
Targeting the Complement Serine Protease MASP-2 as a Therapeutic Strategy for Coronavirus Infections

 , , , , , and
, , , , , and 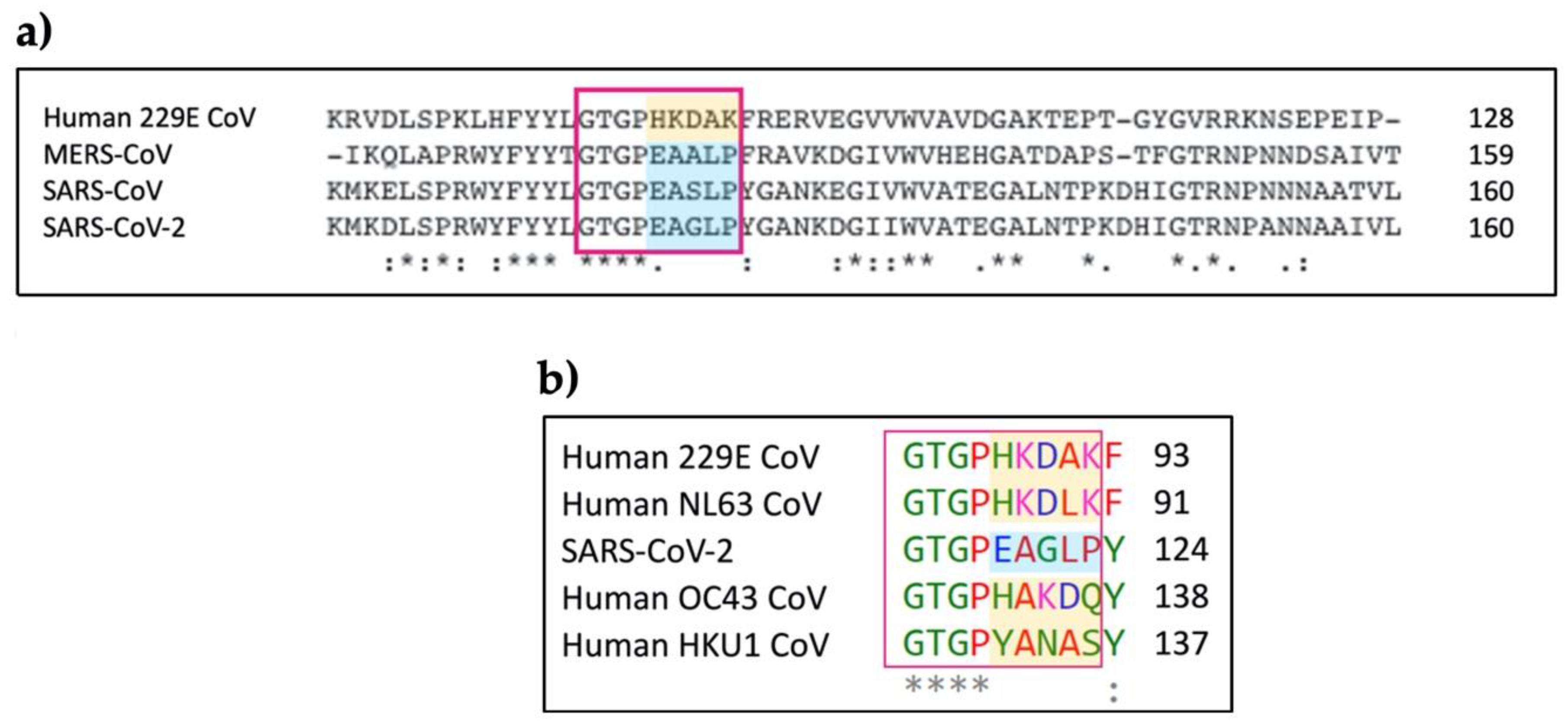{kind=link}
{kind=link}
{kind=link}
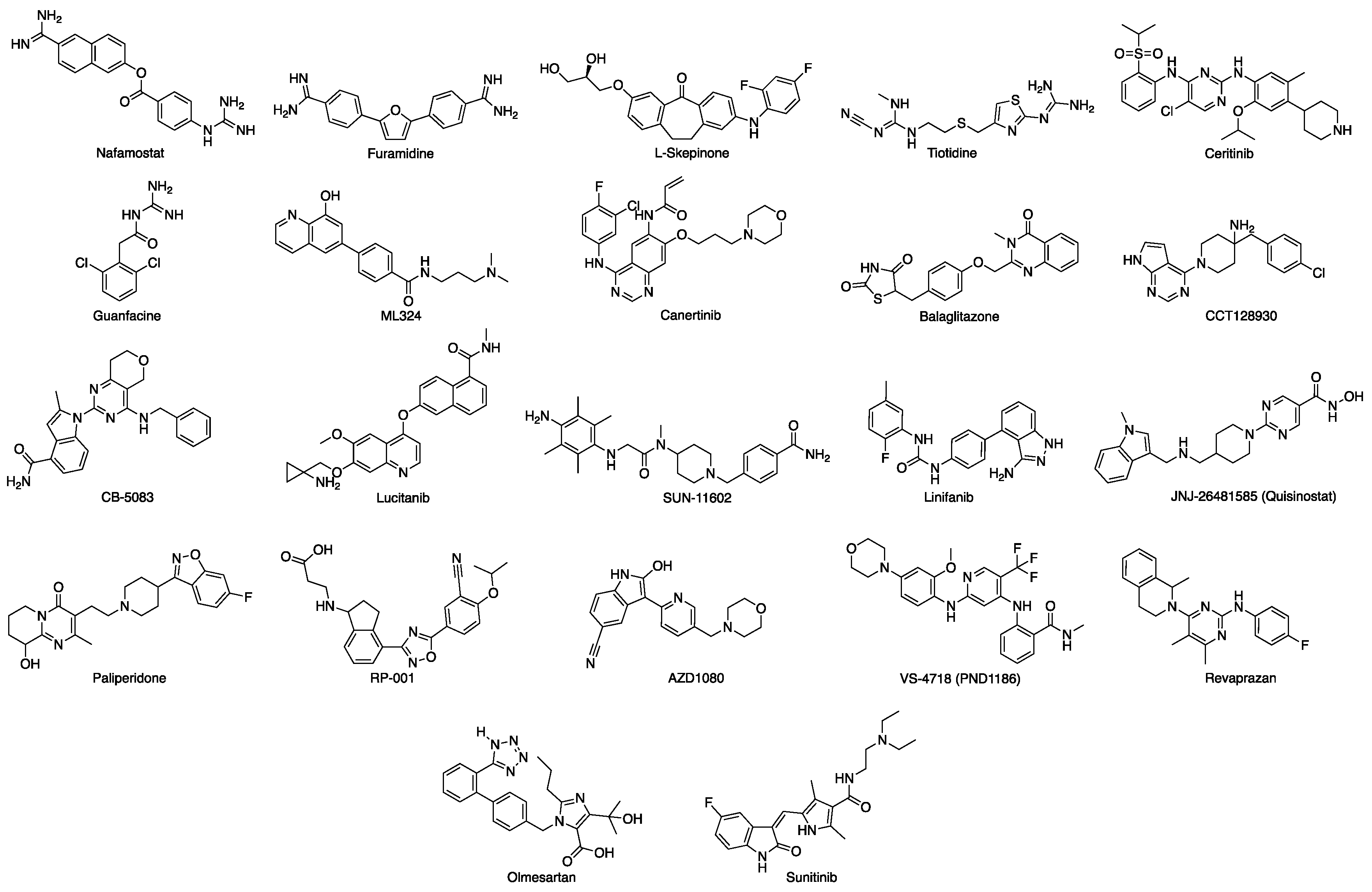{kind=link}
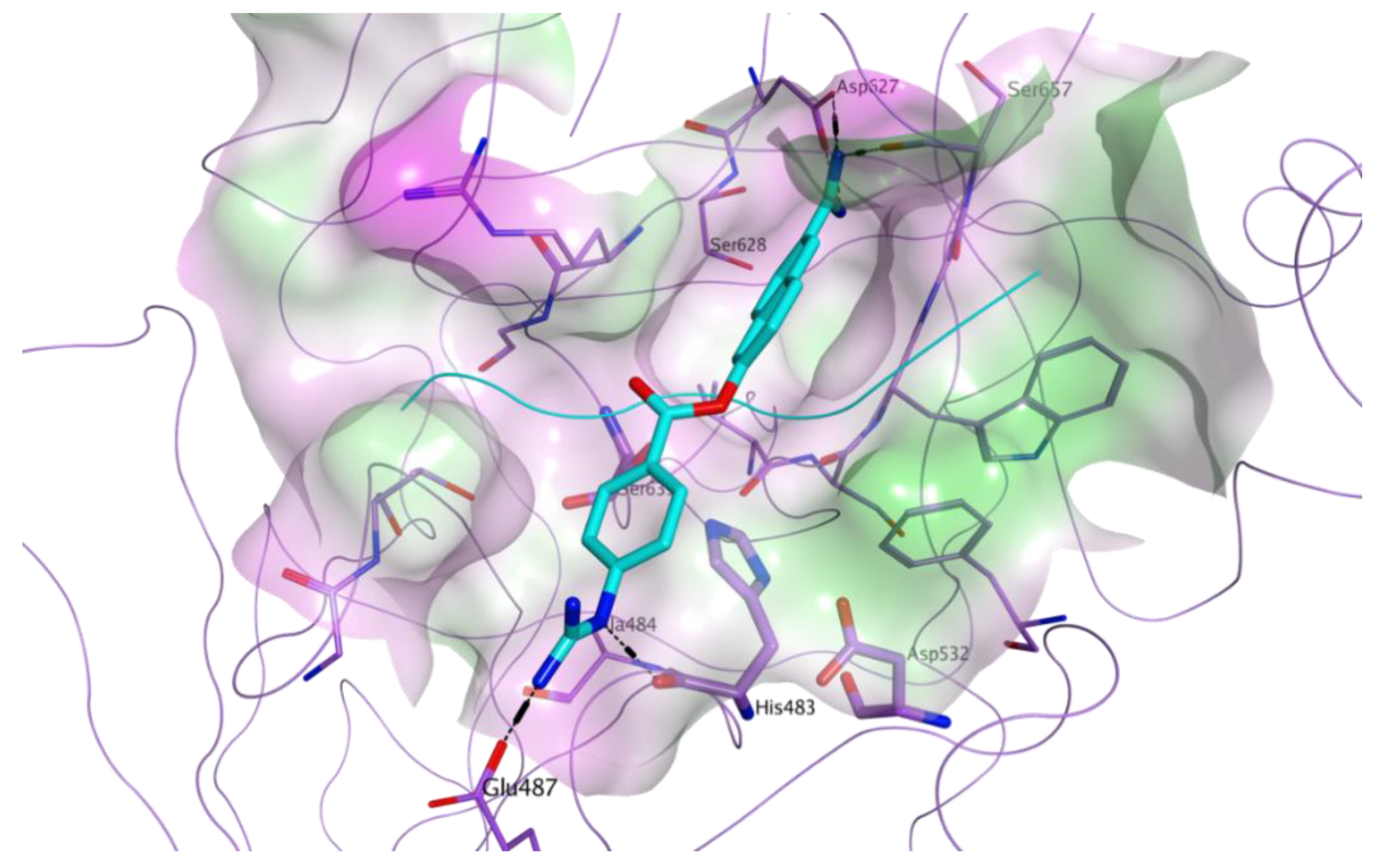{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Modelling
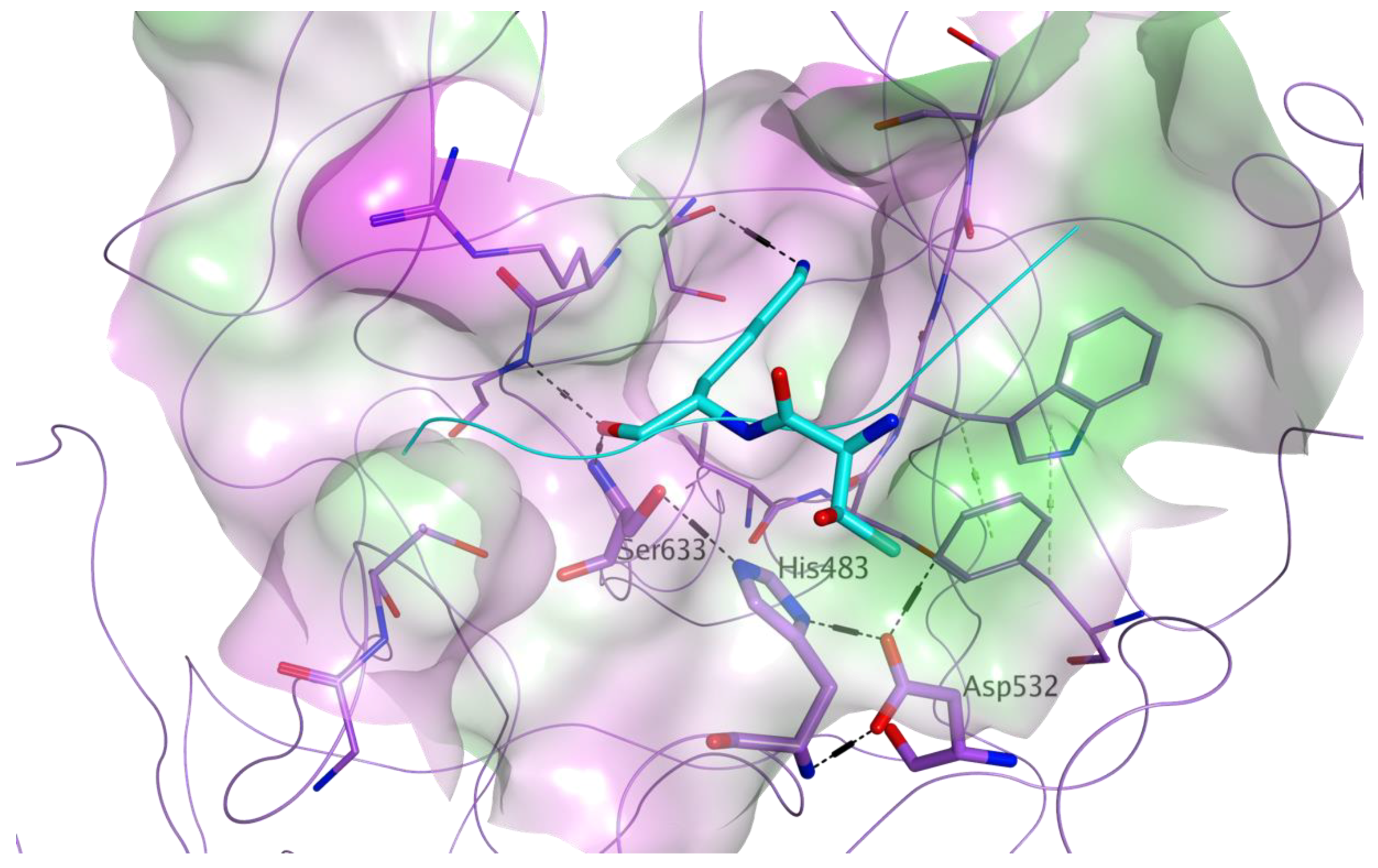
2.1.1. Docking-Based Virtual Screening on MASP-2 Catalytic Site
- ability of a compound to overall occupy MASP-2 active site.
- number of interactions formed between the compound and the protein residues defining the site (H-bonds, pi–pi interactions, etc.).
- coverage of different chemical scaffolds, discarding similar chemical entities.
2.1.2. Protein-Protein Docking between MASP-2 and SARS-CoV-2 N Protein and Druggable Site Analysis
2.1.3. Molecular Dynamics on the MASP-2-N-Protein Interaction Model
2.1.4. Docking-Based Virtual Screening at the Predicted Interaction Site between MASP-2 and SARS-CoV-2 N Protein
2.2. Complement MBL-Pathway Inhibition Assay
2.3. Statistical Analysis
3. Results and Discussion
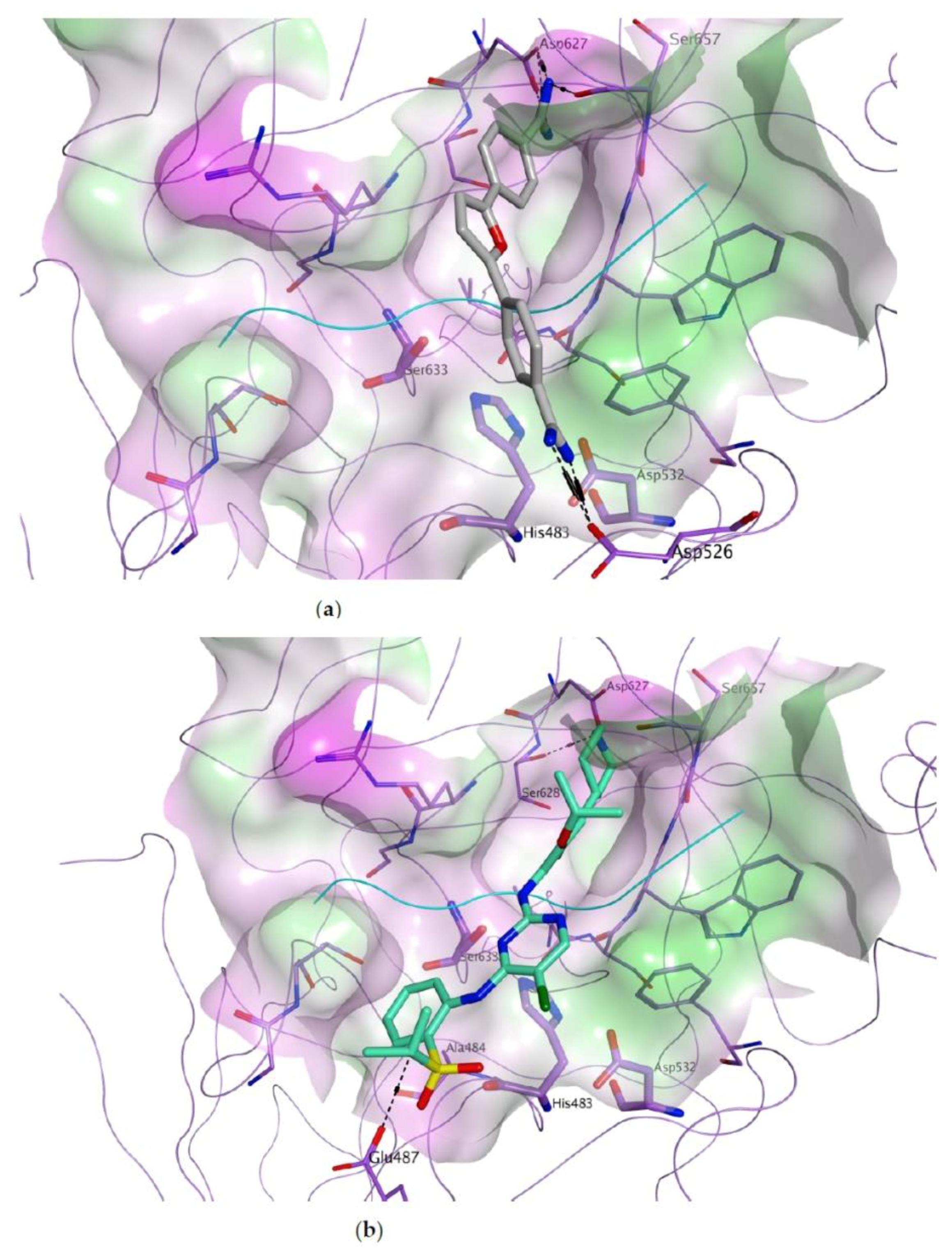
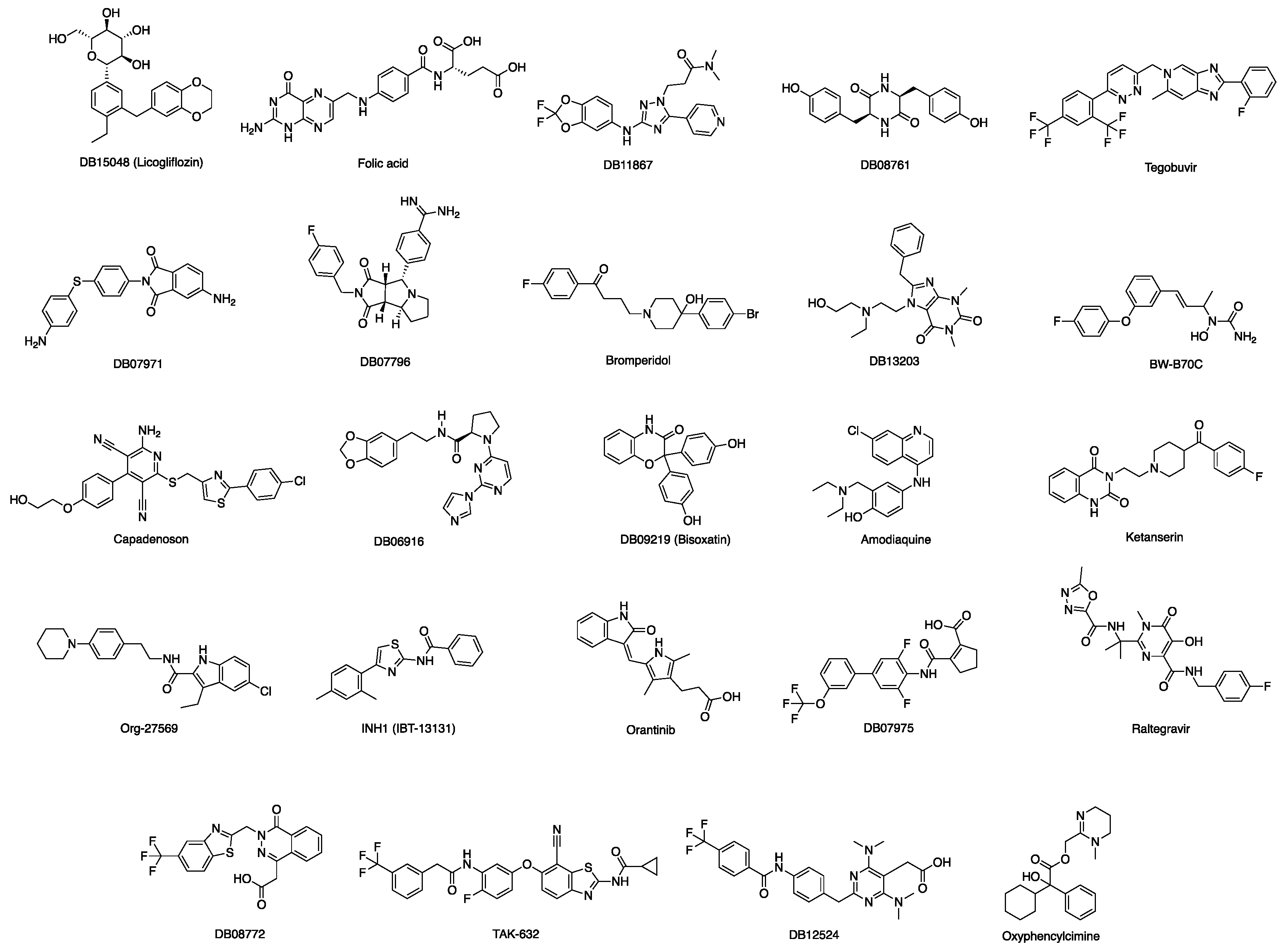
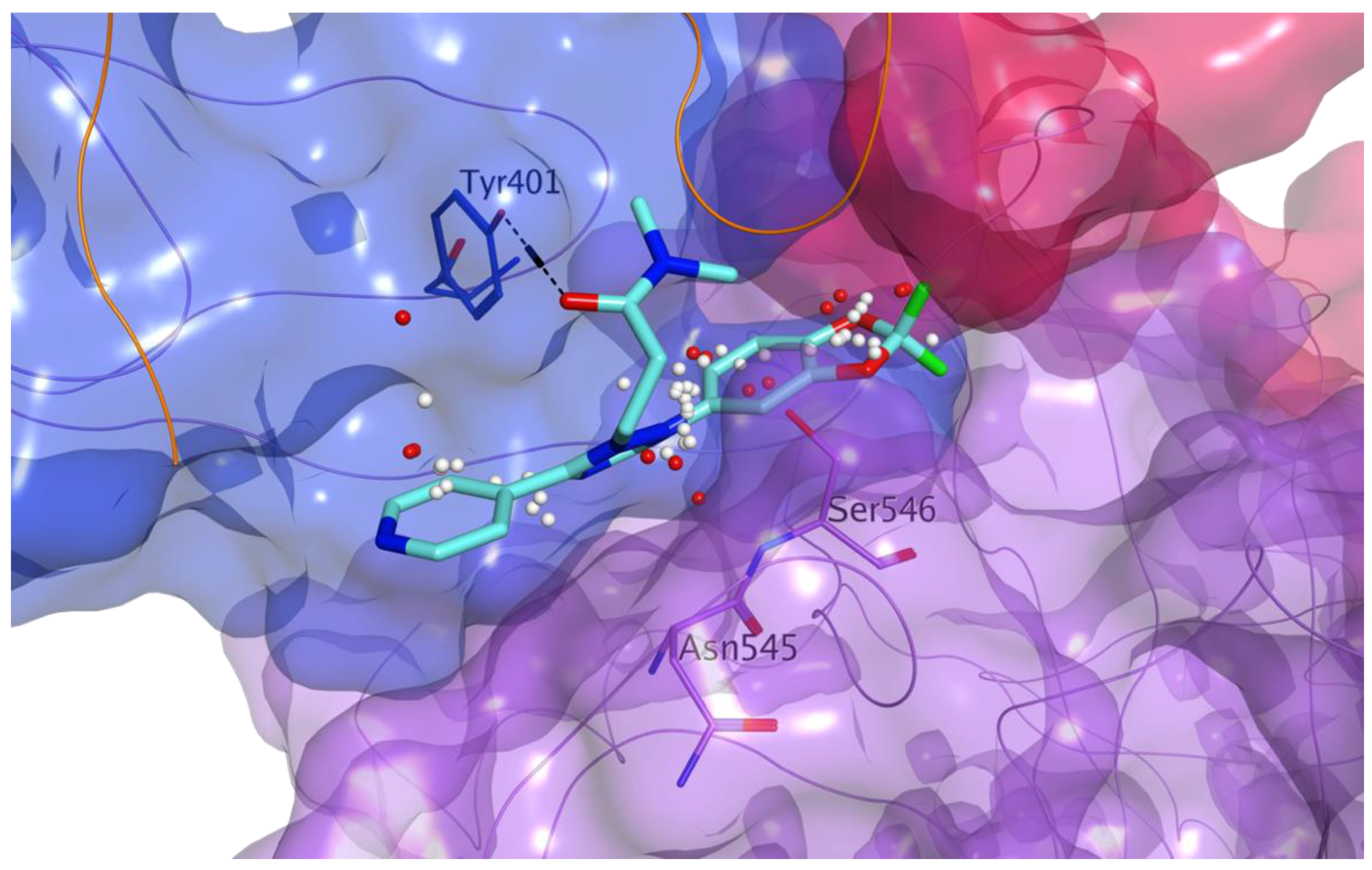
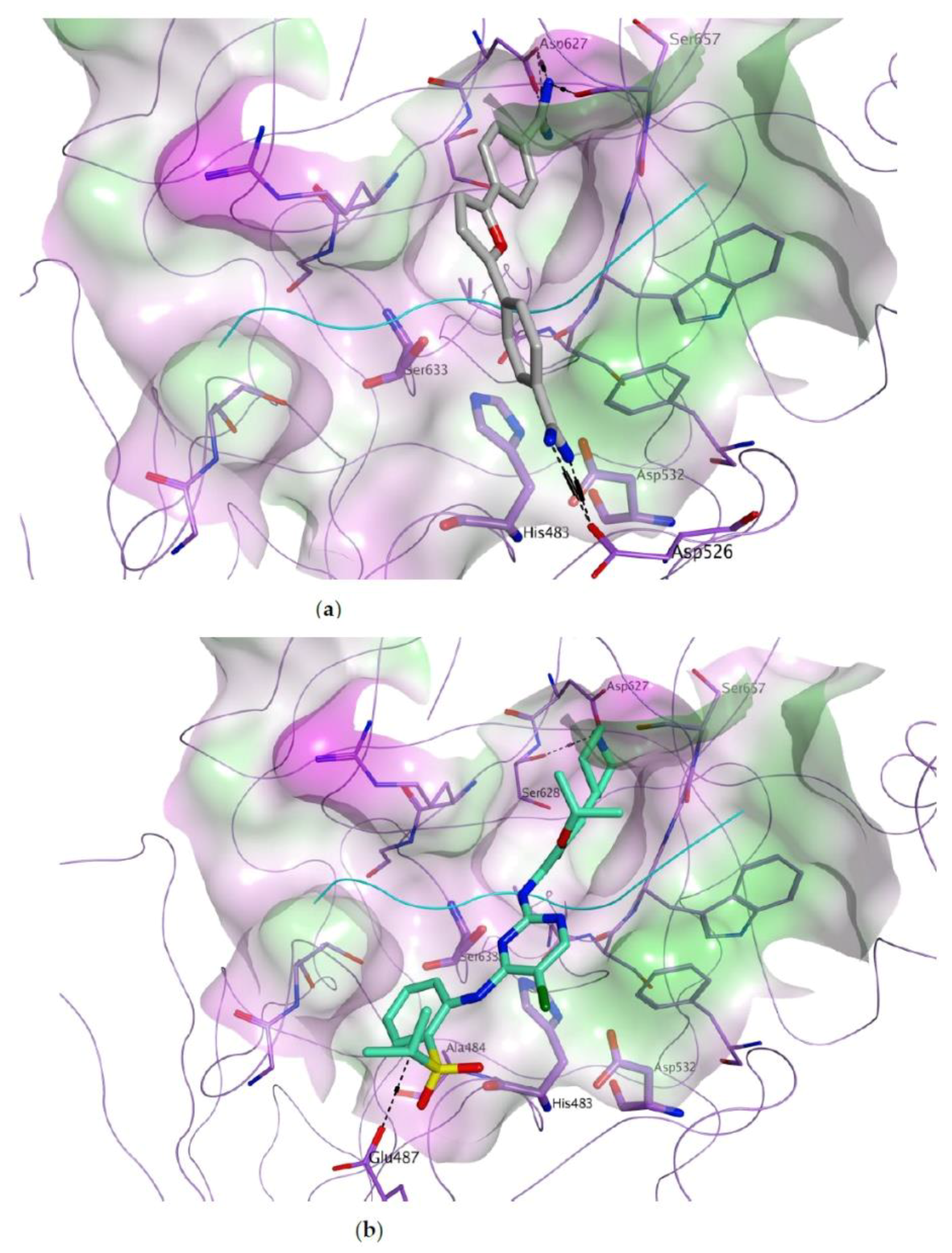
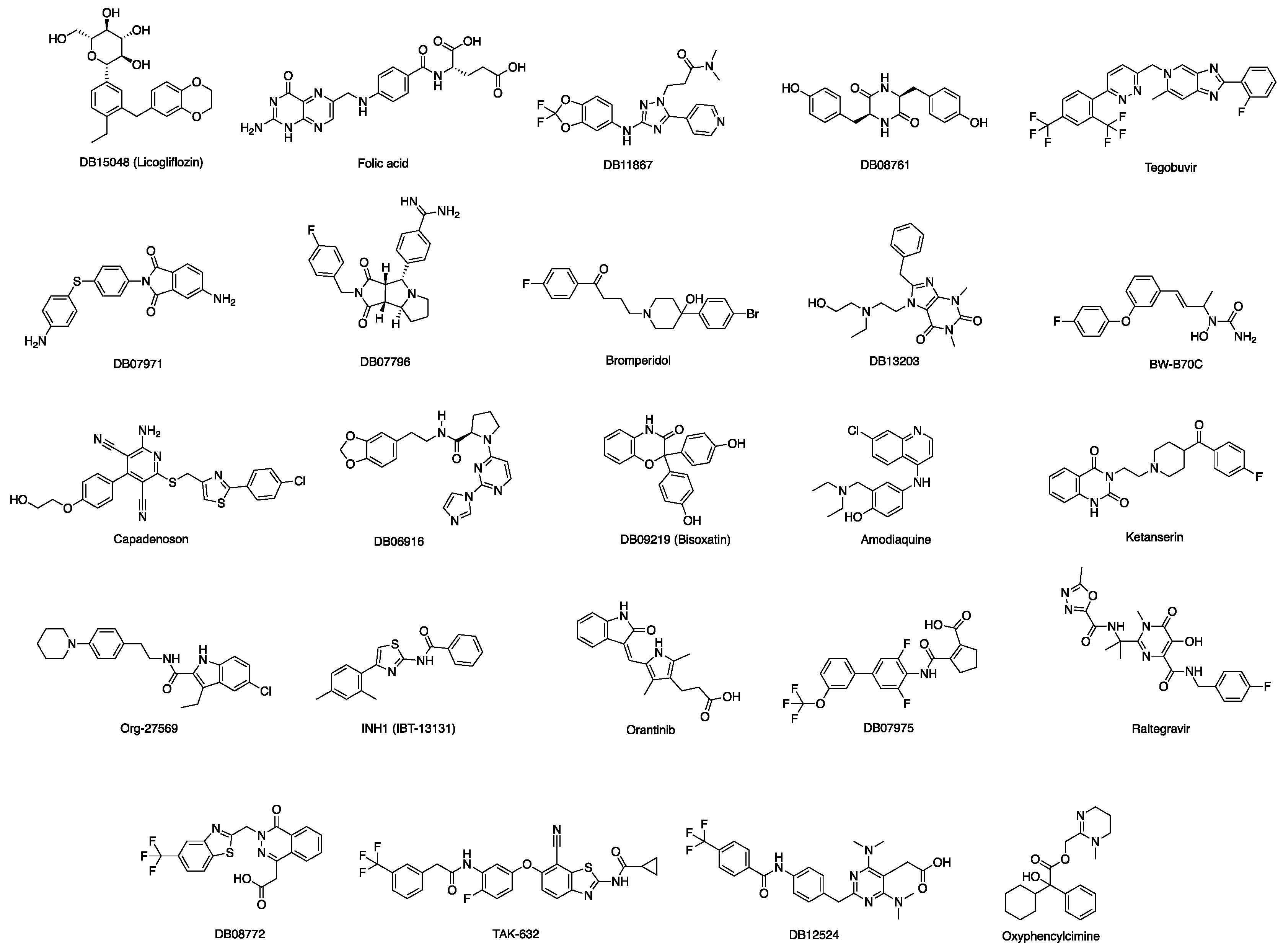
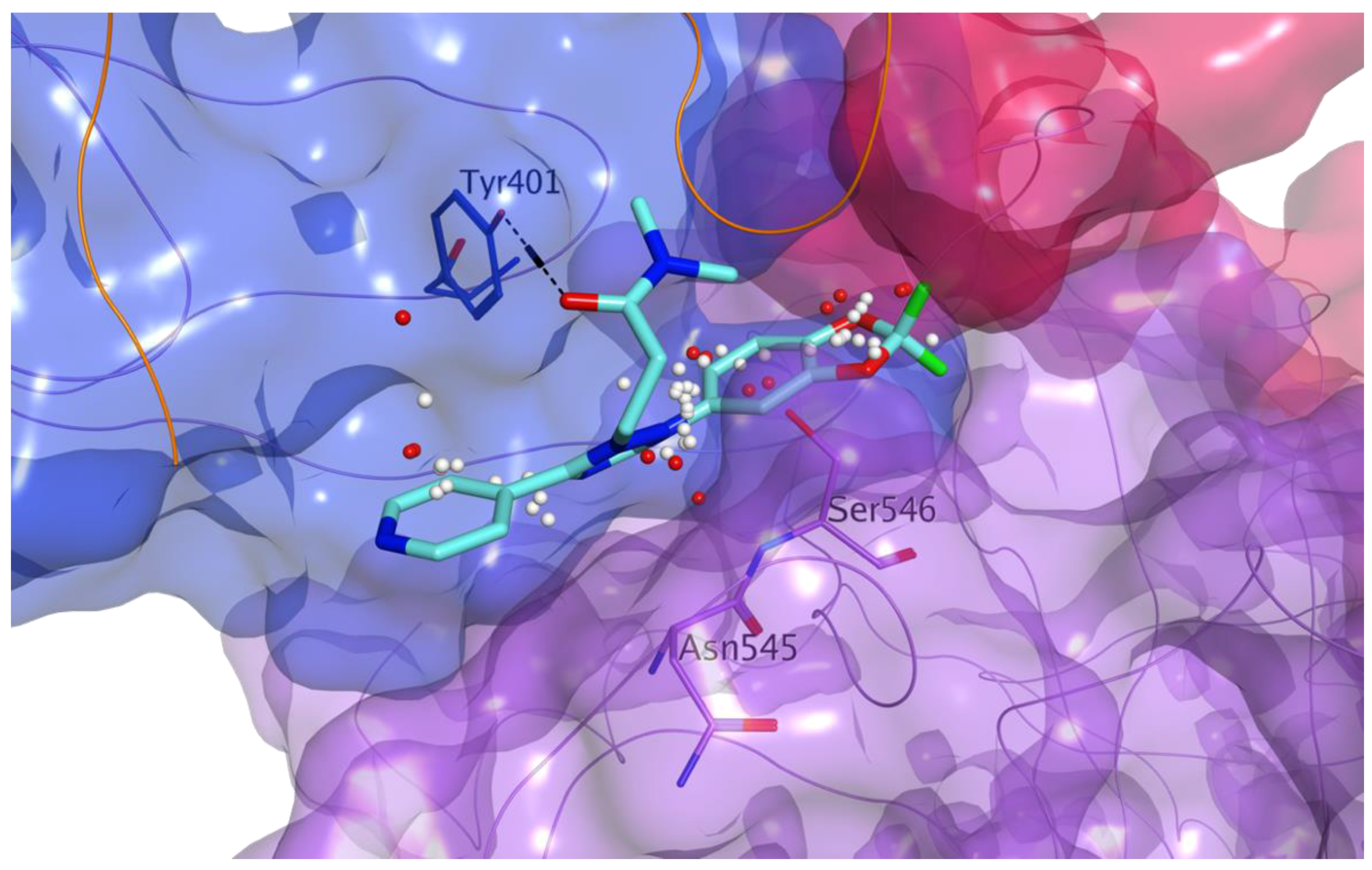
3.1. Docking-Based Virtual Screening of Drug-Repurposing Compounds on MASP-2 Catalytic Site
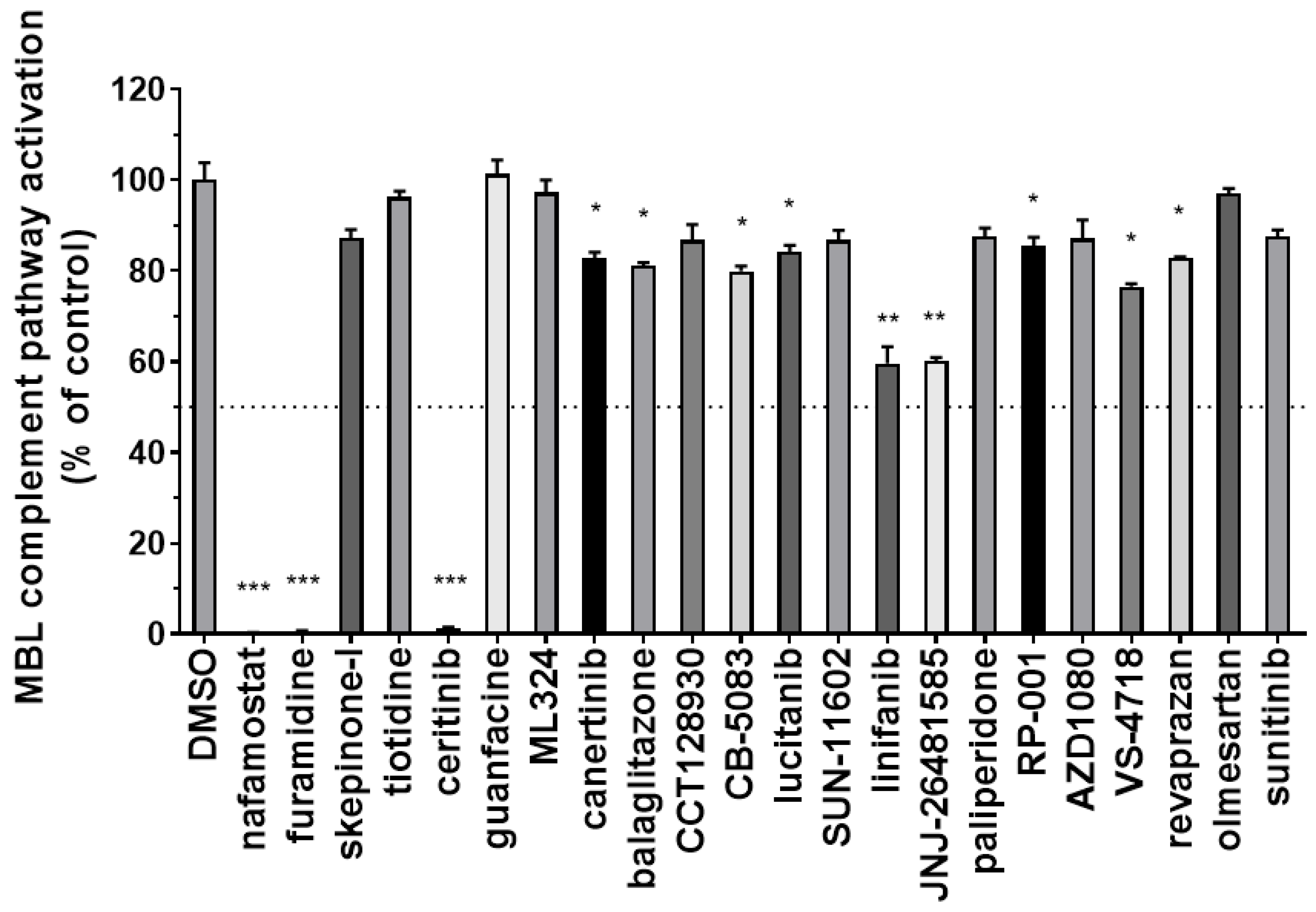
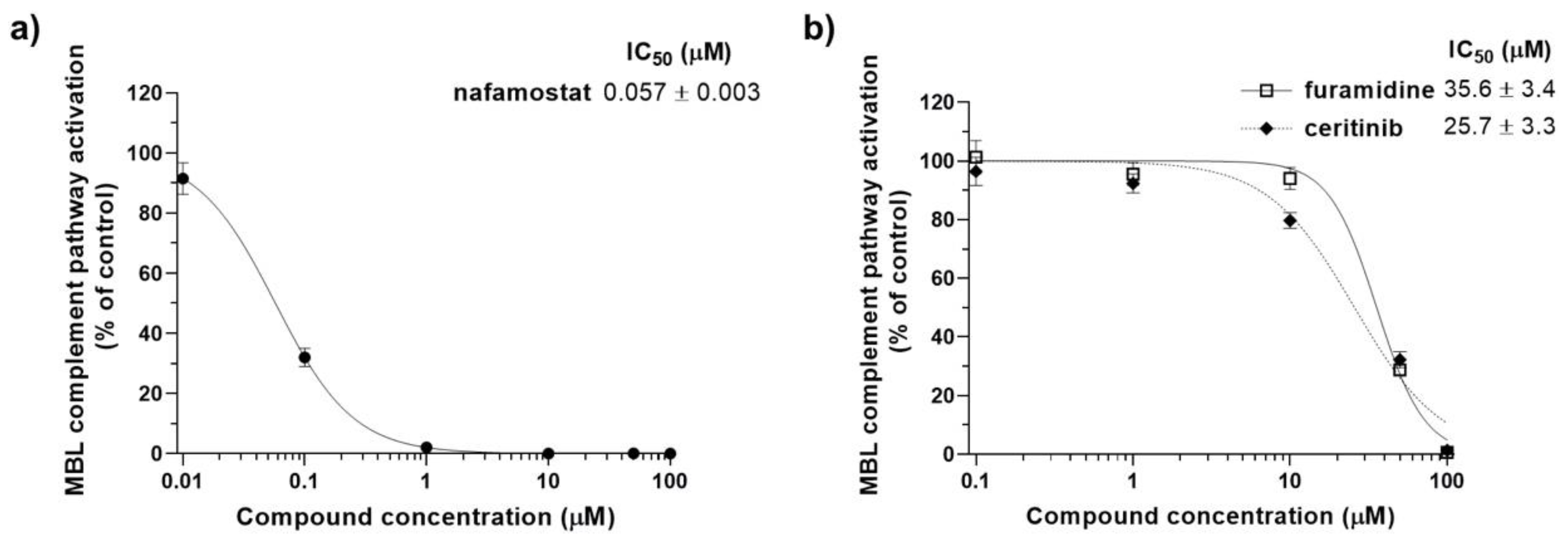
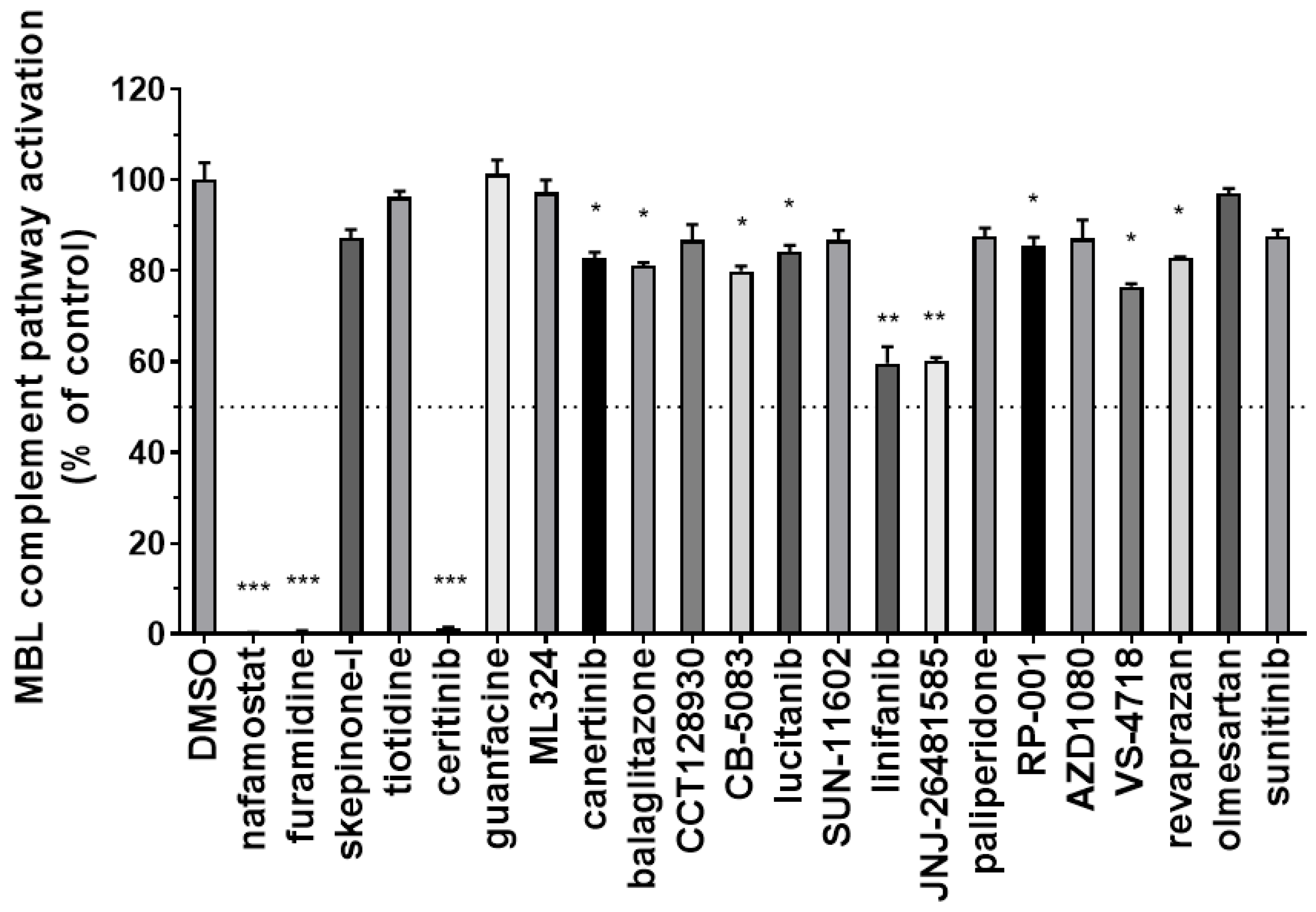
3.2. Identification of Inhibitors of the Complement MBL Pathway
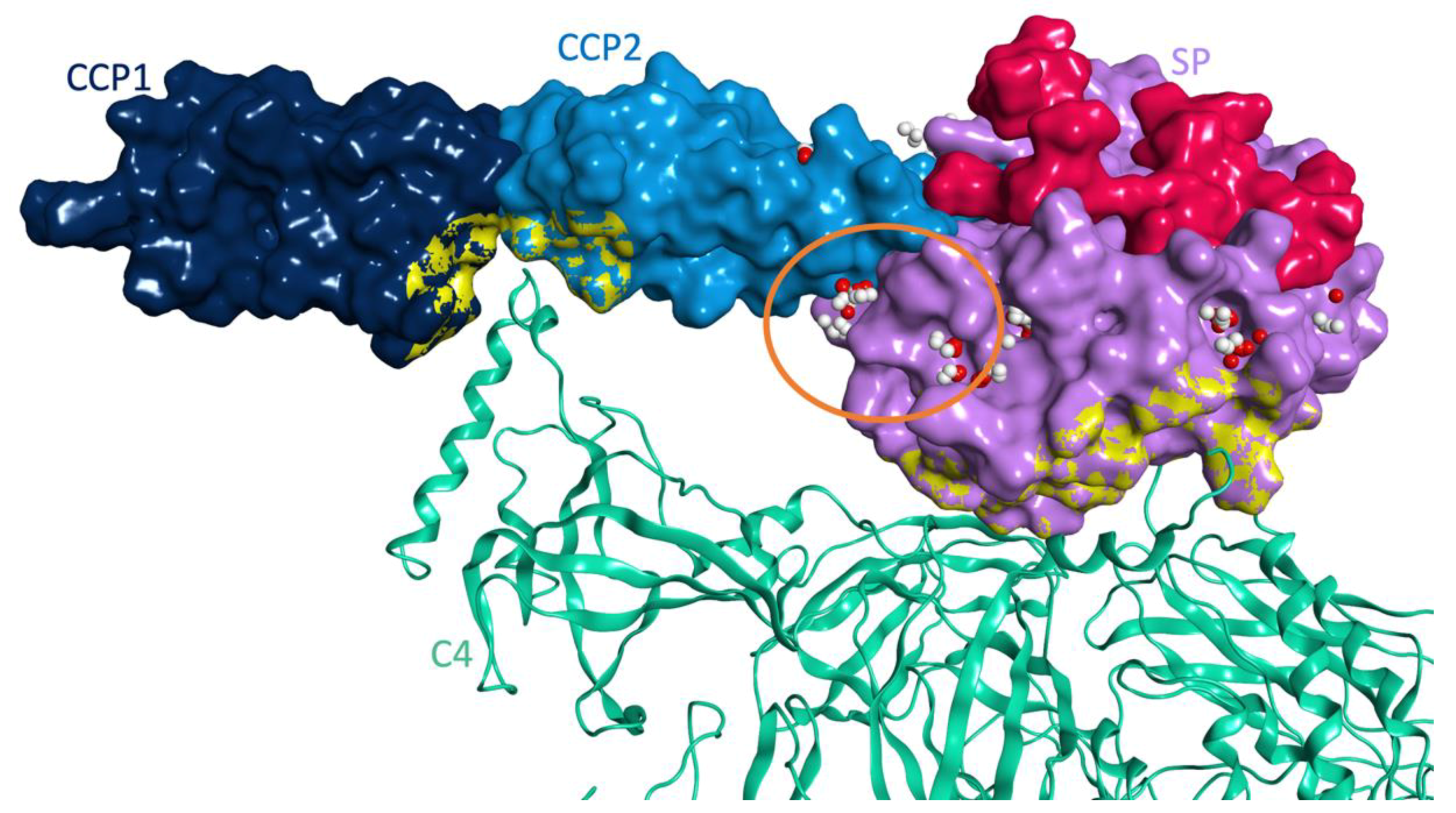
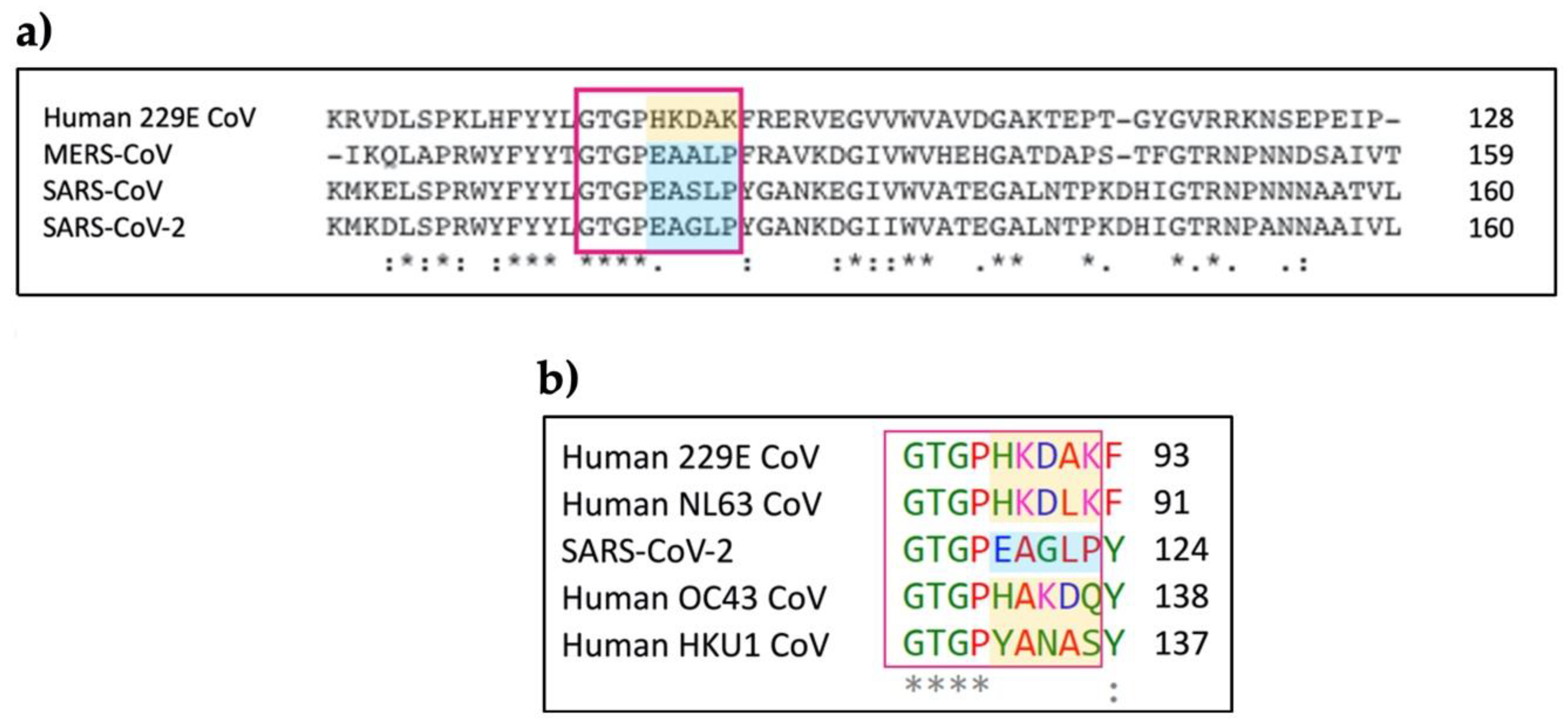
3.3. Molecular Model for the Protein-Protein Interaction between MASP-2 and Coronaviral N-Proteins
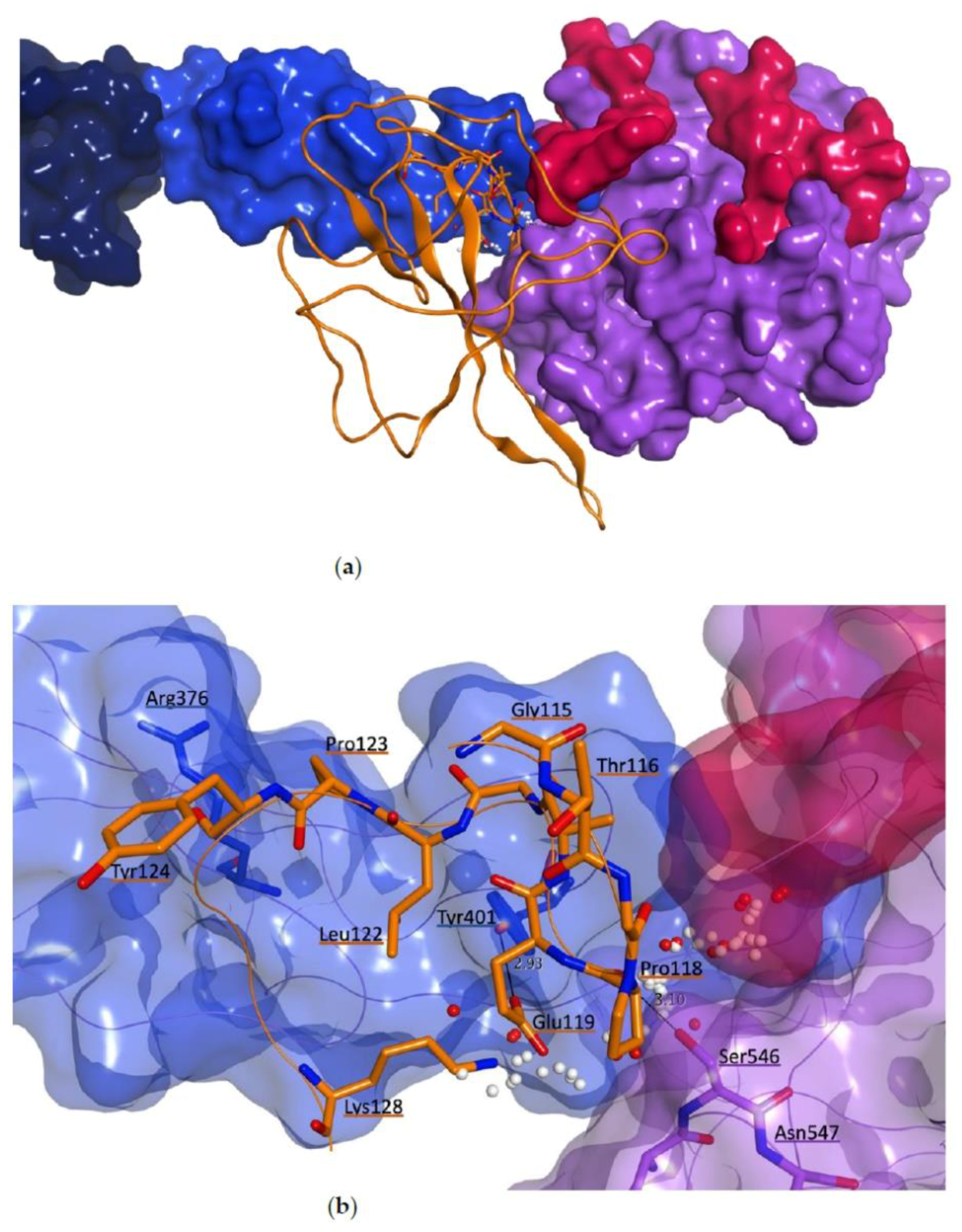
3.3.1. Protein-Protein Docking between MASP-2 and SARS-CoV-2 N Protein
3.3.2. Molecular Dynamics Studies for Structural Optimization of the Interaction Model between MASP-2 and SARS-CoV-2 N Protein
3.3.3. Virtual Screening on the Predicted Interaction Site between MASP-2 and SARS-CoV-2 N Protein
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stoermer, K.A.; Morrison, T.E. Complement and viral pathogenesis. Virology 2011, 411, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarma, V.J.; Huber-Lang, M.; Ward, P.A. Complement in lung disease. Autoimmunity 2006, 39, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, T.R.; Thiel, S.; Andersen, G.R. Toward a structure-based comprehension of the lectin pathway of complement. Mol. Immunol. 2013, 56, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Xiao, H.; Guo, R.; Li, Y.; Shen, B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg. Microbes Infect. 2015, 4, 1–7. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Ram Kumar Pandian, S.; Arunachalam, S.; Deepak, V.; Kunjiappan, S.; Sundar, K. Targeting complement cascade: An alternative strategy for COVID-19. 3 Biotech 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv 2020. [Google Scholar] [CrossRef]
- McBride, R.; Van Zyl, M.; Fielding, B.C. The Coronavirus Nucleocapsid Is a Multifunctional Protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1. Ref. Modul. Life Sci. 2020. [Google Scholar] [CrossRef]
- UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. Available online: https://www.uniprot.org (accessed on 30 November 2020). [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.G.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. Available online: https://www.ebi.ac.uk/Tools/msa/clustalo (accessed on 30 November 2020). [CrossRef] [PubMed]
- Rambaldi, A.; Gritti, G.; Micò, M.C.; Frigeni, M.; Borleri, G.; Salvi, A.; Landi, F.; Pavoni, C.; Sonzogni, A.; Gianatti, A.; et al. Endothelial injury and thrombotic microangiopathy in COVID-19: Treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology 2020, 225, 152001. [Google Scholar] [CrossRef]
- Guillén, L.; Padilla, S.; Fernández, M.; Agulló, V.; García, J.A.; Telenti, G.; García-Abellán, J.; Botella, Á.; Gutiérrez, F.; Masiá, M. Preemptive interleukin-6 blockade in patients with COVID-19. Sci. Rep. 2020, 10, 16826. [Google Scholar] [CrossRef]
- Matthay, M.A.; Thompson, B. Dexamethasone in hospitalised patients with COVID-19: Addressing uncertainties. Lancet 2020, 8, 1170–1172. [Google Scholar] [CrossRef]
- Chemical Computing Group, Inc. Molecular Operating Environment (MOE 2019.10); Chemical Computing Group, Inc.: Montreal, QC, Canada; Available online: http://www.chemcomp.com (accessed on 30 November 2020).
- Glide Schrödinger LLC. Schrödinger Release 2020-2; Glide Schrödinger LLC: New York, NY, USA, 2020; Available online: https://www.schrodinger.com/maestro (accessed on 30 November 2020).
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- BioSolveIT GmbH. SeeSAR, Version 9.2; BioSolveIT GmbH: Sankt Augustin, Germany, 2020; Available online: www.biosolveit.de/SeeSAR (accessed on 30 November 2020).
- OpenEye Scientific Software. OEDOCKING 4.0.0.0; OpenEye Scientific Software: Santa Fe, NM, USA. Available online: http://www.eyesopen.com (accessed on 30 November 2020).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. Available online: https://go.drugbank.com/ (accessed on 27 March 2020). [CrossRef] [PubMed]
- Drug Repurposing Hub, The Broad Institute, Cambridge, MA, USA. Available online: https://clue.io/repurposing (accessed on 27 March 2020).
- Harmat, V.; Gal, P.; Kardos, J.; Szilagyi, K.; Ambrus, G.; Vegh, B.; Naray-Szabo, G.; Zavodszky, P. The Structure of MBL-associated Serine Protease-2 Reveals that Identical Substrate Specificities of C1s and MASP-2 are Realized Through Different Sets of Enzyme–Substrate Interactions. J. Mol. Biol. 2004, 342, 1533–1546. [Google Scholar] [CrossRef] [PubMed]
- Heja, D.; Harmat, V.; Fodor, K.; Wilmanns, M.; Dobo, J.; Kekesi, K.A.; Zavodszky, P.; Gal, P.; Pal, G. Monospecific Inhibitors Show That Both Mannan-binding Lectin-associated Serine Protease-1 (MASP-1) and -2 Are Essential for Lectin Pathway Activation and Reveal Structural Plasticity of MASP-2. J. Biol. Chem. 2012, 287, 20290–20300. [Google Scholar] [CrossRef] [Green Version]
- Kidmose, R.T.; Laursen, N.S.; Dobó, J.; Kjaer, T.R.; Sirotkina, S.; Yatime, L.; Sottrup-Jensen, L.; Thiel, S.; Gál, P.; Andersen, G.R. Structural basis for activation of the complement system by component C4 cleavage. Proc. Natl. Acad. Sci. USA 2012, 109, 15425–15430. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Gal, P.; Harmat, V.; Kocsis, A.; Bian, T.; Barna, L.; Ambrus, G.; Vegh, B.; Balczer, J.; Sim, R.B.; Naray-Szabo, G.; et al. A true autoactivating enzyme: Structural insight into mannose-binding lectin-associated serine protease-2 activations. J. Biol. Chem. 2005, 280, 33435–33444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger. Maestro-Desmond Interoperability Tools, Version 3.1; Schrödinger: New York, NY, USA, 2020; Available online: https://www.schrodinger.com/maestro (accessed on 30 November 2020).
- Seelen, M.A.; Roos, A.; Wieslander, J.; Mollnes, T.E.; Sjfholm, A.G.; Wurzner, R.; Loos, M.; Tedesco, F.; Sim, R.B.; Garred, P.; et al. Functional analysis of the classical, alternative, and MBL pathways of the complement system: Standardization and validation of a simple ELISA. J. Immunol. Methods 2005, 296, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Rhee, J.-Y. Three cases of treatment with nafamostat in elderly patients with COVID-19 pneumonia who need oxygen therapy. Int. J. Infect. Dis. 2020, 96, 500–502. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, e00754-20. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G., Jr.; Sharma, A.; Ramaiah, A.; Sen, C.; Kohn, D.; Gomperts, B.; Svendsen, C.N.; Damoiseaux, R.D.; Arumugaswami, V. Antiviral Drug Screen of Kinase inhibitors Identifies Cellular Signaling Pathways Critical for SARS-CoV-2 Replication. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, P.G.; Tang, D.J.; Hua, Z.; Wang, Z.; An, J. Sunitinib reduces the infection of SARS-CoV, MERS-CoV and SARS-CoV-2 partially by inhibiting AP2M1 phosphorylation. Cell Discov. 2020, 6, 71. [Google Scholar] [CrossRef]
- Tagawa, T. Protease inhibitor nafamostat mesilate attenuates complement activation and improves function of xenografts in a discordant lung perfusion model. Xenotransplantation 2011, 18, 315–319. [Google Scholar] [CrossRef]
- Ko, M.; Jeon, S.; Ryu, W.-S.; Kim, S. Comparative analysis of antiviral efficacy of FDA-approved drugs against SARS-CoV-2 in human lung cells. J. Med. Virol. 2020, 93, 1403–1408. [Google Scholar] [CrossRef]
- Saikatendu, K.S.; Joseph, J.S.; Subramanian, V.; Neuman, B.W.; Buchmeier, M.J.; Stevens, R.C.; Kuhn, P. Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N-terminal domain of N protein. J. Virol. 2007, 81, 3913–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.M.; Lin, S.C.; Hsu, J.N.; Chang, C.K.; Chien, C.M.; Wang, Y.S.; Wu, H.Y.; Jeng, U.S.; Kehn-Hall, K.; Hou, M.H. Structure-Based Stabilization of Non-native Protein-Protein Interactions of Coronavirus Nucleocapsid Proteins in Antiviral Drug Design. J. Med. Chem. 2020, 63, 3131–3141. [Google Scholar] [CrossRef]
- Acosta-Elias, J.; Espinosa-Tanguma, R. The Folate Concentration and/or Folic Acid Metabolites in Plasma as Factor for COVID-19 Infection. Front. Pharmacol. 2020, 11, 1062. [Google Scholar] [CrossRef]
- Rosa, S.G.V.; Santos, W.C. Clinical trials on drug repositioning for COVID-19 treatment. Rev. Panam. Salud Publica 2020, 44, e40. [Google Scholar] [CrossRef] [PubMed]
- Unni, S.; Aouti, S.; Thiyagarajan, S.; Padmanabhan, B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020, 45, 130. [Google Scholar] [CrossRef] [PubMed]
- Bocci, G.; Bradfute, S.B.; Ye, C.; Garcia, M.J.; Parvathareddy, J.; Reichard, W.; Surendranathan, S.; Bansal, S.; Bologa, C.G.; Perkins, D.J.; et al. Virtual and In Vitro Antiviral Screening Revive Therapeutic Drugs for COVID-19. ACS Pharmacol. Transl. Sci. 2020, 3, 1278–1292. [Google Scholar] [CrossRef]
- Kuindersma, M.; Spronk, P.E. Ketanserin as potential additive drug to improve V/Q mismatch in COVID-19? Crit. Care 2020, 24, 526. [Google Scholar] [CrossRef]
- Frediansyah, A.; Tiwari, R.; Sharun, K.; Dhama, K.; Harapan, H. Antivirals for COVID-19: A critical review. Clin. Epidemiol. Glob. Health 2021, 9, 90–98. [Google Scholar] [CrossRef]
- Pellegrino, B.; Facchinetti, F.; Bordi, P.; Silva, M.; Gnetti, L.; Tiseo, M. Lung Toxicity in Non-Small-Cell Lung Cancer Patients Exposed to ALK Inhibitors: Report of a Peculiar Case and Systematic Review of the Literature. Clin. Lung Cancer 2018, 19, e151–e161. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flude, B.M.; Nannetti, G.; Mitchell, P.; Compton, N.; Richards, C.; Heurich, M.; Brancale, A.; Ferla, S.; Bassetto, M. Targeting the Complement Serine Protease MASP-2 as a Therapeutic Strategy for Coronavirus Infections. Viruses 2021, 13, 312. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020312
Flude BM, Nannetti G, Mitchell P, Compton N, Richards C, Heurich M, Brancale A, Ferla S, Bassetto M. Targeting the Complement Serine Protease MASP-2 as a Therapeutic Strategy for Coronavirus Infections. Viruses. 2021; 13(2):312. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020312
Chicago/Turabian StyleFlude, Ben M., Giulio Nannetti, Paige Mitchell, Nina Compton, Chloe Richards, Meike Heurich, Andrea Brancale, Salvatore Ferla, and Marcella Bassetto. 2021. "Targeting the Complement Serine Protease MASP-2 as a Therapeutic Strategy for Coronavirus Infections" Viruses 13, no. 2: 312. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020312