.; Briggs, J.A. Role of the SP2 domain and its proteolytic cleavage in HIV-1 structural maturation and infectivity. J. Virol. 2012, 86, 13708–13716. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Gorelick, R.J.; Levin, J.G. Selection of fully processed HIV-1 nucleocapsid protein is required for optimal nucleic acid chaperone activity in reverse transcription. Virus Res. 2014, 193, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, M.J.; Semo, N.; Freire, E. The structural stability of the HIV-1 protease. J. Mol. Biol. 1998, 283, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Pettit, S.C.; Sheng, N.; Tritch, R.; Erickson-Viitanen, S.; Swanstrom, R. The regulation of sequential processing of HIV-1 Gag by the viral protease. In Aspartic Proteinases; James, M.N.G., Ed.; Springer: Boston, MA, USA, 1998; pp. 15–25. [Google Scholar]
- Sheng, N.; Pettit, S.C.; Tritch, R.J.; Ozturk, D.H.; Rayner, M.M.; Swanstrom, R.; Erickson-Viitanen, S. Determinants of the human immunodeficiency virus type 1 p15NC-RNA interaction that affect enhanced cleavage by the viral protease. J. Virol. 1997, 71, 5723–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarasinghe, G.K.; De Guzman, R.N.; Turner, R.B.; Chancellor, K.J.; Wu, Z.R.; Summers, M.F. NMR structure of the HIV-1 nucleocapsid protein bound to stem-loop SL2 of the Ψ-RNA packaging signal. Implications for genome recognition. J. Mol. Biol. 2000, 301, 491–511. [Google Scholar] [CrossRef] [Green Version]
- De Guzman, R.N.; Wu, Z.R.; Stalling, C.C.; Pappalardo, L.; Borer, P.N.; Summers, M.F. Structure of the HIV-1 nucleocapsid protein bound to the SL3 Ψ-RNA recognition element. Science 1998, 279, 384–388. [Google Scholar] [CrossRef]
- Wu, H.; Mitra, M.; Naufer, M.N.; McCauley, M.J.; Gorelick, R.J.; Rouzina, I.; Musier-Forsyth, K.; Williams, M.C. Differential contribution of basic residues to HIV-1 nucleocapsid protein’s nucleic acid chaperone function and retroviral replication. Nucleic Acids Res. 2014, 42, 2525–2537. [Google Scholar] [CrossRef] [PubMed]
- Le Cam, E.; Coulaud, D.; Delain, E.; Petitjean, P.; Roques, B.P.; Gérard, D.; Stoylova, E.; Vuilleumier, C.; Stoylov, S.P.; Mély, Y. Properties and growth mechanism of the ordered aggregation of a model RNA by the HIV-1 nucleocapsid protein: An electron microscopy investigation. Biopolym. Orig. Res. Biomol. 1998, 45, 217–229. [Google Scholar] [CrossRef]
- Mouhand, A.; Pasi, M.; Catala, M.; Zargarian, L.; Belfetmi, A.; Barraud, P.; Mauffret, O.; Tisné, C. Overview of the nucleic-acid binding properties of the HIV-1 nucleocapsid protein in its different maturation states. Viruses 2020, 12, 1109. [Google Scholar] [CrossRef] [PubMed]
- Retureau, R.; Oguey, C.; Mauffret, O.; Hartmann, B. Structural explorations of NCp7–nucleic acid complexes give keys to decipher the binding process. J. Mol. Biol. 2019, 431, 1966–1980. [Google Scholar] [CrossRef]
- Mouhand, A.; Belfetmi, A.; Catala, M.; Larue, V.; Zargarian, L.; Brachet, F.; Gorelick, R.J.; Van Heijenoort, C.; Mirambeau, G.; Barraud, P.; et al. Modulation of the HIV nucleocapsid dynamics finely tunes its RNA-binding properties during virion genesis. Nucleic Acids Res. 2018, 46, 9699–9710. [Google Scholar] [CrossRef]
- Khan, R.; Giedroc, D.P. Nucleic acid binding properties of recombinant Zn2 HIV-1 nucleocapsid protein are modulated by COOH-terminal processing. J. Biol. Chem. 1994, 269, 22538–22546. [Google Scholar] [CrossRef]
- Wu, H.; Rouzina, I.; Williams, M.C. Single-molecule stretching studies of RNA chaperones. RNA Biol. 2010, 7, 712–723. [Google Scholar] [CrossRef] [Green Version]
- Cruceanu, M.; Urbaneja, M.A.; Hixson, C.V.; Johnson, D.G.; Datta, S.A.; Fivash, M.J.; Stephen, A.G.; Fisher, R.J.; Gorelick, R.J.; Casas-Finet, J.R.; et al. Nucleic acid binding and chaperone properties of HIV-1 Gag and nucleocapsid proteins. Nucleic Acids Res. 2006, 34, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Potempa, M.; Swanstrom, R. The choreography of HIV-1 proteolytic processing and virion assembly. J. Biol. Chem. 2012, 287, 40867–40874. [Google Scholar] [CrossRef] [Green Version]
- Könnyü, B.; Sadiq, S.K.; Turányi, T.; Hírmondó, R.; Müller, B.; Kräusslich, H.G.; Coveney, P.V.; Müller, V. Gag-Pol processing during HIV-1 virion maturation: A systems biology approach. PLoS Comput. Biol. 2013, 9, e1003103. [Google Scholar] [CrossRef] [Green Version]
- Dussupt, V.; Sette, P.; Bello, N.F.; Javid, M.P.; Nagashima, K.; Bouamr, F. Basic residues in the nucleocapsid domain of Gag are critical for late events of HIV-1 budding. J. Virol. 2011, 85, 2304–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, M.D.; Fu, W.; Soheilian, F.; Nagashima, K.; Ptak, R.G.; Pathak, V.K.; Hu, W.S. Suboptimal inhibition of protease activity in human immunodeficiency virus type 1: Effects on virion morphogenesis and RNA maturation. Virology 2008, 379, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coren, L.V.; Thomas, J.A.; Chertova, E.; Sowder, R.C.; Gagliardi, T.D.; Gorelick, R.J.; Ott, D.E. Mutational analysis of the C-terminal gag cleavage sites in human immunodeficiency virus type 1. J. Virol. 2007, 81, 10047–10054. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Wu, T.; Anderson, J.; Kane, B.F.; Johnson, D.G.; Gorelick, R.J.; Henderson, L.E.; Levin, J.G. Zinc finger structures in the human immunodeficiency virus type 1 nucleocapsid protein facilitate efficient minus-and plus-strand transfer. J. Virol. 2000, 74, 8980–8988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart-Maynard, K.M.; Cruceanu, M.; Wang, F.; Vo, M.N.; Gorelick, R.J.; Williams, M.C.; Rouzina, I.; Musier-Forsyth, K. Retroviral nucleocapsid proteins display nonequivalent levels of nucleic acid chaperone activity. J. Virol. 2008, 82, 10129–10142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carteau, S.; Gorelick, R.J.; Bushman, F.D. Coupled integration of human immunodeficiency virus type 1 cDNA ends by purified integrase in vitro: Stimulation by the viral nucleocapsid protein. J. Virol. 1999, 73, 6670–6679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Mitra, M.; McCauley, M.J.; Thomas, J.A.; Rouzina, I.; Musier-Forsyth, K.; Williams, M.C.; Gorelick, R.J. Aromatic residue mutations reveal direct correlation between HIV-1 nucleocapsid protein’s nucleic acid chaperone activity and retroviral replication. Virus Res. 2013, 171, 263–277. [Google Scholar] [CrossRef] [Green Version]
- Billich, A.; Hammerschmid, F.; Winkler, G. Purification, assay and kinetic features of HIV-1 proteinase. Biol. Chem. Hoppe Seyler 1990, 371, 265–272. [Google Scholar] [CrossRef]
- Bannwarth, L.; Rose, T.; Dufau, L.; Vanderesse, R.; Dumond, J.; Jamart-Grégoire, B.; Pannecouque, C.; De Clercq, E.; Reboud-Ravaux, M. Dimer disruption and monomer sequestration by alkyl tripeptides are successful strategies for inhibiting wild-type and multidrug-resistant mutated HIV-1 proteases. Biochemistry 2009, 48, 379–387. [Google Scholar] [CrossRef]
- Davis, D.A.; Brown, C.A.; Newcomb, F.M.; Boja, E.S.; Fales, H.M.; Kaufman, J.; Stahl, S.J.; Wingfield, P.; Yarchoan, R. Reversible oxidative modification as a mechanism for regulating retroviral protease dimerization and activation. J. Virol. 2003, 77, 3319–3325. [Google Scholar] [CrossRef] [Green Version]
- Rothemund, P.W. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinck, L.; Richer, D.; Howard, J.; Alexander, M.; Purcell, D.F.; Marquet, R.; Paillart, J.C. In vitro dimerization of human immunodeficiency virus type 1 (HIV-1) spliced RNAs. RNA 2007, 13, 2141–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd El-Wahab, E.W.; Smyth, R.P.; Mailler, E.; Bernacchi, S.; Vivet-Boudou, V.; Hijnen, M.; Jossinet, F.; Mak, J.; Paillart, J.C.; Marquet, R. Specific recognition of the HIV-1 genomic RNA by the Gag precursor. Nat. Commun. 2014, 5, 4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldschmidt, V.; Paillart, J.C.; Rigourd, M.; Ehresmann, B.; Aubertin, A.M.; Ehresmann, C.; Marquet, R. Structural variability of the initiation complex of HIV-1 reverse transcription. J. Biol. Chem. 2004, 279, 35923–35931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamon, L.; Pastre, D.; Dupaigne, P.; Breton, C.L.; Cam, E.L.; Pietrement, O. High-resolution AFM imaging of single-stranded DNA-binding (SSB) protein—DNA complexes. Nucleic Acids Res. 2007, 35, e58. [Google Scholar] [CrossRef] [Green Version]
- Gonda, M.A.; Aaronson, S.A.; Ellmore, N.; Zeve, V.H.; Nagashima, K. Ultrastructural studies of surface features of human normal and tumor cells in tissue culture by scanning and transmission electron microscopy. J. Natl. Cancer Inst. 1976, 56, 245–263. [Google Scholar] [CrossRef]
- Ott, D.E.; Chertova, E.N.; Busch, L.K.; Coren, L.V.; Gagliardi, T.D.; Johnson, D.G. Mutational analysis of the hydrophobic tail of the human immunodeficiency virus type 1 p6Gag protein produces a mutant that fails to package its envelope protein. J. Virol. 1999, 73, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Clouse, K.A.; Powell, D.; Washington, I.; Poli, G.; Strebel, K.; Farrar, W.; Barstad, P.; Kovacs, J.; Fauci, A.; Folks, T. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J. Immunol. 1989, 142, 431–438. [Google Scholar]
- Lopez-Iglesias, C.; Puvion-Dutilleul, F. Visualization of glycoproteins after tunicamycin and monensin treatment of herpes simplex virus infected cells. J. Ultrastruct. Mol. Struct. Res. 1988, 101, 75–91. [Google Scholar] [CrossRef]
- Sadiq, S.K. Fine-tuning of sequence specificity by near attack conformations in enzyme-catalyzed peptide hydrolysis. Catalysts 2020, 10, 684. [Google Scholar] [CrossRef]
- Sadiq, S.K.; Coveney, P.V. Computing the role of near attack conformations in an enzyme-catalyzed nucleophilic bimolecular reaction. J. Chem. Theory Comput. 2015, 11, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.A.; King, N.M.; Schiffer, C.A. Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J. Virol. 2004, 78, 12446–12454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; Fabritiis, G.D. ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [Green Version]
- Vo, M.N.; Barany, G.; Rouzina, I.; Musier-Forsyth, K. Effect of Mg2+ and Na+ on the nucleic acid chaperone activity of HIV-1 nucleocapsid protein: Implications for reverse transcription. J. Mol. Biol. 2009, 386, 773–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.C.; Rouzina, I.; Wenner, J.R.; Gorelick, R.J.; Musier-Forsyth, K.; Bloomfield, V.A. Mechanism for nucleic acid chaperone activity of HIV-1 nucleocapsid protein revealed by single molecule stretching. Proc. Natl. Acad. Sci. USA 2001, 98, 6121–6126. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.J.; Fivash, M.J.; Stephen, A.G.; Hagan, N.A.; Shenoy, S.R.; Medaglia, M.V.; Smith, L.R.; Worthy, K.M.; Simpson, J.T.; Shoemaker, R.; et al. Complex interactions of HIV-1 nucleocapsid protein with oligonucleotides. Nucleic Acids Res. 2006, 34, 472–484. [Google Scholar] [CrossRef]
- Qualley, D.F.; Stewart-Maynard, K.M.; Wang, F.; Mitra, M.; Gorelick, R.J.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K. C-terminal domain modulates the nucleic acid chaperone activity of human T-cell leukemia virus type 1 nucleocapsid protein via an electrostatic mechanism. J. Biol. Chem. 2010, 285, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Sadiq, S.K.; Könnyü, B.; Müller, V.; Coveney, P.V. Reaction kinetics of catalyzed competitive heteropolymer cleavage. J. Phys. Chem. B 2011, 115, 11017–11027. [Google Scholar] [CrossRef]
- Venken, T.; Voet, A.; De Maeyer, M.; De Fabritiis, G.; Sadiq, S.K. Rapid conformational fluctuations of disordered HIV-1 fusion peptide in solution. J. Chem. Theory Comput. 2013, 9, 2870–2874. [Google Scholar] [CrossRef]
- Roberts, M.M.; Copeland, T.D.; Oroszlan, S. In situ processing of a retroviral nucleocapsid protein by the viral proteinase. Protein Eng. Des. Sel. 1991, 4, 695–700. [Google Scholar] [CrossRef]
- Tinland, B.; Pluen, A.; Sturm, J.; Weill, G. Persistence length of single-stranded DNA. Macromolecules 1997, 30, 5763–5765. [Google Scholar] [CrossRef]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Visualizing HIV-1 assembly. J. Mol. Biol. 2011, 410, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Prescher, J.; Baumgärtel, V.; Ivanchenko, S.; Torrano, A.A.; Bräuchle, C.; Müller, B.; Lamb, D.C. Super-resolution imaging of ESCRT-proteins at HIV-1 assembly sites. PLoS Pathog. 2015, 11, e1004677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welker, R.; Hohenberg, H.; Tessmer, U.; Huckhagel, C.; Kräusslich, H.G. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J. Virol. 2000, 74, 1168–1177. [Google Scholar] [CrossRef] [Green Version]
- Sadiq, S.K.; Muñiz Chicharro, A.; Friedrich, P.; Wade, R.C. Multiscale approach for computing gated ligand binding from molecular dynamics and Brownian dynamics simulations. J. Chem. Theory Comput. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mattei, S.; Flemming, A.; Anders-Össwein, M.; Kräusslich, H.G.; Briggs, J.A.; Müller, B. RNA and nucleocapsid are dispensable for mature HIV-1 capsid assembly. J. Virol. 2015, 89, 9739–9747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharska, I.; Ding, P.; Zadrozny, K.K.; Dick, R.A.; Summers, M.F.; Ganser-Pornillos, B.K.; Pornillos, O. Biochemical reconstitution of HIV-1 assembly and maturation. J. Virol. 2020, 94, e01844-19. [Google Scholar] [CrossRef]
- Fontana, J.; Jurado, K.A.; Cheng, N.; Ly, N.L.; Fuchs, J.R.; Gorelick, R.J.; Engelman, A.N.; Steven, A.C. Distribution and redistribution of HIV-1 nucleocapsid protein in immature, mature, and integrase-inhibited virions: A role for integrase in maturation. J. Virol. 2015, 89, 9765–9780. [Google Scholar] [CrossRef] [Green Version]
- Kessl, J.J.; Kutluay, S.B.; Townsend, D.; Rebensburg, S.; Slaughter, A.; Larue, R.C.; Shkriabai, N.; Bakouche, N.; Fuchs, J.R.; Bieniasz, P.D.; et al. HIV-1 integrase binds the viral RNA genome and is essential during virion morphogenesis. Cell 2016, 166, 1257–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, J.L.; Eschbach, J.E.; Koneru, P.C.; Li, W.; Puray-Chavez, M.; Townsend, D.; Lawson, D.Q.; Engelman, A.N.; Kvaratskhelia, M.; Kutluay, S.B. Integrase-RNA interactions underscore the critical role of integrase in HIV-1 virion morphogenesis. eLife 2020, 9, e54311. [Google Scholar] [CrossRef]
- Cuccurullo, E.C.; Valentini, C.; Pizzato, M. Retroviral factors promoting infectivity. Prog. Mol. Biol. Transl. Sci. 2015, 129, 213–251. [Google Scholar] [PubMed]
- Cen, S.; Niu, M.; Saadatmand, J.; Guo, F.; Huang, Y.; Nabel, G.J.; Kleiman, L. Incorporation of Pol into human immunodeficiency virus type 1 Gag virus-like particles occurs independently of the upstream Gag domain in Gag-Pol. J. Virol. 2004, 78, 1042–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, A.; Moore, K.L.; Mak, J.; Sluis-Cremer, N.; de Bethune, M.P.; Tachedjian, G. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLoS Pathog. 2006, 2, e119. [Google Scholar] [CrossRef] [Green Version]
- Bendjennat, M.; Saffarian, S. The race against protease activation defines the role of ESCRTs in HIV budding. PLoS Pathog. 2016, 12, e1005657. [Google Scholar] [CrossRef] [PubMed]
- Chamontin, C.; Rassam, P.; Ferrer, M.; Racine, P.J.; Neyret, A.; Lainé, S.; Milhiet, P.E.; Mougel, M. HIV-1 nucleocapsid and ESCRT-component Tsg101 interplay prevents HIV from turning into a DNA-containing virus. Nucleic Acids Res. 2015, 43, 336–347. [Google Scholar] [CrossRef]
- Popova, E.; Popov, S.; Göttlinger, H.G. Human immunodeficiency virus type 1 nucleocapsid p1 confers ESCRT pathway dependence. J. Virol. 2010, 84, 6590–6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Kovalenko, L.; Lyonnais, S.; Antaki, D.; Torbett, B.E.; Botta, M.; Mirambeau, G.; Mély, Y. Nucleocapsid protein: A desirable target for future therapies against HIV-1. In The Future of HIV-1 Therapeutics; Torbett, B., Goodsell, D., Richman, D., Eds.; Springer: Cham, Switzerland, 2015; pp. 53–92. [Google Scholar]
- Dumont, S.; Prakash, M. Emergent mechanics of biological structures. Mol. Biol. Cell 2014, 25, 3461–3465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poudyal, R.R.; Pir Cakmak, F.; Keating, C.D.; Bevilacqua, P.C. Physical principles and extant biology reveal roles for RNA-containing membraneless compartments in origins of life chemistry. Biochemistry 2018, 57, 2509–2519. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyonnais, S.; Sadiq, S.K.; Lorca-Oró, C.; Dufau, L.; Nieto-Marquez, S.; Escribà, T.; Gabrielli, N.; Tan, X.; Ouizougun-Oubari, M.; Okoronkwo, J.; et al. The HIV-1 Nucleocapsid Regulates Its Own Condensation by Phase-Separated Activity-Enhancing Sequestration of the Viral Protease during Maturation. Viruses 2021, 13, 2312. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112312
Lyonnais S, Sadiq SK, Lorca-Oró C, Dufau L, Nieto-Marquez S, Escribà T, Gabrielli N, Tan X, Ouizougun-Oubari M, Okoronkwo J, et al. The HIV-1 Nucleocapsid Regulates Its Own Condensation by Phase-Separated Activity-Enhancing Sequestration of the Viral Protease during Maturation. Viruses. 2021; 13(11):2312. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112312
Chicago/Turabian StyleLyonnais, Sébastien, S. Kashif Sadiq, Cristina Lorca-Oró, Laure Dufau, Sara Nieto-Marquez, Tuixent Escribà, Natalia Gabrielli, Xiao Tan, Mohamed Ouizougun-Oubari, Josephine Okoronkwo, and et al. 2021. "The HIV-1 Nucleocapsid Regulates Its Own Condensation by Phase-Separated Activity-Enhancing Sequestration of the Viral Protease during Maturation" Viruses 13, no. 11: 2312. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112312