
Cryo-EM Structures of Two Bacteriophage Portal Proteins Provide Insights for Antimicrobial Phage Engineering

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning, Expression, and Purification
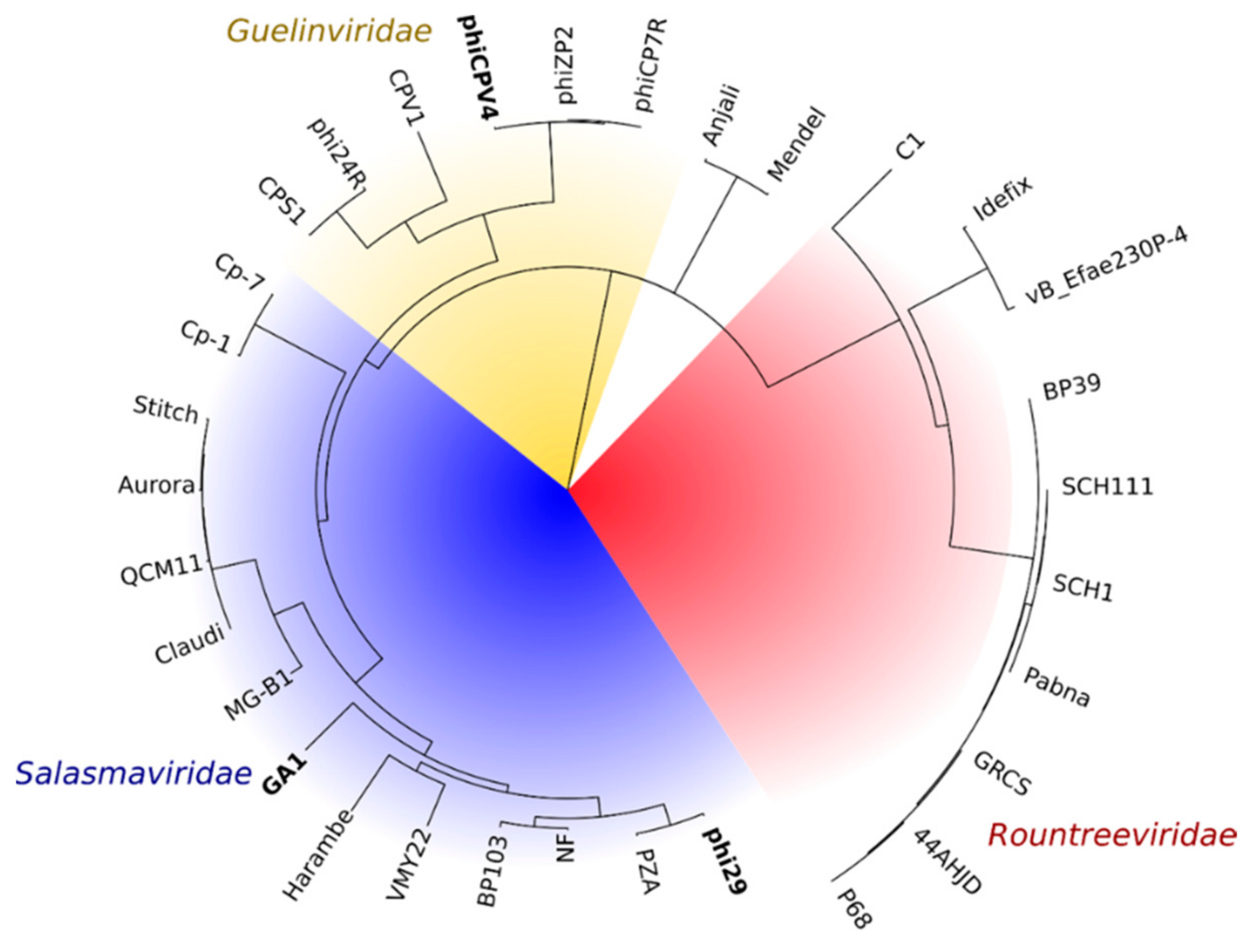
2.2. Bioinformatics
2.3. Grid Preparation and Data Collection
2.4. Image Processing
2.5. Model Building and Refinement
3. Results
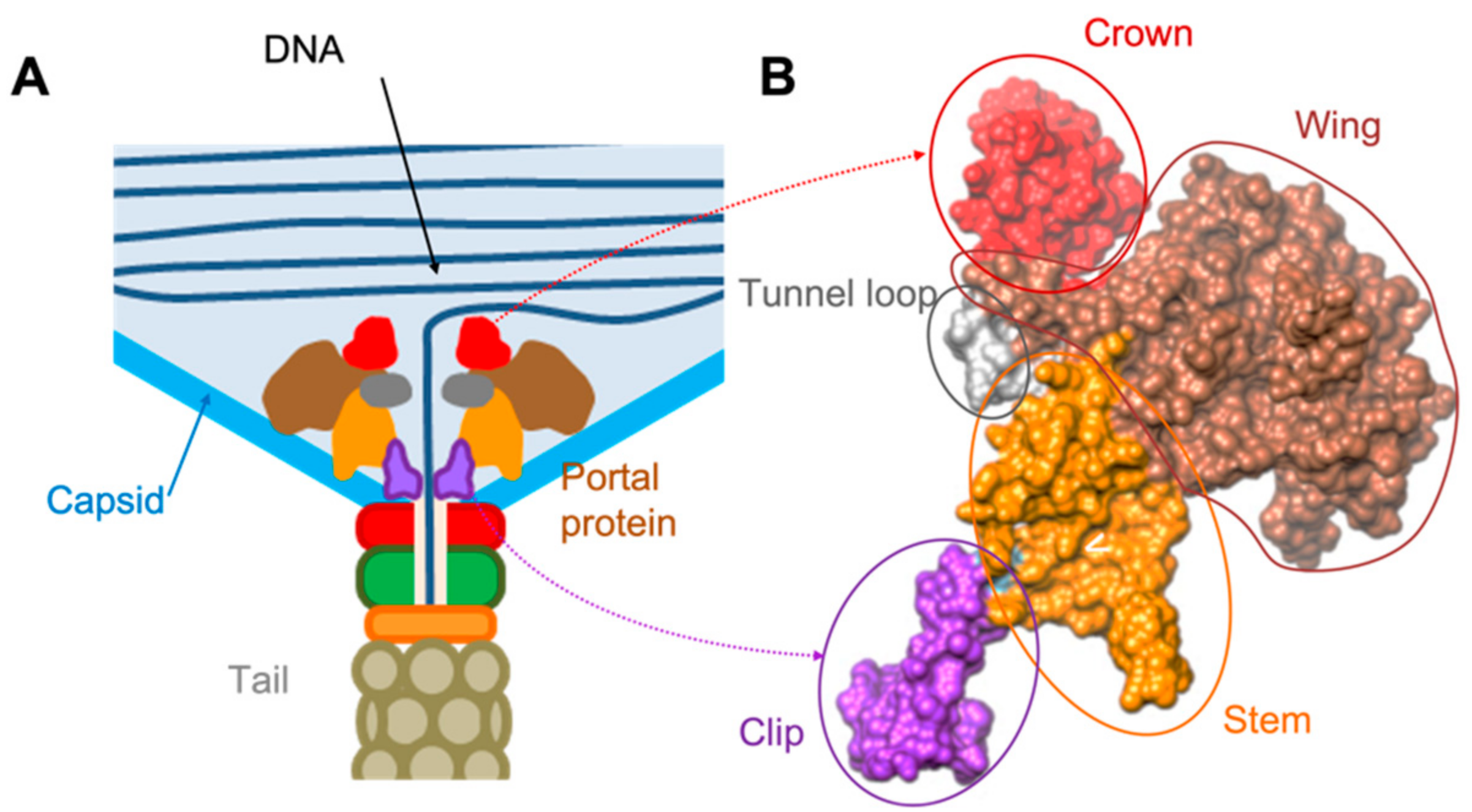
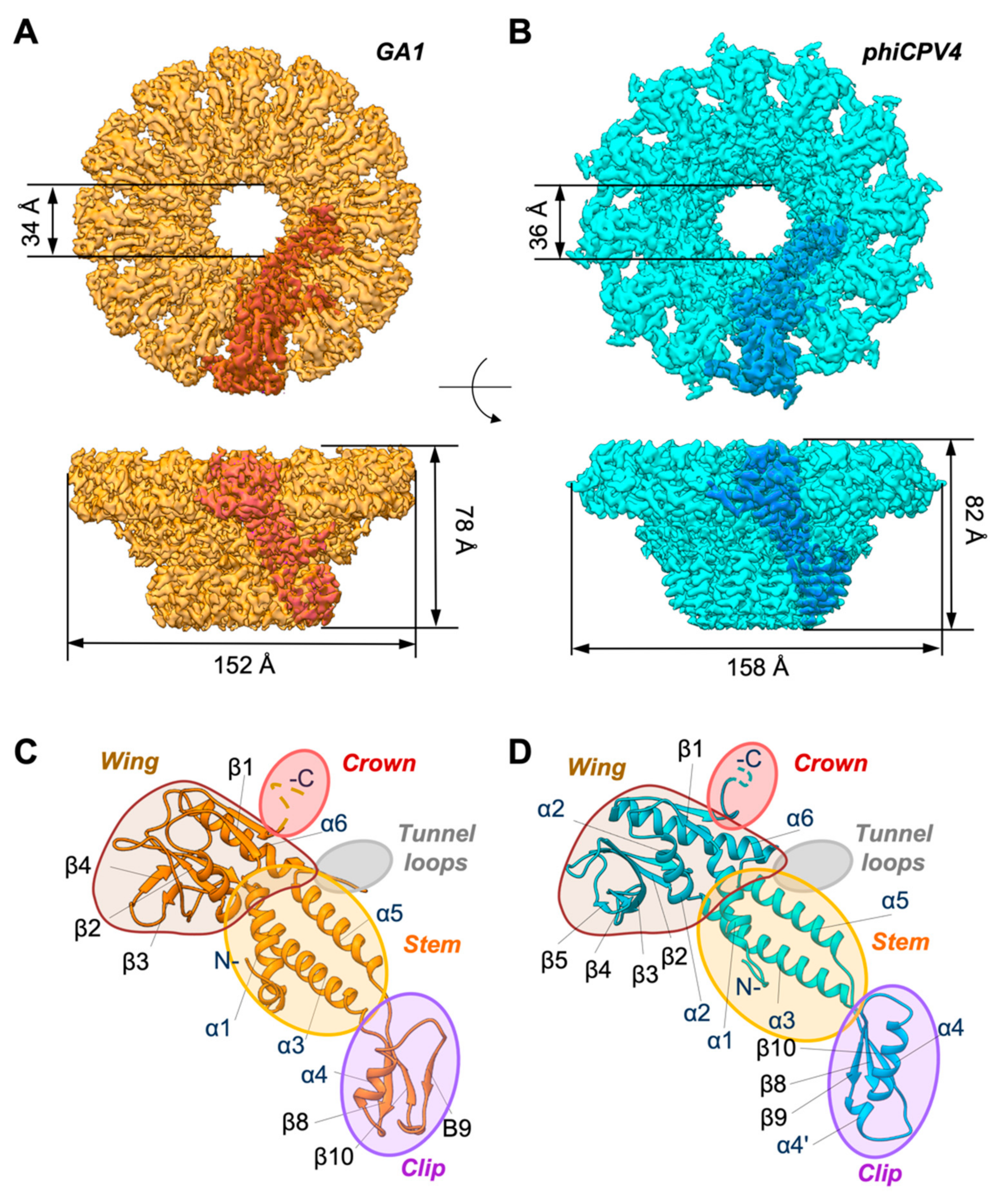
3.1. Overall Organisation of GA1 gp10 and phiCPV4 gp17 PPs
3.2. Crown Domain
3.3. Wing Domain
3.4. Tunnel Loop
3.5. Stem Domain
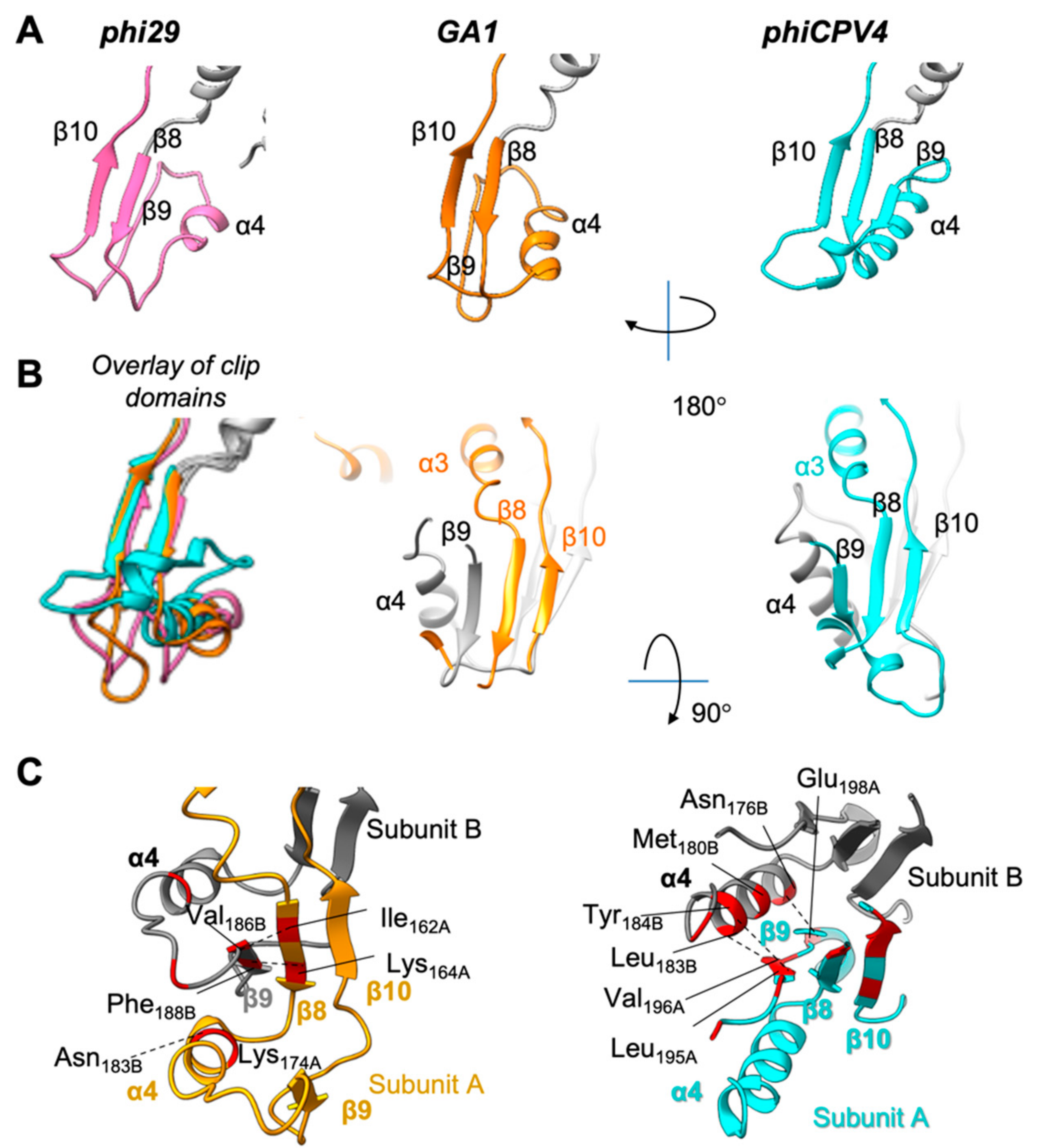
3.6. Clip Domain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Sumrall, E.; Plückthun, L.H.A.; Kilcher, M.J.L.S. Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 2019, 29, 1336–1350. [Google Scholar] [CrossRef] [Green Version]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Farley, M.M.; Tu, J.; Kearns, D.B.; Molineux, I.J.; Liu, J. Ultrastructural analysis of bacteriophage phi29 during infection of Bacillus subtilis. J. Struct. Biol. 2017, 197, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Longás, E.; Villar, L.; Lázaro, J.M.; de Vega, M.; Salas, M. Phage phi29 and Nf terminal protein-priming domain specifies the internal template nucleotide to initiate DNA replication. Proc. Natl. Acad. Sci. USA 2008, 105, 18290–18295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrascosa, J.L.; Camacho, A.; Moreno, F.; Jimenez, F.; Mellado, R.P.; Vinuela, E.; Salas, M. Bacillus subtilis phage phi29. Characterization of gene products and functions. Eur. J. Biochem. 1976, 66, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.A.; Tao, Y.; Leiman, P.G.; Badasso, M.O.; He, Y.; Jardine, P.J.; Olson, N.H.; Morais, M.C.; Grimes, S.; Anderson, D.L.; et al. Structure of the bacteriophage φ29 DNA packaging motor. Nature 2000, 408, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.C. The dsDNA packaging motor in bacteriophage phi29. Adv. Exp. Med. Biol. 2012, 726, 511–547. [Google Scholar]
- Xu, J.; Wang, D.; Gui, M.; Xiang, Y. Structural assembly of the tailed bacteriophage phi29. Nat. Commun. 2019, 10, 2366. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.Y.; Prevelige, P.E. In-Vitro Incorporation of the Phage phi29 Connector Complex. Virology 2019, 394, 149–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebedev, A.A.; Krause, M.H.; Isidro, A.L.; Vagin, A.A.; Orlova, E.V.; Turner, J.; Dodson, E.J.; Tavares, A.A. Structural framework for DNA translocation via the viral portal protein. EMBO J. 2007, 26, 1984–1994. [Google Scholar] [CrossRef]
- Padilla-Sanchez, V.; Gao, S.; Kim, H.R.; Kihara, D.; Sun, L.; Rossmann, M.G.; Rao, V.B. Structure-function analysis of the DNA translocating portal of the bacteriophage T4 packaging machine. J. Mol. Biol. 2014, 426, 1019–1038. [Google Scholar] [CrossRef] [Green Version]
- Corynne, L.D.; Cingolani, G.; Teschke, C.M. Portal Protein: The Orchestrator of Capsid Assembly for the dsDNA Tailed Bacteriophages and Herpesviruses. Ann. Rev. Virol. 2019, 6, 141–160. [Google Scholar]
- Rao, V.B.; Feiss, M. The bacteriophage DNA packaging motor. Ann. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, S.; Gallopin, M.; Gilquin, B.; Brasiles, S.; Lancelot, N.; Letellier, G.; Gilles, M.; Dethan, G.; Orlova, E.V.; Couprie, J.; et al. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc. Natl. Acad. Sci. USA 2009, 106, 8507–8512. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Saha, M.; Reyes-Aldrete, E.; Sherman, M.B.; Woodson, M.; Atz, R.; Grimes, S.; Jardine, P.J.; Morais, M.C. Structural and Molecular Basis for Coordination in a Viral DNA Packaging Motor. Cell Rep. 2016, 14, 2017–2029. [Google Scholar] [CrossRef] [Green Version]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrebík, D.; Štveráková, D.; Škubník, K.; Füzik, T.; Pantůček, R.; Plevka, P. Structure and genome ejection mechanism of Staphylococcus aureus phage P68. Sci. Adv. 2019, 5, eaaw7414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. Methods Mol. Biol. 2014, 1079, 105–116. [Google Scholar] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Palovcak, E.; Armache, J.-P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zivanov, J.; Nakane, T.; Forsberg, O.B.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 2018, 7, e42166. [Google Scholar] [CrossRef]
- Zhang, K. Gautomatch 0.56. 2020. Available online: https://www2.mrc-lmb.cam.ac.uk/download/gautomatch-056/ (accessed on 23 September 2021).
- Orlova, E.V.; Gowen, B.; Dröge, A.; Stiege, A.; Weise, F.; Lurz, R.; van Heel, M.; Tavares, P. Structure of a viral DNA gatekee per at 10 A resolution by cryo-electron microscopy. EMBO J. 2003, 22, 1255–1262. [Google Scholar] [CrossRef]
- Trus, B.L.; Cheng, N.; Newcomb, W.W.; Homa, F.L.; Brown, J.C.; Steven, A.C. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 2004, 78, 12668–12671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Heel, M.; Harauz, G.; Orlova, E.V.; Schmidt, R.; Schatz, M. A new generation of the IMAGIC image processing system. J. Struct. Biol. 1996, 116, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization andanalysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. 2004, D60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Croll, T.I. ISOLDE: A physically realistic environment for model building into low-resolution electron-density maps. Acta Cryst. 2018, D74, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Klaholz, B.P.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D.; Urzhumtsev, A. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 814–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. 2010, D66, 12–21. [Google Scholar]
- Xiao, F.; Demeler, B.; Guo, P. Assembly Mechanism of the Sixty-Subunit Nanoparticles via Interaction of RNA with the Reengineered Protein Connector of phi29 DNA-Packaging Motor. ACS Nano 2010, 4, 3293–3301. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Gao, S.; Rao, P.A.; Padilla-Sanchez, V.; Chen, Z.; Sun, S.; Xiang, Y.; Subramaniam, S.; Rao, V.B.; et al. Cryo-EM structure of the bacteriophage T4 portal protein assembly at near-atomic resolution. Nat. Commun. 2015, 6, 7548. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Jing, P.; Haque, F.; Guo, P. Role of Channel Lysines and the “Push Through a One-Way Valve” Mechanism of the Viral DNA Packaging Motor. Biophys. J. 2012, 102, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Grubmüller, H. Phi29 Connector-DNA Interactions Govern DNA Crunching and Rotation, Supporting the Check-Valve Model. Biophys. J. 2016, 110, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Morais, M.C.; Battisti, A.J.; Grimes, S.; Jardine, P.J.; Anderson, D.L.; Rossmann, M.G. Structural changes of bacteriophage phi29 upon DNA packaging and release. EMBO J. 2006, 25, 5229–5239. [Google Scholar]
- Simpson, A.A.; Leiman, P.G.; Tao, Y.; He, Y.; Badasso, M.O.; Jardine, P.J.; Anderson, D.L.; Rossmann, M.G. Structure determination of the head-tail connector of bacteriophage phi29. Acta Crystallogr. D Biol. Crystallogr. 2001, 57 Pt 9, 1260–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.; Fàbrega-Ferrer, M.; Machón, C.; Conesa, J.J.; Fernández, F.J.; Pérez-Luque, R.; Pérez-Ruiz, M.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; et al. Structures of T7 bacteriophage portal and tail suggest a viral DNA retention and ejection mechanism. Nat. Commun. 2019, 10, 3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olia, A.S.; Prevelige, P.E.; Johnson, J.E.; Cingolani, G. Three-dimensional structure of a viral genome-delivery portal vertex. Nat. Struct. Mol. Biol. 2011, 18, 597–603. [Google Scholar] [CrossRef]
- Bayfield, O.W.; Klimuk, E.; Winkler, D.C.; Hesketh, E.L.; Chechik, M.; Cheng, N.; Dykeman, E.C.; Minakhin, L.; Ranson, N.A.; Severinov, K.; et al. Cryo-EM structure and in vitro DNA packaging of a thermophilic virus with supersized T=7 capsids. Proc. Natl. Acad. Sci. USA 2019, 116, 3556–3561. [Google Scholar] [CrossRef] [Green Version]
- Holguera, I.; Redrejo-Rodriguez, M.; Salas, M.; Munoz-Espin, D. New insights in the Phi29 terminal protein DNA-binding and host nucleoid localization functions. Mol. Microbiol. 2013, 91, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.R.; Salas, M. Multiple roles of genome-attached bacteriophage terminal proteins. Virology 2014, 468–470, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Volozhantsev, N.V.; Oakley, B.B.; Morales, C.A.; Verevkin, V.V.; Bannov, V.A.; Krasilnikova, V.M.; Popova, A.V.; Zhilenkov, E.L.; Garrish, J.K.; Schegg, K.M.; et al. Molecular characterization of podoviral bacteriophages virulent for Clostridium perfringens and their comparison with members of the Picovirinae. PLoS ONE 2012, 7, e38283. [Google Scholar]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef]
- Dunne, M.; Prokhorov, N.S.; Loessner, M.J.; Leiman, P.G. Reprogramming bacteriophage host range: Design principles and strategies for engineering receptor binding proteins. Curr. Opin. Biotechnol. 2021, 68, 272–281. [Google Scholar] [CrossRef]
- Van Nies, P.; Westerlaken, I.; Blanken, D.; Salas, M.; Mencia, M.; Danelon, C. Self-replication of DNA by its encoded proteins in liposome-based synthetic cells. Nat. Commun. 2018, 9, 1583. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Grimes, S.; Anderson, D. A defined system for in vitro packaging of DNA-gp3 of the Bacillus subtilis bacteriophage phi 29. Proc. Natl. Acad. Sci. USA 1986, 83, 3505–3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Portal Protein (PP) | Molecular Wight of PP (kDa) | The Length of the Polypeptide Chain (aa) | Narrowest Diameter within Clip Domain (Å) | Genome Size kb | References |
|---|---|---|---|---|---|
| phi29 gp10 | 35.9 | 309 | 34 | 19.2 | [6] |
| GA1 gp10 | 35.3 | 306 | 34 | 21.2 | This MS |
| phiCPV4 gp17 | 34.6 | 301 | 36 | 17.9 | This MS |
| SPP1 gp6 | 57.3 | 503 | 30 | 45 | [27] |
| T4 gp9 | 61 | 524 | 32 | 168.9 | [38] |
| GA1 vs. phi29 | phiCPV4 vs. phi29 | GA1 vs. phiCPV4 | |
|---|---|---|---|
| Wing domain | 1.8 | 7.3 | 5.2 |
| Stem domain | 0.69 | 0.93 | 0.61 |
| Clip domain | 2.85 | 8.4 | 8.98 |
| Overall | 3.06 | 5.86 | 5.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javed, A.; Villanueva, H.; Shataer, S.; Vasciaveo, S.; Savva, R.; Orlova, E.V. Cryo-EM Structures of Two Bacteriophage Portal Proteins Provide Insights for Antimicrobial Phage Engineering. Viruses 2021, 13, 2532. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122532
Javed A, Villanueva H, Shataer S, Vasciaveo S, Savva R, Orlova EV. Cryo-EM Structures of Two Bacteriophage Portal Proteins Provide Insights for Antimicrobial Phage Engineering. Viruses. 2021; 13(12):2532. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122532
Chicago/Turabian StyleJaved, Abid, Hugo Villanueva, Shadikejiang Shataer, Sara Vasciaveo, Renos Savva, and Elena V. Orlova. 2021. "Cryo-EM Structures of Two Bacteriophage Portal Proteins Provide Insights for Antimicrobial Phage Engineering" Viruses 13, no. 12: 2532. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122532