
Analysis of the Physicochemical Properties, Replication and Pathophysiology of a Massively Glycosylated Hepatitis B Virus HBsAg Escape Mutant




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction and Cells
2.2. Transfection
2.3. TunicamycinTM Treatment and Cell Viability Assay
2.4. Virus Preparation and Cesium Chloride (CsCl) Density Gradient Centrifugation
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Western Blotting
2.7. Immunofluorescence Analysis (IFA)
2.8. DNA Extraction and Quantitative Real-Time PCR (qPCR)
2.9. Southern Blot Analysis of Viral DNA
2.10. Statistical Analyses
3. Results
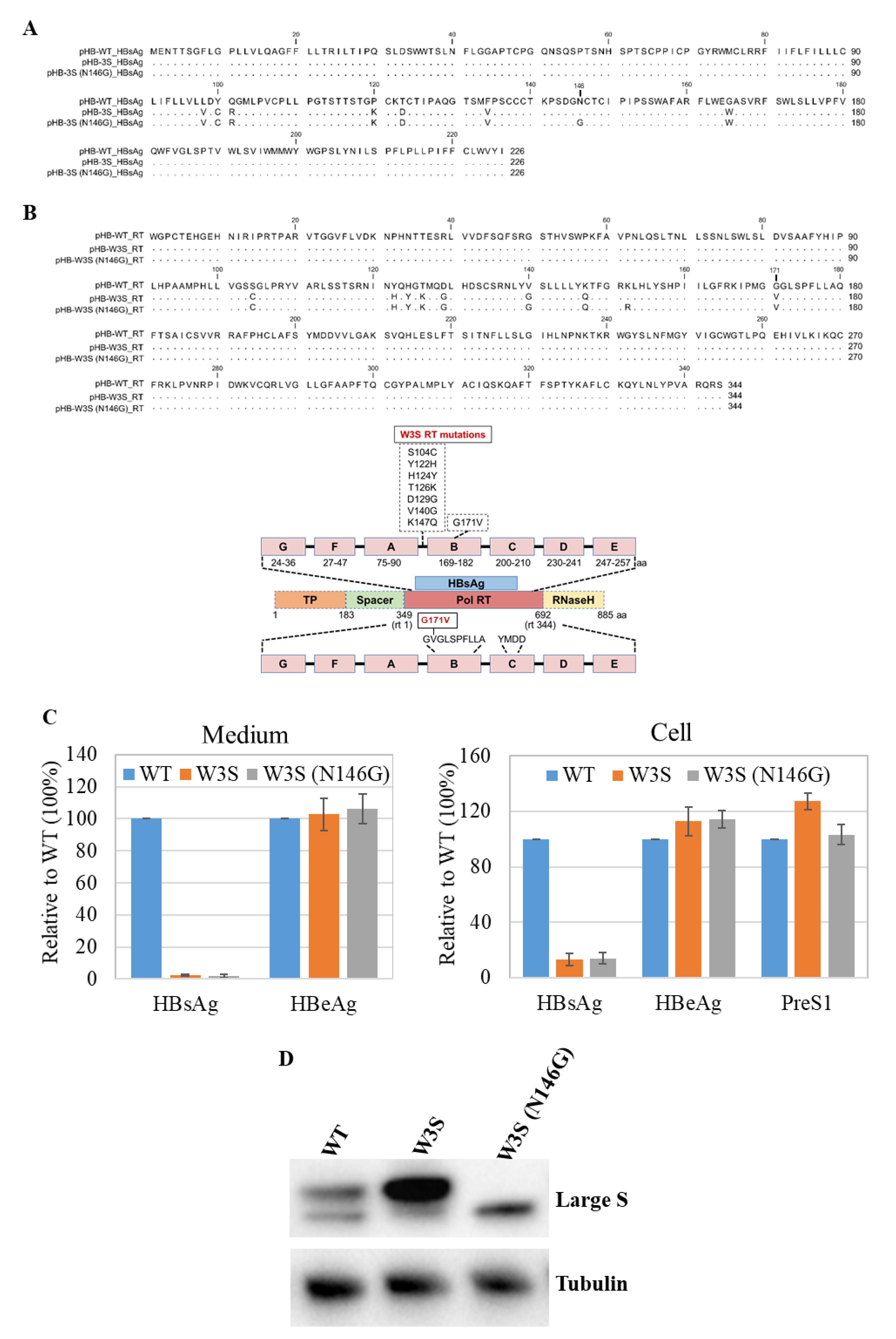
3.1. Comparison of HBs and Pol Amino Acids Sequences of HBV-WT (adr4) and W3S
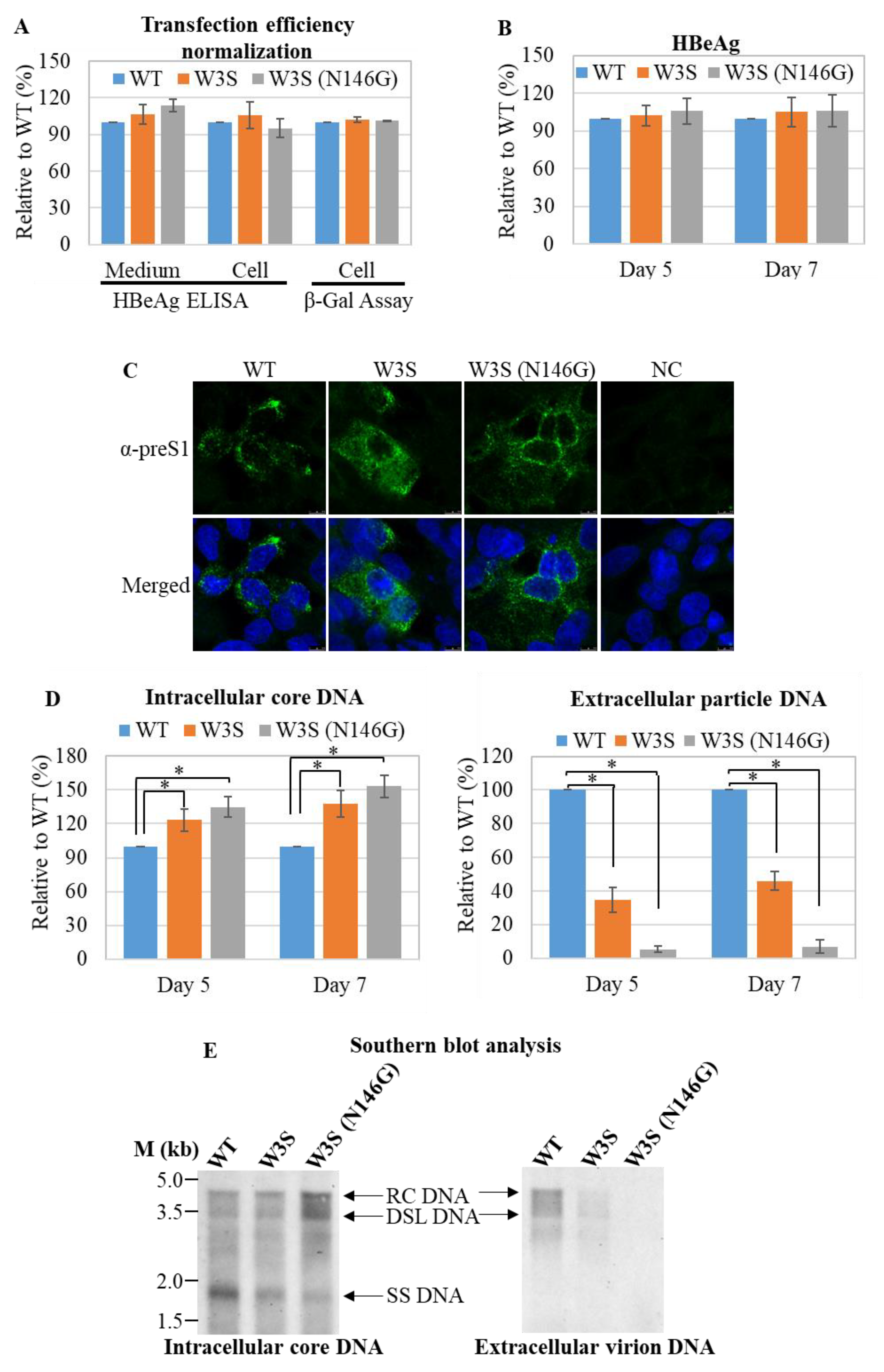
3.2. Analysis of the Replication Status of HBs/RT Mutant HBV
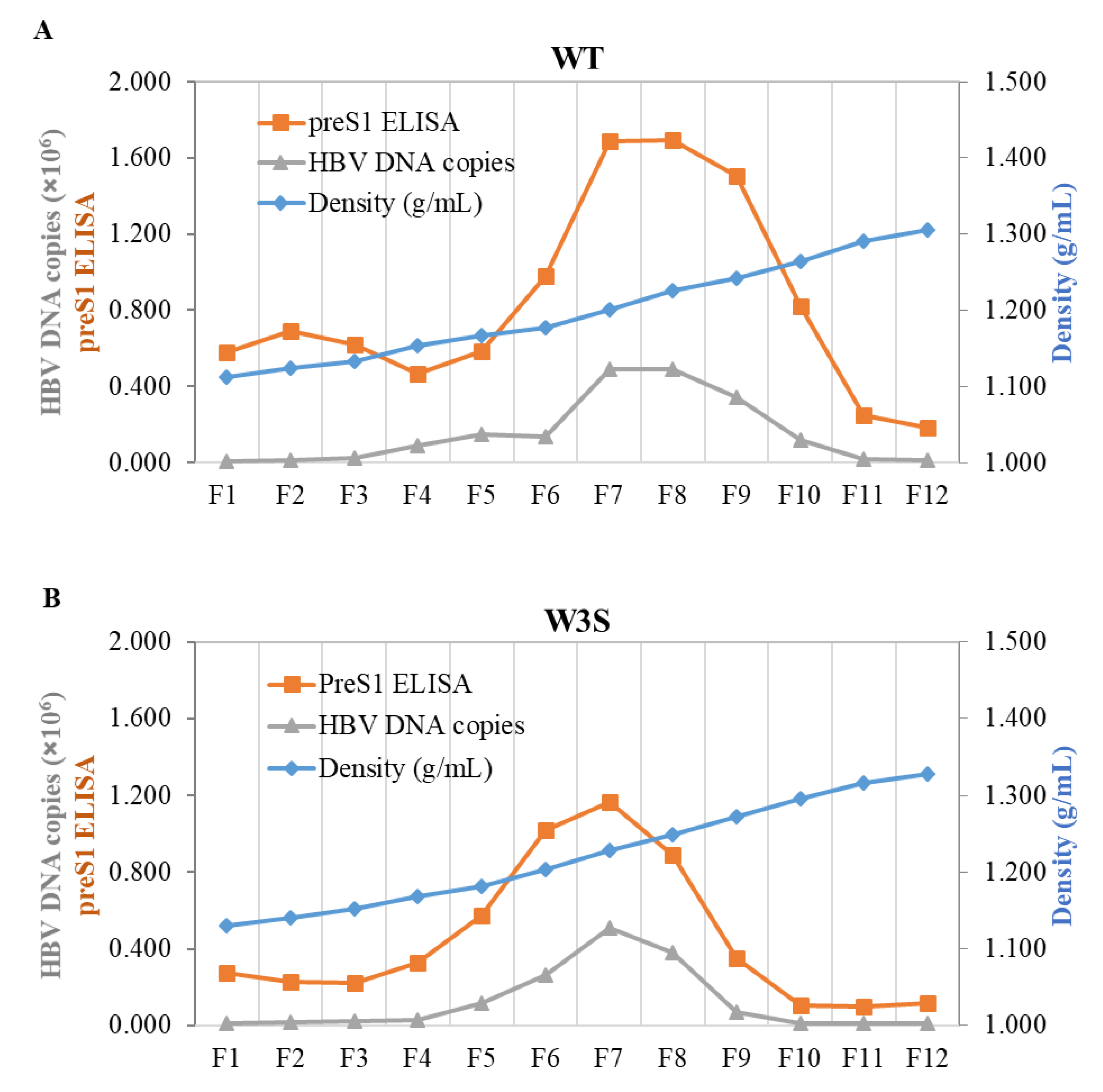
3.3. The Physicochemical Properties of the WT and W3S Dane Particles Are Basically Identical
3.4. Effect of Tunicamycin on HBV Replication and Virion Production
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shedain, P.R.; Devkota, M.D.; Banjara, M.R.; Ling, H.; Dhital, S. Prevalence and risk factors of hepatitis B infection among mothers and children with hepatitis B infected mother in upper Dolpa, Nepal. BMC Infect. Dis. 2017, 17, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, K.; Tsurimoto, T.; Matsubara, K. Three envelope proteins of hepatitis B virus: Large S, middle S, and major S proteins needed for the formation of Dane particles. J. Virol. 1991, 65, 3521–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delius, H.; Gough, N.M.; Cameron, C.H.; Murray, K. Structure of the hepatitis B virus genome. J. Virol. 1983, 47, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; Bremer, C.M. The molecular virology of hepatitis B virus. Semin. Liver Dis. 2013, 33, 103–112. [Google Scholar]
- Grimm, D.; Thimme, R.; Blum, H.E. HBV life cycle and novel drug targets. Hepatol. Int. 2011, 5, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef] [Green Version]
- Park, S.G.; Kim, Y.; Park, E.; Ryu, H.M.; Jung, G. Fidelity of hepatitis B virus polymerase. Eur. J. Biochem. 2003, 270, 2929–2936. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, J.; Rodès, B.; Zoulim, F.; Bartholomeusz, A.; Soriano, V. Mutations affecting the replication capacity of the hepatitis B virus. J. Viral Hepat. 2006, 13, 427–434. [Google Scholar] [CrossRef]
- Croagh, C.M.; Desmond, P.V.; Bell, S.J. Genotypes and viral variants in chronic hepatitis B: A review of epidemiology and clinical relevance. World J. Hepatol. 2015, 7, 289–303. [Google Scholar] [CrossRef]
- Caligiuri, P.; Cerruti, R.; Icardi, G.; Bruzzone, B. Overview of hepatitis B virus mutations and their implications in the management of infection. World J. Gastroenterol. 2016, 22, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F. Mechanism of viral persistence and resistance to nucleoside and nucleotide analogs in chronic hepatitis B virus infection. Antivir. Res. 2004, 64, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, D.R.; Walsh, A.W.; Baldick, C.J.; Eggers, B.J.; Rose, R.E.; Levine, S.M.; Kapur, A.J.; Colonno, R.J.; Tenney, D.J. Inhibition of Hepatitis B Virus Polymerase by Entecavir. J. Virol. 2007, 81, 3992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, P.K.; Wang, R.Y.; Lin, M.H.; Masuda, T.; Suk, F.M.; Shih, C. Reduced secretion of virions and hepatitis B virus (HBV) surface antigen of a naturally occurring HBV variant correlates with the accumulation of the small S envelope protein in the endoplasmic reticulum and Golgi apparatus. J. Virol. 2005, 79, 13483–13496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poovorawan, Y.; Sanpavat, S.; Pongpunglert, W.; Chumdermpadetsuk, S.; Sentrakul, P.; Vandepapeliere, P.; Safary, A. Long term efficacy of hepatitis B vaccine in infants born to hepatitis B e antigen-positive mothers. Pediatr. Infect. Dis. J. 1992, 11, 816–821. [Google Scholar] [CrossRef]
- Miyanohara, A.; Toh-e, A.; Nozaki, C.; Hamada, F.; Ohtomo, N.; Matsubara, K. Expression of hepatitis B surface antigen gene in yeast. Proc. Natl. Acad. Sci. USA 1983, 80, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, P.; Medina, A.; Rutter, W.J.; Ammerer, G.; Hall, B.D. Synthesis and assembly of hepatitis B virus surface antigen particles in yeast. Nature 1982, 298, 347–350. [Google Scholar] [CrossRef]
- Harford, N.; Cabezon, T.; Crabeel, M.; Simoen, E.; Rutgers, A.; De Wilde, M. Expression of hepatitis B surface antigen in yeast. Dev. Biol. Stand. 1983, 54, 125–130. [Google Scholar]
- Carman, W.F. The clinical significance of surface antigen variants of hepatitis B virus. J. Viral Hepat. 1997, 4 (Suppl. 1), 11–20. [Google Scholar] [CrossRef]
- Seddigh-Tonekaboni, S.; Waters, J.A.; Jeffers, S.; Gehrke, R.; Ofenloch, B.; Horsch, A.; Hess, G.; Thomas, H.C.; Karayiannis, P. Effect of variation in the common ”a“ determinant on the antigenicity of hepatitis B surface antigen. J. Med. Virol. 2000, 60, 113–121. [Google Scholar] [CrossRef]
- Roznovsky, L.; Harrison, T.J.; Fang, Z.L.; Ling, R.; Lochman, I.; Orsagova, I.; Pliskova, L. Unusual hepatitis B surface antigen variation in a child immunised against hepatitis B. J. Med. Virol. 2000, 61, 11–14. [Google Scholar] [CrossRef]
- Ni, F.; Fang, D.; Gan, R.; Li, Z.; Duan, S.; Xu, Z. A new immune escape mutant of hepatitis B virus with an Asp to Ala substitution in aa144 of the envelope major protein. Res. Virol. 1995, 146, 397–407. [Google Scholar] [CrossRef]
- Julithe, R.; Abou-Jaoudé, G.; Sureau, C. Modification of the hepatitis B virus envelope protein glycosylation pattern interferes with secretion of viral particles, infectivity, and susceptibility to neutralizing antibodies. J. Virol. 2014, 88, 9049–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Qin, Y.; Guarnieri, M.; Garcia, T.; Kwei, K.; Mizokami, M.; Zhang, J.; Li, J.; Wands, J.R.; Tong, S. Impairment of hepatitis B virus virion secretion by single-amino-acid substitutions in the small envelope protein and rescue by a novel glycosylation site. J. Virol. 2010, 84, 12850–12861. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Zhang, X.; Tian, Y.; Song, J.; Yang, D.; Roggendorf, M.; Lu, M.; Chen, X. Biological significance of amino acid substitutions in hepatitis B surface antigen (HBsAg) for glycosylation, secretion, antigenicity and immunogenicity of HBsAg and hepatitis B virus replication. J. Gen. Virol. 2010, 91 Pt 2, 483–492. [Google Scholar] [CrossRef]
- Qiao, Y.; Lu, S.; Xu, Z.; Li, X.; Zhang, K.; Liu, Y.; Zhao, L.; Chen, R.; Si, L.; Lin, S.; et al. Additional N-glycosylation mutation in the major hydrophilic region of hepatitis B virus S gene is a risk indicator for hepatocellular carcinoma occurrence in patients with coexistence of HBsAg/anti-HBs. Oncotarget 2017, 8, 61719–61730. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.G.; Ueda, K. Investigation of a Novel Hepatitis B Virus Surface Antigen (HBsAg) Escape Mutant Affecting Immunogenicity. PLoS ONE 2017, 12, e0167871. [Google Scholar] [CrossRef]
- Neumann-Fraune, M.; Beggel, B.; Kaiser, R.; Obermeier, M. Hepatitis B virus drug resistance tools: One sequence, two predictions. Intervirology 2014, 57, 232–236. [Google Scholar] [CrossRef]
- Ueda, K.; Omori, H. Successful Generation of Hepatitis B virus (HBV) Pseudotype Particle; A Versatile Tool for Identification of the HBV Receptor and Investigation of HBV Infectivity. J. Liver 2015, 04, 1000169. [Google Scholar]
- Hossain, M.G.; Mahmud, M.M.; Nazir, K.H.M.N.H.; Ueda, K. PreS1 Mutations Alter the Large HBsAg Antigenicity of a Hepatitis B Virus Strain Isolated in Bangladesh. Int. J. Mol. Sci. 2020, 21, 546. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.G.; Ohsaki, E.; Honda, T.; Ueda, K. Importance of Promyelocytic Leukema Protein (PML) for Kaposi's Sarcoma-Associated Herpesvirus Lytic Replication. Front. Microbiol. 2018, 9, 2324. [Google Scholar] [CrossRef]
- Okuyama-Dobashi, K.; Kasai, H.; Tanaka, T.; Yamashita, A.; Yasumoto, J.; Chen, W.; Okamoto, T.; Maekawa, S.; Watashi, K.; Wakita, T.; et al. Hepatitis B virus efficiently infects non-adherent hepatoma cells via human sodium taurocholate cotransporting polypeptide. Sci. Rep. 2015, 5, 17047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurimoto, T.; Fujiyama, A.; Matsubara, K. Stable expression and replication of hepatitis B virus genome in an integrated state in a human hepatoma cell line transfected with the cloned viral DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 444–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiyama, A.; Miyanohara, A.; Nozaki, C.; Yoneyama, T.; Ohtomo, N.; Matsubara, K. Cloning and structural analyses of hepatitis B virus DNAs, subtype adr. Nucleic Acids Res. 1983, 11, 4601–4610. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef]
- Jin, S.P.; Chung, J.H. Inhibition of N-glycosylation by tunicamycin attenuates cell-cell adhesion via impaired desmosome formation in normal human epidermal keratinocytes. Biosci. Rep. 2018, 38, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Mehta, A.; Dwek, R.; Butters, T.; Block, T. Evidence that N-linked glycosylation is necessary for hepatitis B virus secretion. Virology 1995, 213, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.G.; Ueda, K. A meta-analysis on genetic variability of RT/HBsAg overlapping region of hepatitis B virus (HBV) isolates of Bangladesh. Infect. Agents Cancer 2019, 14, 33. [Google Scholar] [CrossRef] [Green Version]
- Leong, J.; Lin, D.; Nguyen, M.H. Hepatitis B surface antigen escape mutations: Indications for initiation of antiviral therapy revisited. World J. Clin. Cases 2016, 4, 71–75. [Google Scholar] [CrossRef]
- Purdy, M.A. Hepatitis B virus S gene escape mutants. Asian J. Transfus. Sci. 2007, 1, 62–70. [Google Scholar] [CrossRef]
- Hossain, M.G.; Akter, S.; Ohsaki, E.; Ueda, K. Impact of the Interaction of Hepatitis B Virus with Mitochondria and Associated Proteins. Viruses 2020, 12, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, W.L.; Wu, J.F.; Ni, Y.H.; Chen, H.L.; Chiang, C.L.; Chang, M.H.; Hsu, H.Y. Occult hepatitis B virus and surface antigen mutant infection in healthy vaccinated cohorts and children with various forms of hepatitis and multiple transfusions. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, P.; Zeng, J.; Du, P.; Zheng, X.; Ye, X.; Zhu, W.; Fu, Y.; Candotti, D.; Allain, J.P.; et al. Occurrence of occult hepatitis B virus infection associated with envelope protein mutations according to anti-HBs carriage in blood donors. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2020, 92, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokaya, J.; McNaughton, A.L.; Hadley, M.J.; Beloukas, A.; Geretti, A.M.; Goedhals, D.; Matthews, P.C. A systematic review of hepatitis B virus (HBV) drug and vaccine escape mutations in Africa: A call for urgent action. PLoS Negl. Trop. Dis. 2018, 12, e0006629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Zhang, X.; Kozlowski, M.; Li, W.; Wu, M.; Liu, J.; Chen, L.; Zhang, J.; Huang, Y.; Yuan, Z. Extracellular Hepatitis B Virus RNAs Are Heterogeneous in Length and Circulate as Capsid-Antibody Complexes in Addition to Virions in Chronic Hepatitis B Patients. J. Virol. 2018, 92, e00798-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasukawa, T.; Uchiyama, J.; Taharaguchi, S.; Ota, S.; Ujihara, T.; Matsuzaki, S.; Murakami, H.; Mizukami, K.; Sakaguchi, M. Virus purification by CsCl density gradient using general centrifugation. Arch. Virol. 2017, 162, 3523–3528. [Google Scholar] [CrossRef]
- Choi, Y.-M.; Lee, S.-Y.; Kim, B.-J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 597. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.L.; Chen, C.C.; Huang, Y.L.; Hsieh, D.J.; Hu, C.P.; Chen, C.F.; Chang, C. Osthole increases glycosylation of hepatitis B surface antigen and suppresses the secretion of hepatitis B virus in vitro. Hepatology 1996, 24, 508–515. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, M.G.; Suwanmanee, Y.; Du, K.; Ueda, K. Analysis of the Physicochemical Properties, Replication and Pathophysiology of a Massively Glycosylated Hepatitis B Virus HBsAg Escape Mutant. Viruses 2021, 13, 2328. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112328
Hossain MG, Suwanmanee Y, Du K, Ueda K. Analysis of the Physicochemical Properties, Replication and Pathophysiology of a Massively Glycosylated Hepatitis B Virus HBsAg Escape Mutant. Viruses. 2021; 13(11):2328. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112328
Chicago/Turabian StyleHossain, Md. Golzar, Yadarat Suwanmanee, Kaili Du, and Keiji Ueda. 2021. "Analysis of the Physicochemical Properties, Replication and Pathophysiology of a Massively Glycosylated Hepatitis B Virus HBsAg Escape Mutant" Viruses 13, no. 11: 2328. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112328