
Integrase-Defective Lentiviral Vector Is an Efficient Vaccine Platform for Cancer Immunotherapy


 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
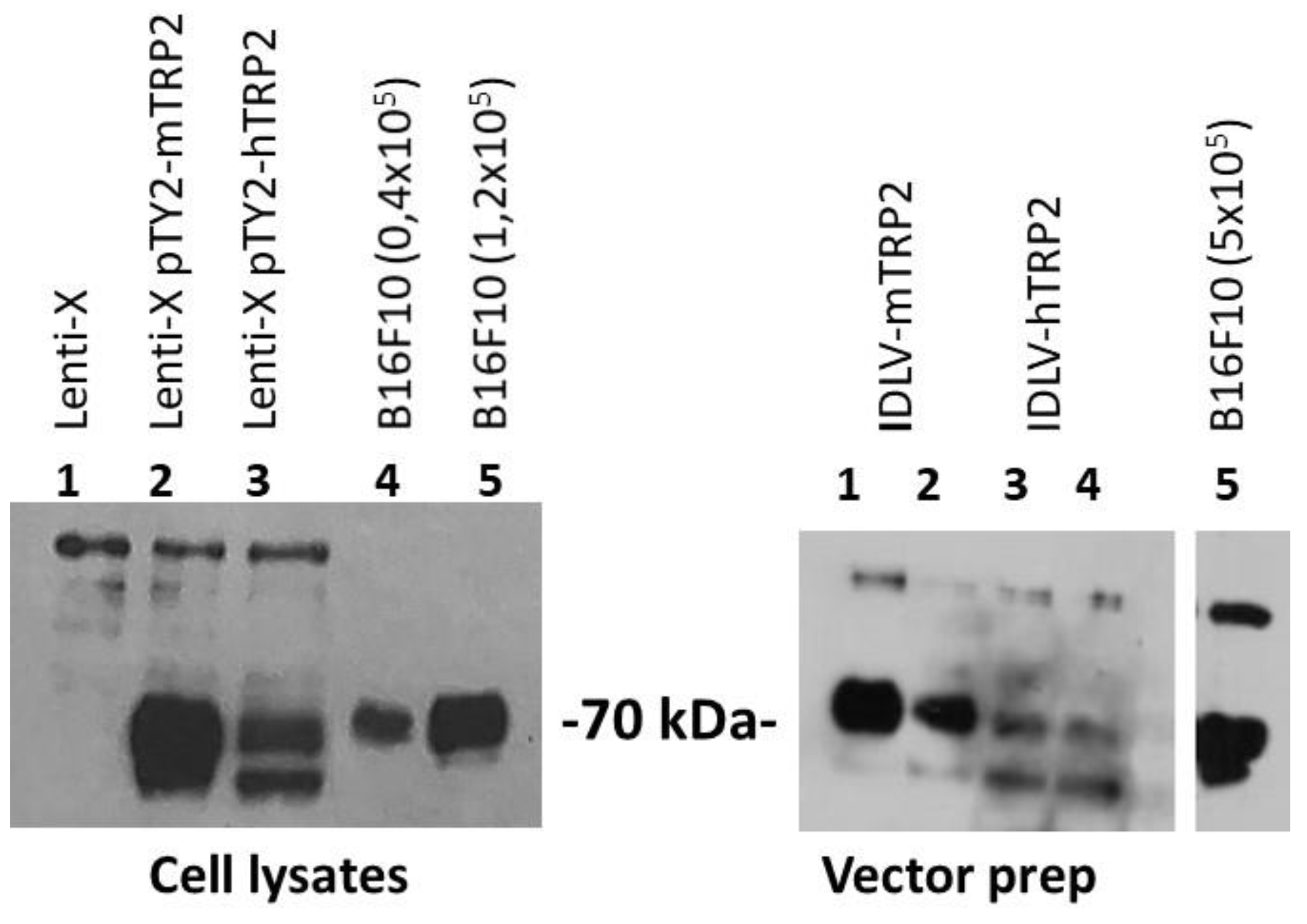
2.2. Construction and Production of IDLV
2.3. Western Blotting
2.4. In Vivo Experiments
2.5. Tissue Processing
2.6. IFNγ ELISpot Assay
2.7. Measurement of Specific IgG Antibodies by ELISA
2.8. Intracellular Staining to Detect Cytokines Production
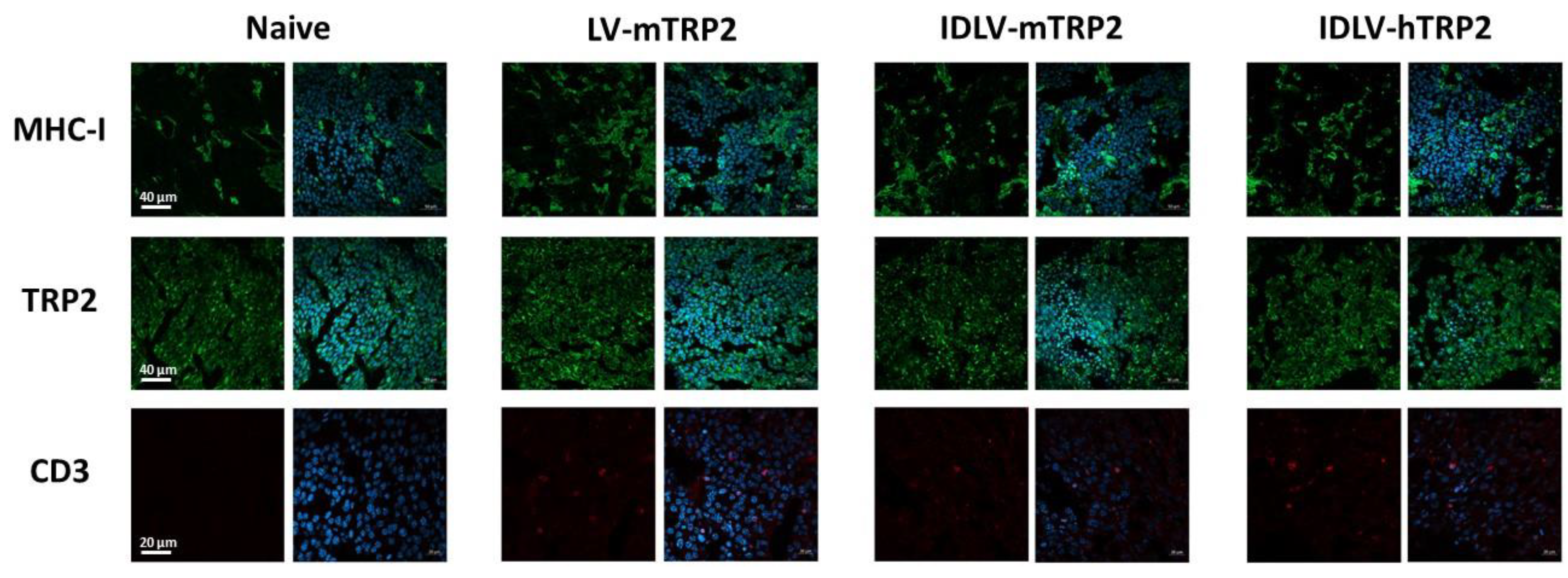
2.9. Confocal Laser Scanning Microscopy (CLSM) Analyses
2.10. Statistical Analysis
3. Results
3.1. A Single Immunization with IDLV-OVA in E.G7-OVA-Bearing Mice Eradicated the Large Tumor Mass and Generated a Protective Anti-Tumor Memory Response
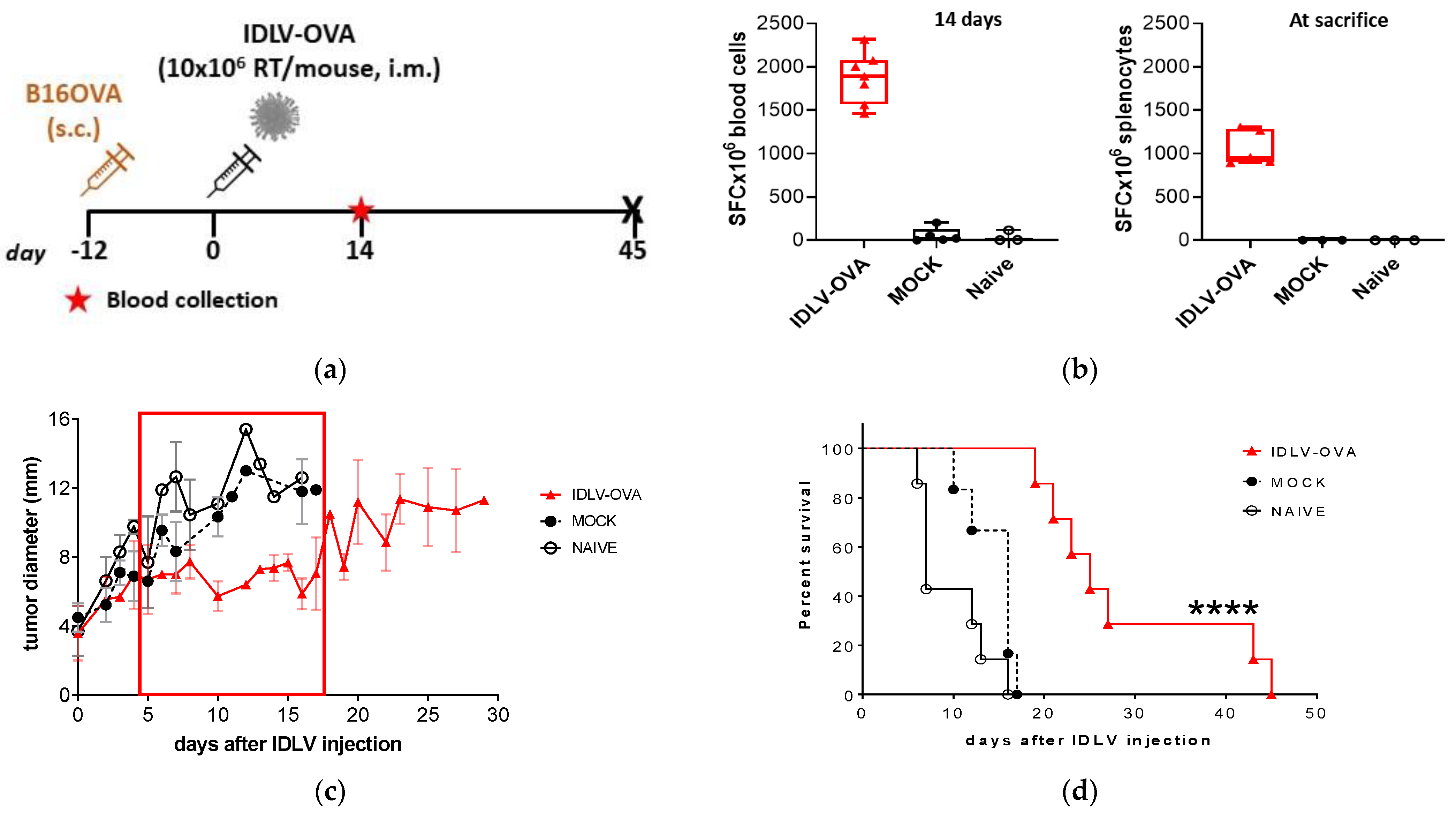
3.2. IDLV-OVA Vaccination Controlled B16OVA Tumor Growth and Survival in Both Preventive and Therapeutic Settings
3.3. Self-TAA in the Melanoma Model
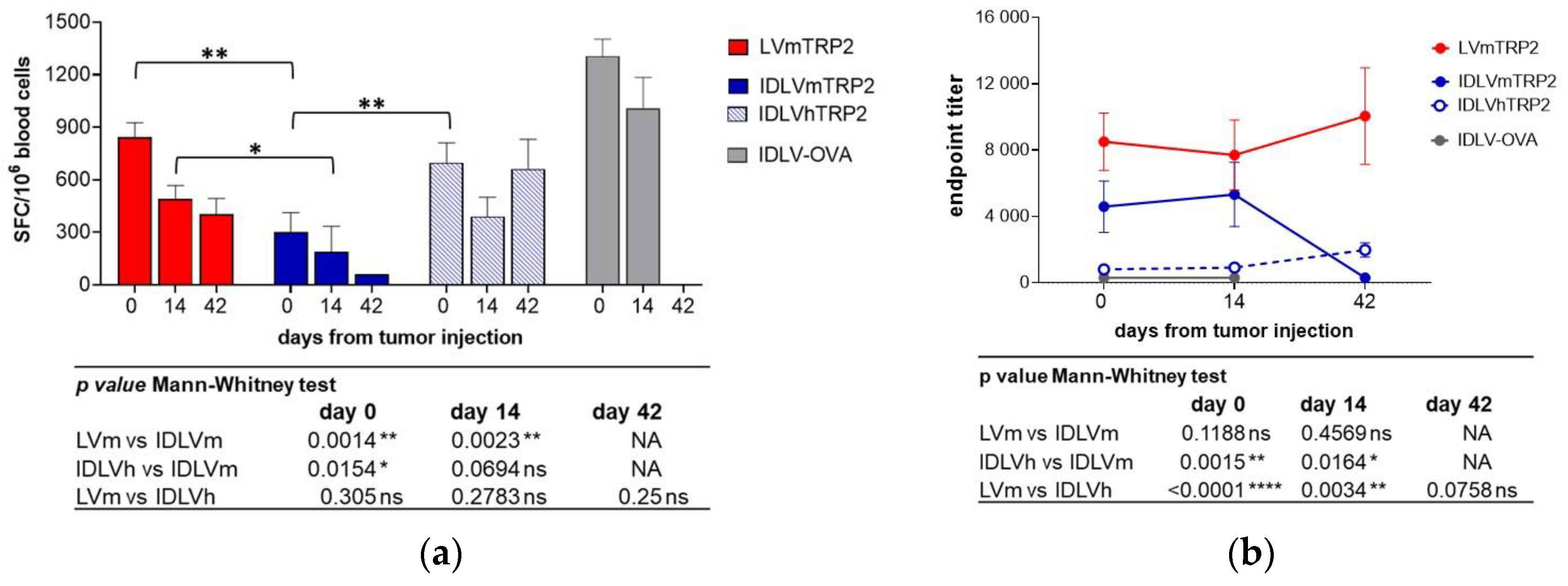
3.4. The human TRP2 as an Alternative Antigen Together with an Increase of Dose Resulted in a Better Efficacy of IDLV Vaccination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N. Engl. J. Med. 2007, 356, 1915–1927. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef]
- Park, H.Y.; Kosmadaki, M.; Yaar, M.; Gilchrest, B.A. Cellular mechanisms regulating human melanogenesis. Cell Mol. Life Sci. 2009, 66, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Negroiu, G.; Dwek, R.A.; Petrescu, S.M. The inhibition of early N-glycan processing targets TRP-2 to degradation in B16 melanoma cells. J. Biol. Chem. 2003, 278, 27035–27042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, W.; Pak, B.J.; Bani, M.R.; Kapoor, M.; Lu, S.J.; Tamir, A.; Kerbel, R.S.; Ben-David, Y. Tyrosinase-related protein 2 as a mediator of melanoma specific resistance to cis-diamminedichloroplatinum (II): Therapeutic implications. Oncogene 2000, 19, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overwijk, W.W.; Restifo, N.P. B16 as a mouse model for human melanoma. Curr. Protocol. Immunol. 2001, 20. [Google Scholar] [CrossRef]
- Bloom, M.B.; Perry-Lalley, D.; Robbins, P.F.; Li, Y.; el-Gamil, M.; Rosenberg, S.A.; Yang, J.C. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J. Exp. Med. 1997, 185, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Castelli, C.; Tarsini, P.; Mazzocchi, A.; Rini, F.; Rivoltini, L.; Ravagnani, F.; Gallino, F.; Belli, F.; Parmiani, G. Novel HLA-Cw8-restricted T cell epitopes derived from tyrosinase-related protein-2 and gp100 melanoma antigens. J. Immunol. 1999, 162, 1739–1748. [Google Scholar]
- Muntasell, A.; Ochoa, M.C.; Cordeiro, L.; Berraondo, P.; López-Díaz de Cerio, A.; Cabo, M.; López-Botet, M.; Melero, I. Targeting NK-cell checkpoints for cancer immunotherapy. Curr. Opin. Immunol. 2017, 45, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Smith, T.; Grey, F.; Hill, A.B. Cytomegalovirus-based cancer vaccines expressing TRP2 induce rejection of melanoma in mice. Biochem. Biophys. Res. Commun. 2013, 437, 287–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.A. Gene-based vaccines: Recent developments. Curr. Opin. Mol. Ther. 2010, 12, 86–93. [Google Scholar] [PubMed]
- Koup, R.A.; Douek, D.C. Vaccine design for CD8 T lymphocyte responses. Cold Spring Harb. Perspect. Med. 2011, 1, a007252. [Google Scholar] [CrossRef] [Green Version]
- Hematti, P.; Hong, B.K.; Ferguson, C.; Adler, R.; Hanawa, H.; Sellers, S.; Holt, I.E.; Eckfeldt, C.E.; Sharma, Y.; Schmidt, M.; et al. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004, 2, e423. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, E234. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [Green Version]
- Vargas, J., Jr.; Gusella, G.L.; Najfeld, V.; Klotman, M.E.; Cara, A. Novel integrase-defective lentiviral episomal vectors for gene transfer. Hum. Gene Ther. 2004, 15, 361–372. [Google Scholar] [CrossRef]
- Cara, A.; Klotman, M.E. Retroviral E-DNA: Persistence and gene expression in non dividing immune cells. J. Leukoc. Biol. 2006, 80, 1013–1017. [Google Scholar] [CrossRef]
- Lin, Y.Y.; Belle, I.; Blasi, M.; Huang, M.N.; Buckley, A.F.; Rountree, W.; Klotman, M.E.; Cara, A.; Negri, D. Skeletal Muscle Is an Antigen Reservoir in Integrase-Defective Lentiviral Vector-Induced Long-Term Immunity. Mol. Ther. Methods Clin. Dev. 2020, 17, 532–544. [Google Scholar] [CrossRef]
- Negri, D.R.; Michelini, Z.; Bona, R.; Blasi, M.; Filati, P.; Leone, P.; Rossi, A.; Franco, M.; Cara, A. Integrase-defective lentiviral vector- based vaccine: A new vector for induction of T cell immunity. Expert Opin. Biol. Ther. 2011, 1, 739–750. [Google Scholar] [CrossRef]
- Apolonia, L. The Old and the New: Prospects for Non-Integrating Lentiviral Vector Technology. Viruses 2020, 12, 1103. [Google Scholar] [CrossRef]
- Negri, D.R.; Michelini, Z.; Baroncelli, S.; Spada, M.; Vendetti, S.; Buffa, V.; Bona, R.; Leone, P.; Klotman, M.E.; Cara, A. Successful Immunization with a Single Injection of Non-integrating Lentiviral Vector. Mol. Ther. 2007, 15, 1716–1723. [Google Scholar] [CrossRef]
- Michelini, Z.; Negri, D.R.; Baroncelli, S.; Spada, M.; Leone, P.; Bona, R.; Klotman, M.E.; Cara, A. Development and use of SIV-based Integrase defective lentiviral vector for immunization. Vaccine 2009, 27, 4622–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negri, D.; Blasi, M.; LaBranche, C.; Parks, R.; Balachandran, H.; Lifton, M.; Shen, X.; Denny, T.; Ferrari, G.; Vescio, M.F.; et al. Immunization with an SIV-based IDLV Expressing HIV-1 Env 1086 Clade C Elicits Durable Humoral and Cellular Responses in Rhesus Macaques. Mol. Ther. 2016, 24, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Grasso, F.; Negri, D.R.; Mochi, S.; Rossi, A.; Cesolini, A.; Giovannelli, A.; Chiantore, M.V.; Leone, P.; Giorgi, C.; Cara, A. Successful therapeutic vaccination with integrase defective lentiviral vector expressing nononcogenic human papillomavirus E7 protein. Int. J. Cancer 2013, 132, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Pollack, S.M.; Lu, H.; Gnjatic, S.; Somaiah, N.; O’Malley, R.B.; Jones, R.L.; Hsu, F.J.; Ter Meulen, J. First-in-Human Treatment With a Dendritic Cell-targeting Lentiviral Vector-expressing NY-ESO-1, LV305, Induces Deep, Durable Response in Refractory Metastatic Synovial Sarcoma Patient. J. Immunother. 2017, 40, 302–306. [Google Scholar] [CrossRef]
- Cousin, C.; Oberkampf, M.; Felix, T.; Rosenbaum, P.; Weil, R.; Fabrega, S.; Morante, V.; Negri, D.; Cara, A.; Dadaglio, G.; et al. Persistence of Integrase-Deficient Lentiviral Vectors Correlates with the Induction of STING-Independent CD8+ T Cell Responses. Cell Rep. 2019, 26, 1242–1257. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, J.M.; Christos, P.J.; Michelini, Z.; Negri, D.; Cara, A.; Salvatore, M. Mucosal immunization with integrase-defective lentiviral vectors protects against influenza virus challenge in mice. PLoS ONE 2014, 9, e97270. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.J.; Cottingham, M.G.; Jenks, J.A.; Longley, R.J.; Capone, S.; Colloca, S.; Folgori, A.; Cortese, R.; Nicosia, A.; Bregu, M.; et al. Enhanced vaccine-induced CD8+ T cell responses to malaria antigen ME-TRAP by fusion to MHC class ii invariant chain. PLoS ONE 2014, 9, e100538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steitz, J.; Brück, J.; Steinbrink, K.; Enk, A.; Knop, J.; Tüting, T. Genetic immunization of mice with human tyrosinase-related protein 2: Implications for the immunotherapy of melanoma. Int. J. Cancer. 2000, 86, 89–94. [Google Scholar] [CrossRef]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Ke, Y.; Ma, H.; Kapp, J.A. Antigen is required for the activation of effector activities, whereas interleukin 2 Is required for the maintenance of memory in ovalbumin-specific, CD8+ cytotoxic T lymphocytes. J. Exp. Med. 1998, 187, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Murillo, O.; Dubrot, J.; Palazón, A.; Arina, A.; Azpilikueta, A.; Alfaro, C.; Solano, S.; Ochoa, M.C.; Berasain, C.; Gabari, I.; et al. In vivo depletion of DC impairs the anti-tumor effect of agonistic anti-CD137 mAb. Eur. J. Immunol. 2009, 39, 2424–2436. [Google Scholar] [CrossRef]
- Albershardt, T.C.; Campbell, D.J.; Parsons, A.J.; Slough, M.M.; Ter Meulen, J.; Berglund, P. LV305, a dendritic cell-targeting integration-deficient ZVex(TM)-based lentiviral vector encoding NY-ESO-1, induces potent anti-tumor immune response. Mol. Ther. Oncolytics 2016, 3, 16010. [Google Scholar] [CrossRef] [Green Version]
- Gallinaro, A.; Borghi, M.; Bona, R.; Grasso, F.; Calzoletti, L.; Palladino, L.; Cecchetti, S.; Vescio, M.F.; Macchia, D.; Morante, V.; et al. Integrase Defective Lentiviral Vector as a Vaccine Platform for Delivering Influenza Antigens. Front. Immunol. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.S.; Ferrone, C.R.; Guevara-Patiño, J.A.; Hawkins, W.G.; Dyall, R.; Engelhorn, M.E.; Wolchok, J.D.; Lewis, J.J.; Houghton, A.N. A single heteroclitic epitope determines cancer immunity after xenogeneic DNA immunization against a tumor differentiation antigen. J. Immunol. 2003, 170, 5188–5194. [Google Scholar] [CrossRef] [Green Version]
- Duperret, E.K.; Yan, J.; Weiner, D.B. Designing consensus immunogens to break tolerance to self-antigens for cancer therapy. Oncotarget 2018, 9, 35513–35514. [Google Scholar] [CrossRef] [PubMed]
- Avogadri, F.; Merghoub, T.; Maughan, M.F.; Hirschhorn-Cymerman, D.; Morris, J.; Ritter, E.; Olmsted, R.; Houghton, A.N.; Wolchok, J.D. Alphavirus replicon particles expressing TRP-2 provide potent therapeutic effect on melanoma through activation of humoral and cellular immunity. PLoS ONE 2010, 5, e12670. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joglekar, A.V.; Stein, L.; Ho, M.; Hoban, M.D.; Hollis, R.P.; Kohn, D.B. Dissecting the mechanism of histone deacetylase inhibitors to enhance the activity of zinc finger nucleases delivered by integrase-defective lentiviral vectors. Hum. Gene Ther. 2014, 25, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelascini, L.P.; Janssen, J.M.; Gonçalves, M.A. Histone deacetylase inhibition activates transgene expression from integration-defective lentiviral vectors in dividing and non-dividing cells. Hum. Gene Ther. 2013, 24, 78–96. [Google Scholar] [CrossRef]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weeks after Immunization | Mean (SFC/106 Cells) | ±SD |
|---|---|---|
| 2 | 957 | 28 |
| 8 | 1020 | 255 |
| 26 (tumor injection) | 947 | 98 |
| 32 (6 after tumor injection) | 1037 | 288 |
| 45 (19 after tumor injection) | 774 | 212 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morante, V.; Borghi, M.; Farina, I.; Michelini, Z.; Grasso, F.; Gallinaro, A.; Cecchetti, S.; Di Virgilio, A.; Canitano, A.; Pirillo, M.F.; et al. Integrase-Defective Lentiviral Vector Is an Efficient Vaccine Platform for Cancer Immunotherapy. Viruses 2021, 13, 355. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020355
Morante V, Borghi M, Farina I, Michelini Z, Grasso F, Gallinaro A, Cecchetti S, Di Virgilio A, Canitano A, Pirillo MF, et al. Integrase-Defective Lentiviral Vector Is an Efficient Vaccine Platform for Cancer Immunotherapy. Viruses. 2021; 13(2):355. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020355
Chicago/Turabian StyleMorante, Valeria, Martina Borghi, Iole Farina, Zuleika Michelini, Felicia Grasso, Alessandra Gallinaro, Serena Cecchetti, Antonio Di Virgilio, Andrea Canitano, Maria Franca Pirillo, and et al. 2021. "Integrase-Defective Lentiviral Vector Is an Efficient Vaccine Platform for Cancer Immunotherapy" Viruses 13, no. 2: 355. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020355